A Novel Inhibitor of STAT5 Signaling Overcomes Chemotherapy Resistance in Myeloid Leukemia Cells

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
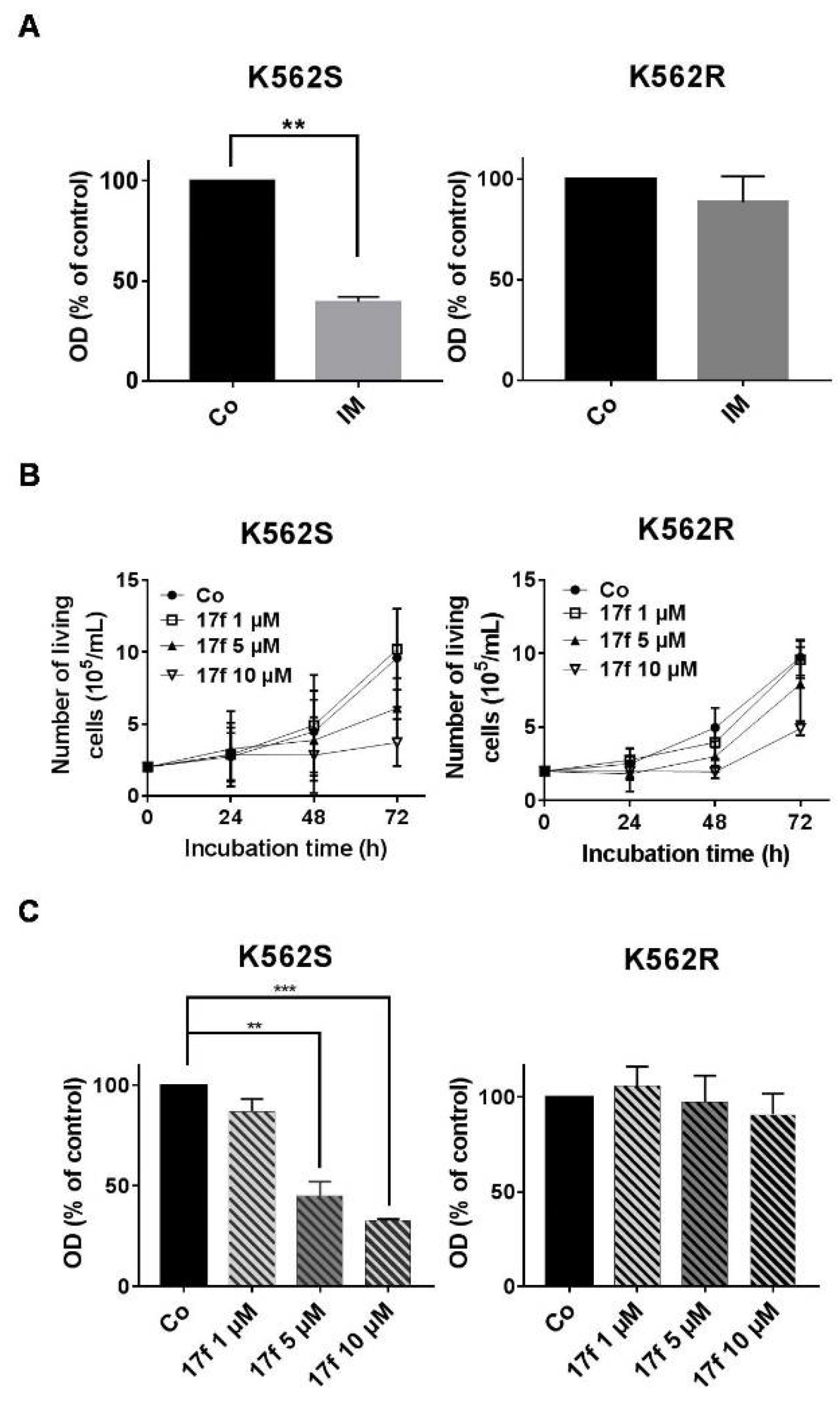
2.1. Effects of 17f Compound on Growth and Viability of IM-Sensitive and IM-Resistant BCR-ABL+ Cells
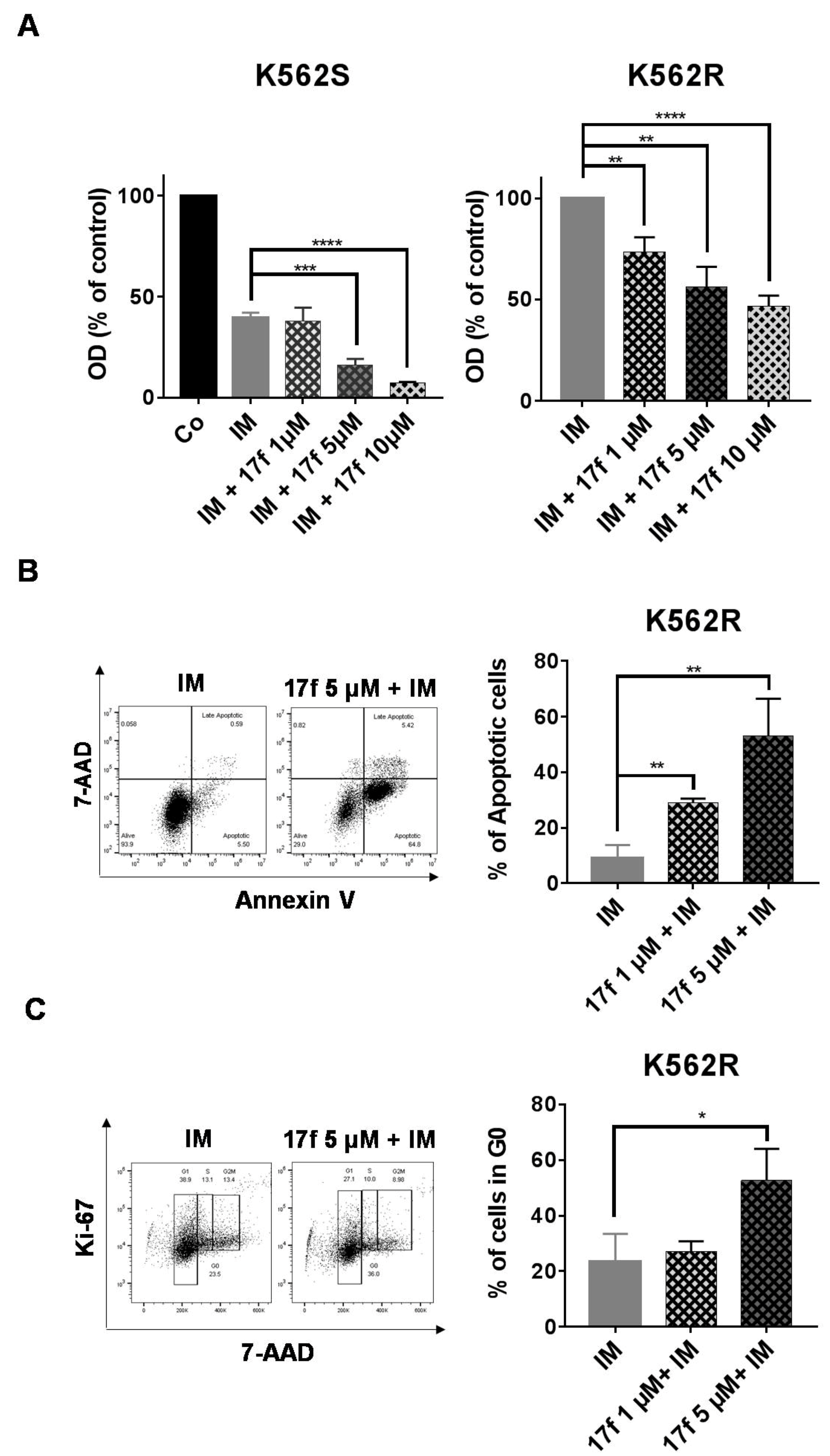
2.2. 17f Induces Apoptosis and Cell Cycle Arrest in K562R Cells and Relieves the Resistance to IM
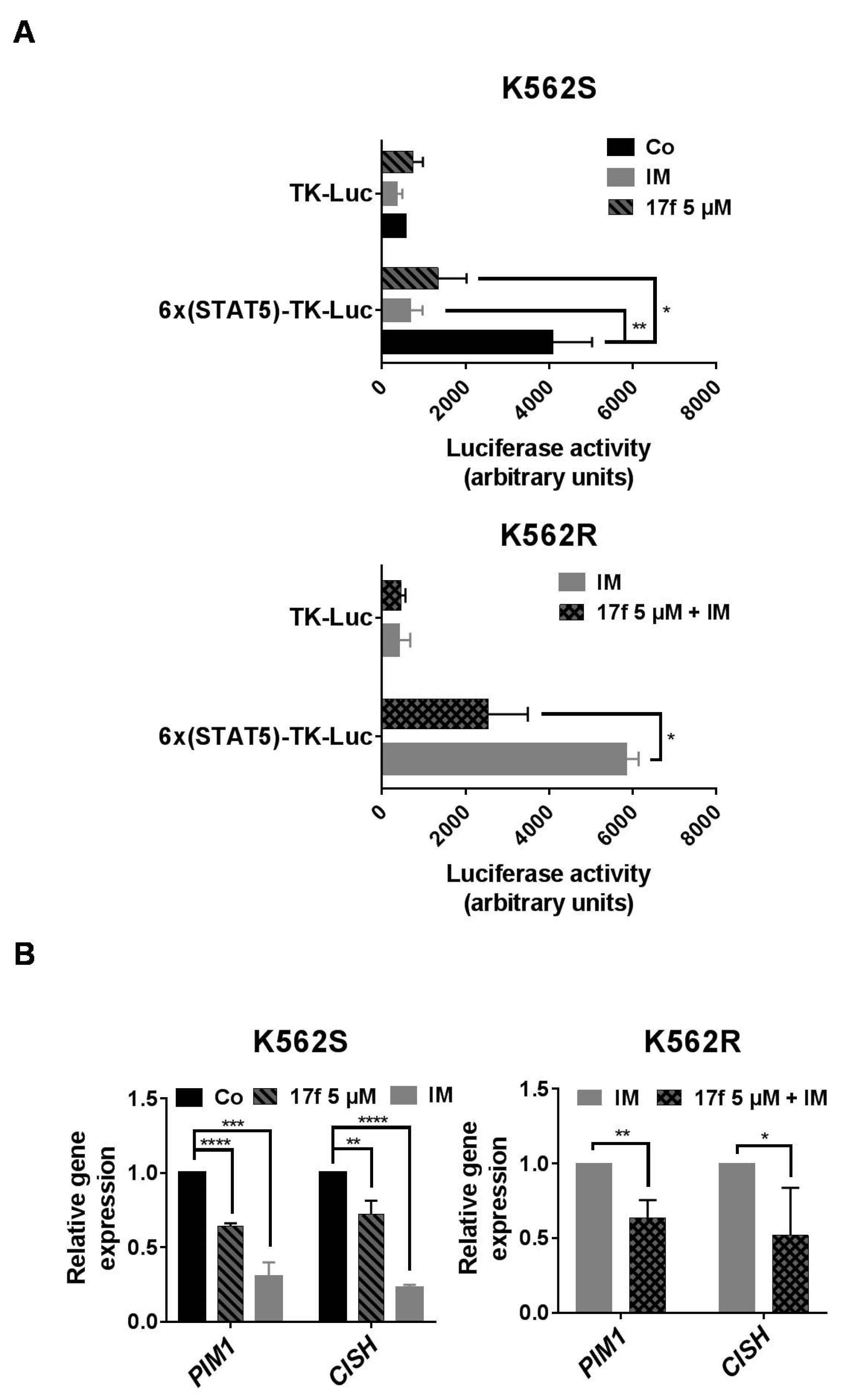
2.3. 17f Inhibits STAT5-Dependent Transcriptional Activity in K562R Cells
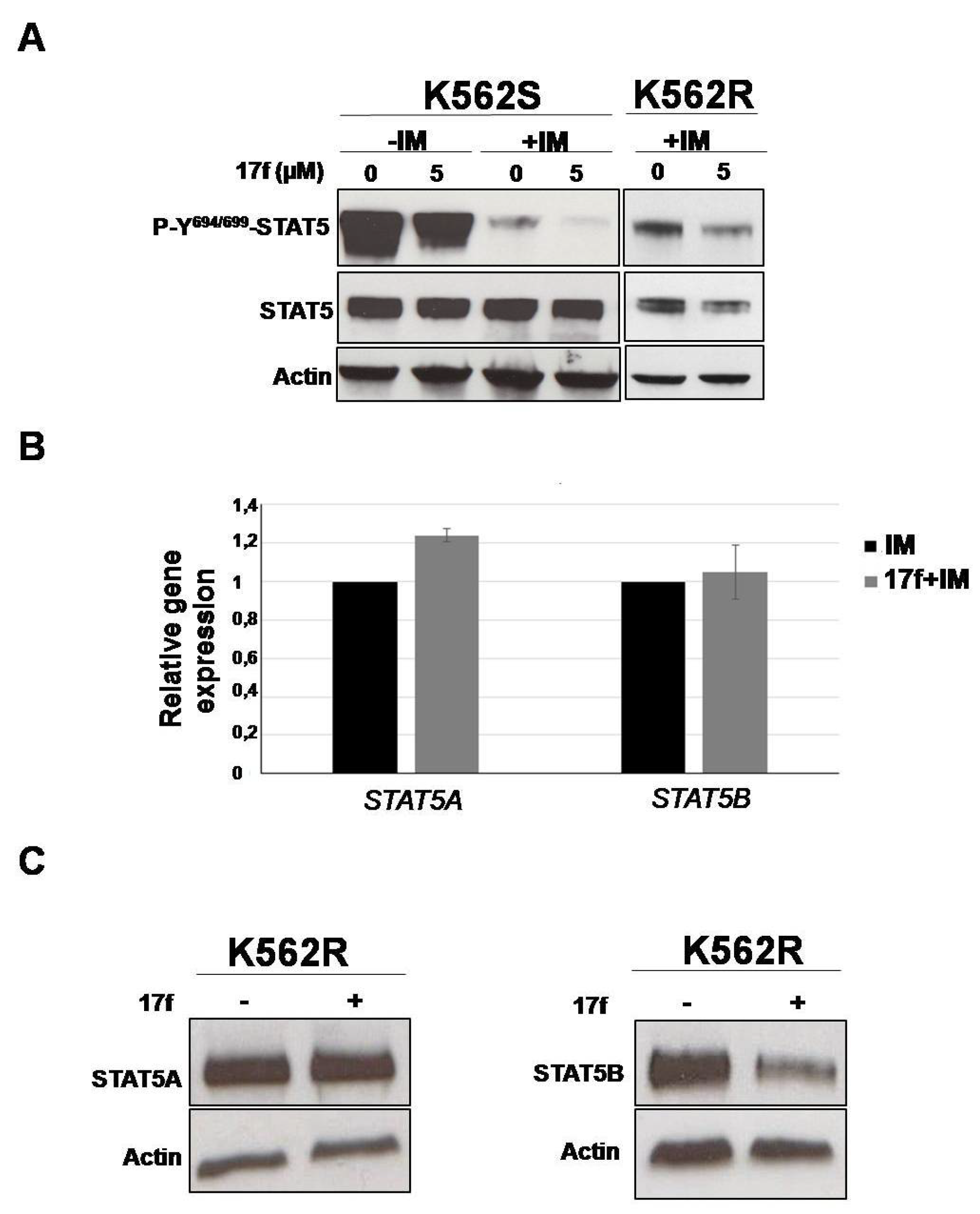
2.4. 17f Inhibits STAT5B Protein Expression in IM-Resistant K562 Cells
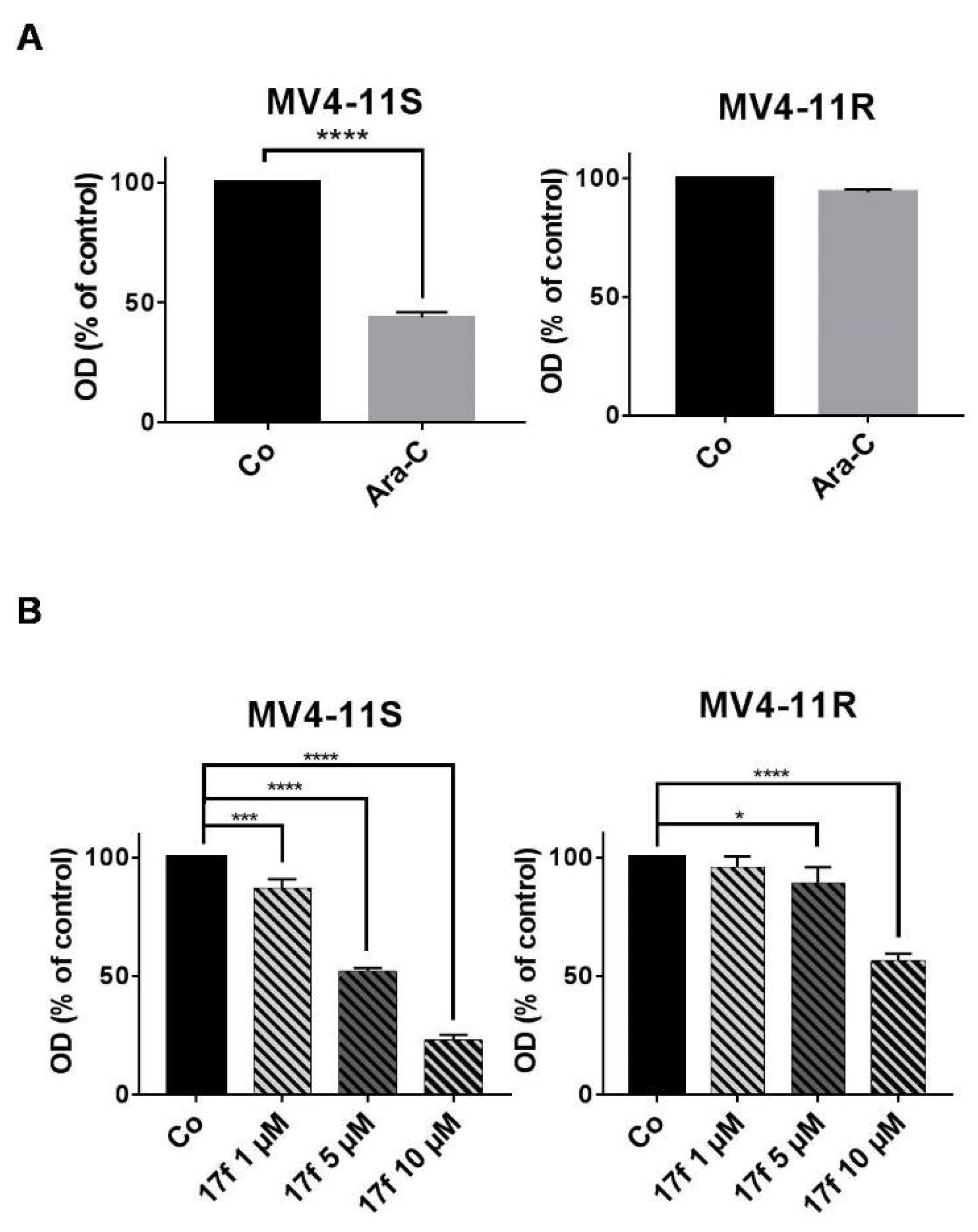
2.5. Effects of 17f on Growth and Viability of Ara-C-Sensitive and Ara-C-Resistant FLT3-ITD Expressing Leukemic Cells
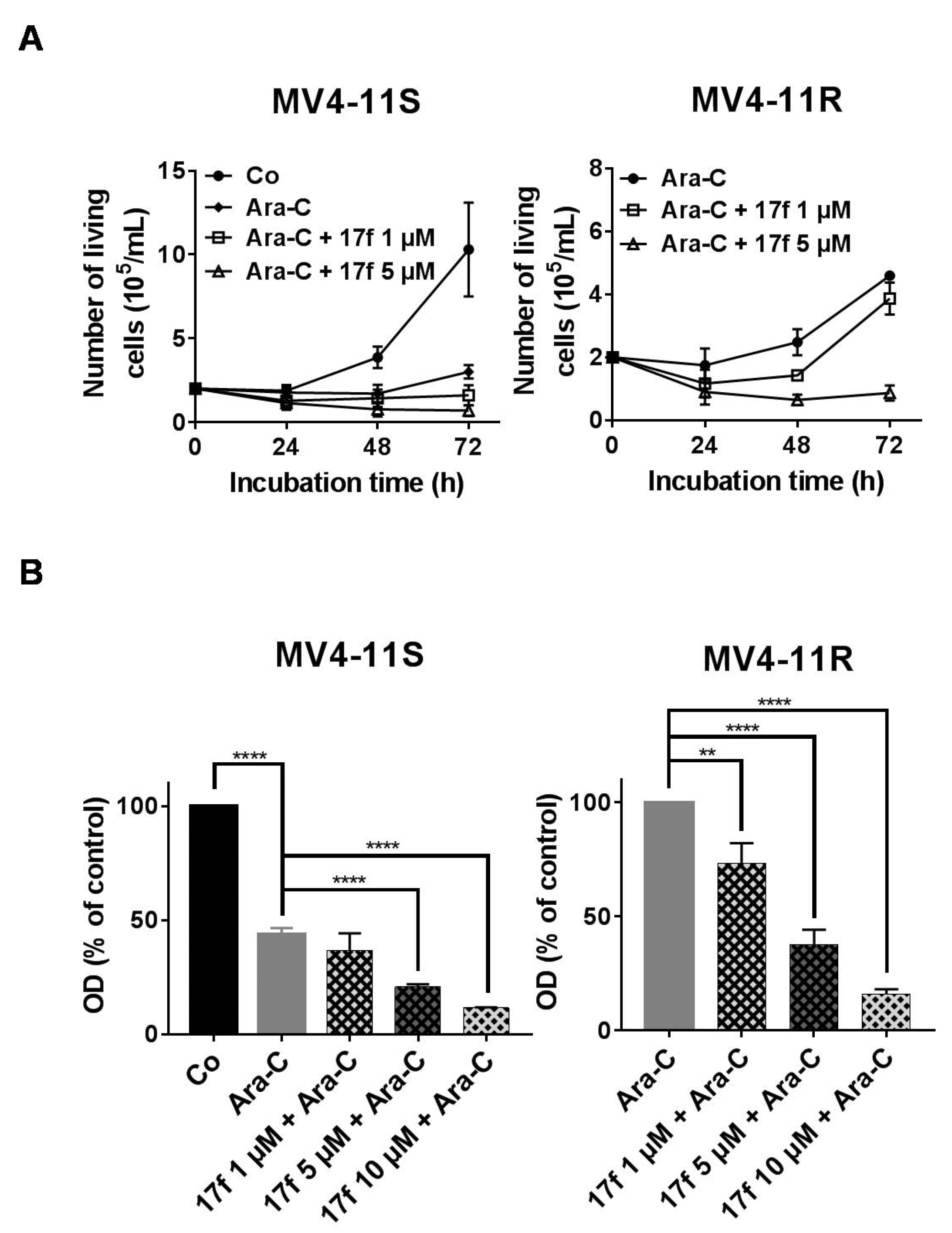
2.6. 17f Sensitizes MV4-11R Cells to Ara-C Treatment
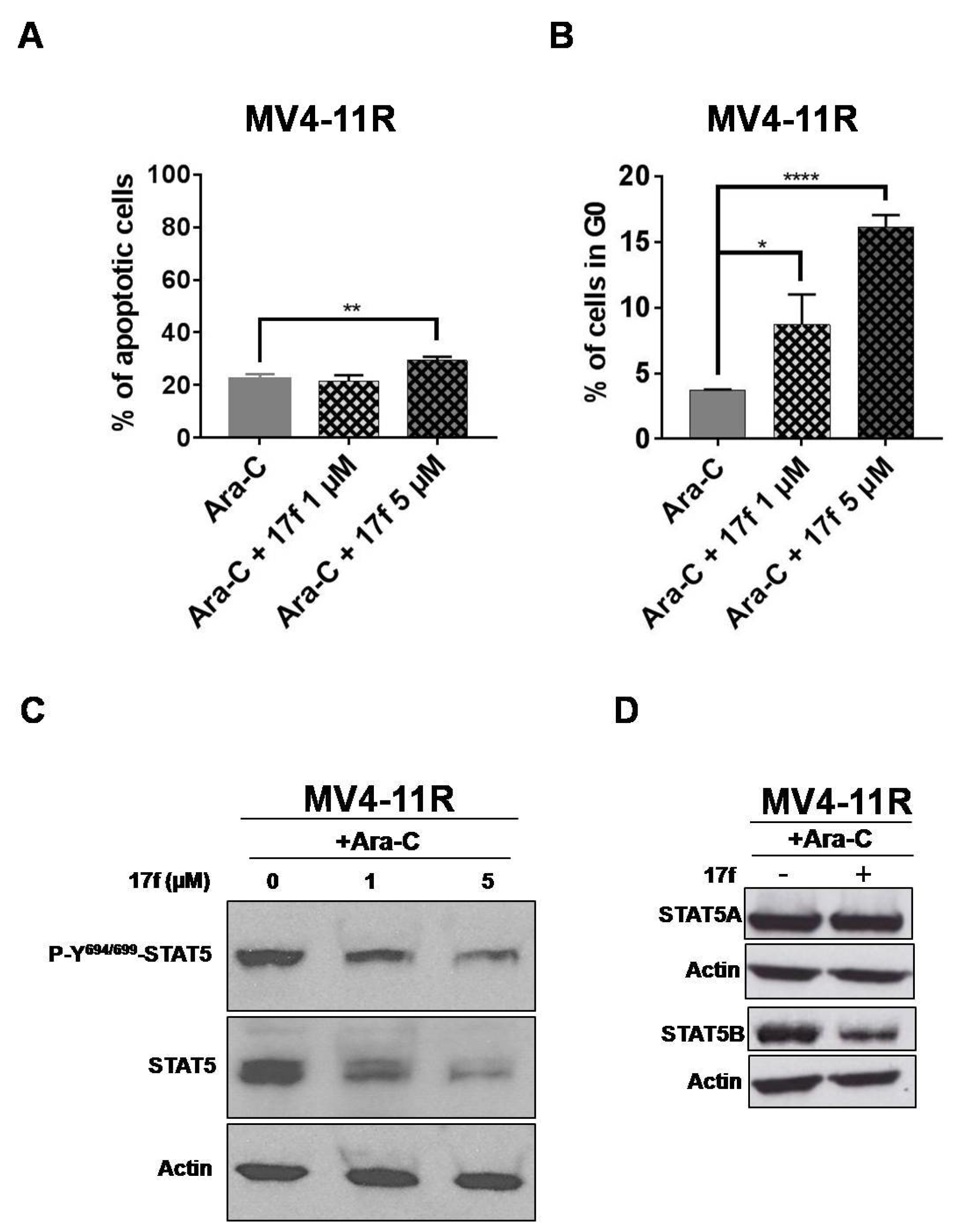
2.7. 17f Triggers Apoptosis, Cell Cycle Arrest and Inhibition of STAT5B Expression in MV4-11R Cells
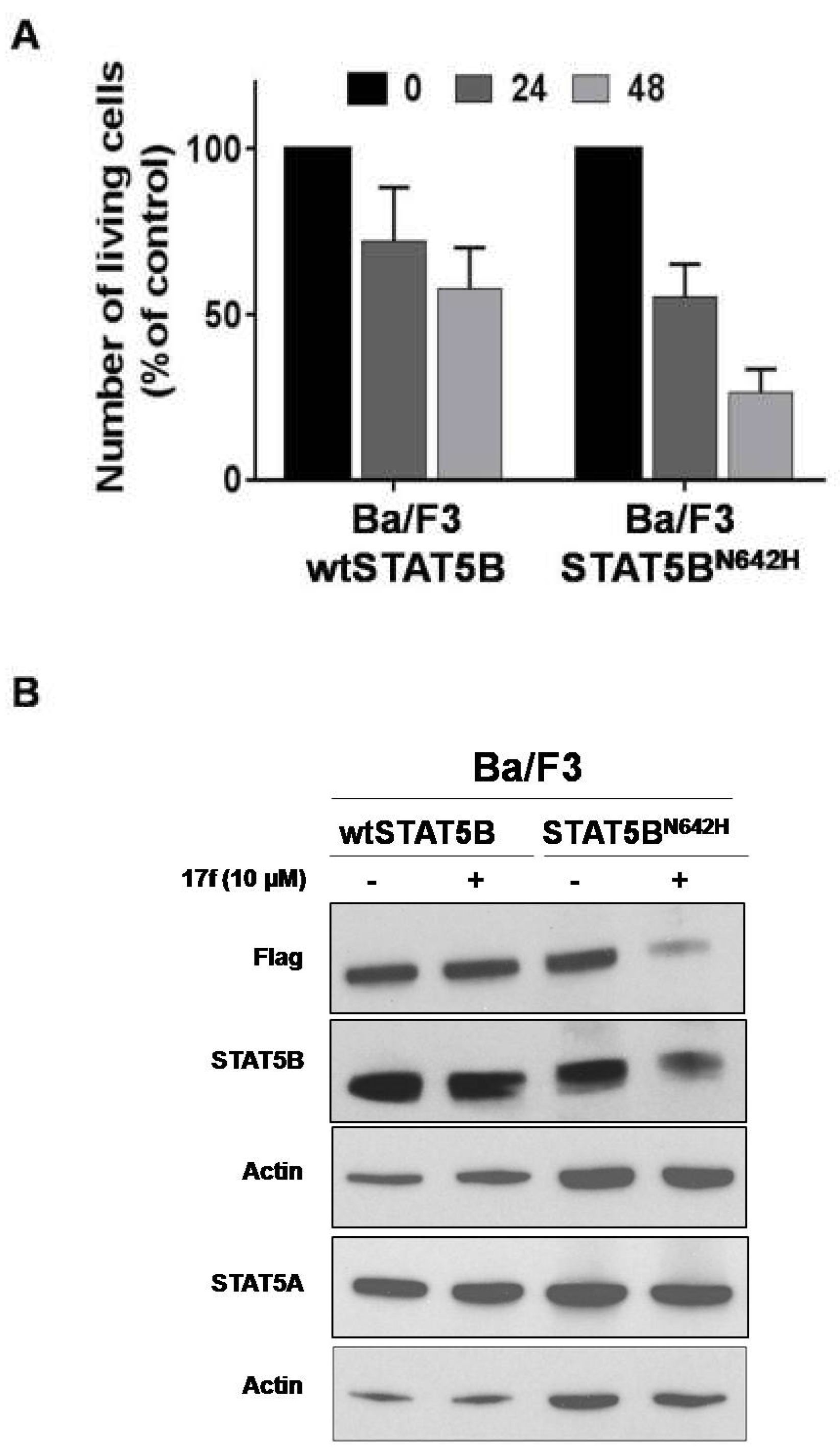
2.8. 17f Inhibits Expression of Oncogenic STAT5BN642H Mutant
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Reagents
4.2. Cell Proliferation Assays
4.3. Apoptosis and Cell Cycle Analysis
4.4. Plasmids, Transfection and Luciferase Reporter Assays
4.5. Western Blot
4.6. P-Y694/699-STAT5 Flow Cytometry Analysis
4.7. qRT-PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bunting, K.D. STAT5 signaling in normal and pathologic hematopoiesis. Front. Biosci. 2007, 12, 2807–2820. [Google Scholar] [CrossRef] [PubMed]
- Hoelbl, A.; Schuster, C.; Kovacic, B.; Zhu, B.; Wickre, M.; Hoelzl, M.A.; Fajmann, S.; Grebien, F.; Warsch, W.; Stengl, G.; et al. Stat5 is indispensable for the maintenance of Bcr/abl-positive leukemia. Eur. Mol. Biol. Organ. Mol. Med. 2010, 2, 98–110. [Google Scholar]
- Ye, D.; Wolff, N.; Li, L.; Zhang, S.; Ilaria, R.L., Jr. STAT5 signaling is required for the efficient induction and maintenance of CML in mice. Blood 2006, 107, 4917–4925. [Google Scholar] [CrossRef] [PubMed]
- Mizuki, M.; Fenski, R.; Halfter, H.; Matsumura, I.; Schmidt, R.; Müller, C.; Grüning, W.; Kratz-Albers, K.; Serve, S.; Steur, C.; et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood 2000, 96, 3907–3914. [Google Scholar] [CrossRef]
- Chomel, J.C.; Bonnet, M.L.; Sorel, N.; Sloma, I.; Bennaceur-Griscelli, A.; Rea, D.; Legros, L.; Marfaing-Koka, A.; Bourhis, J.H.; Ame, S.; et al. Leukemic stem cell persistence in chronic myeloid leukemia patients in deep molecular response induced by tyrosine kinase inhibitors and the impact of therapy discontinuation. Oncotarget 2016, 7, 35293–35301. [Google Scholar] [CrossRef]
- Gambacorti-Passerini, C.B.; Gunby, R.H.; Piazza, R.; Galietta, A.; Rostagno, R.; Scapozza, L. Molecular mechanisms of resistance to imatinib in Philadelphia-chromosome-positive leukaemias. Lancet Oncol. 2003, 4, 75–85. [Google Scholar] [CrossRef]
- Cumaraswamy, A.A.; Todic, A.; Resetca, D.; Minden, M.D.; Gunning, P.T. Inhibitors of Stat5 protein signaling. Med. Chem. Commun. 2012, 3, 22–27. [Google Scholar] [CrossRef]
- Warsch, W.; Kollmann, K.; Eckelhart, E.; Fajmann, S.; Cerny-Reiterer, S.; Hölbl, A.; Gleixner, K.V.; Dworzak, M.; Mayerhofer, M.; Hoermann, G.; et al. High STAT5 levels mediate imatinib resistance and indicate disease progression in chronic myeloid leukemia. Blood 2011, 117, 3409–3420. [Google Scholar] [CrossRef]
- Warsch, W.; Grundschober, E.; Berger, A.; Gille, L.; Cerny-Reiterer, S.; Tigan, A.-S.; Hoelbl-Kovacic, A.; Valent, P.; Moriggl, R.; Sexl, V. STAT5 triggers BCR-ABL1 mutation by mediating ROS production in chronic myeloid leukemia. Oncotarget 2012, 3, 1669–1687. [Google Scholar] [CrossRef]
- Prost, S.; Relouzat, F.; Spentchian, M.; Ouzegdouh, Y.; Saliba, J.; Massonnet, G.; Beressi, J.P.; Verhoeyen, E.; Raggueneau, V.; Maneglier, B.; et al. Erosion of the chronic myeloid leukemia stem cell pool by PPARγ agonists. Nature 2015, 525, 380–383. [Google Scholar] [CrossRef]
- Nelson, E.A.; Walker, S.R.; Weisberg, E.; Bar-Natan, M.; Barrett, R.; Gashin, L.B.; Terrell, S.; Klitgaard, J.L.; Santo, L.; Addorio, M.R.; et al. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood 2011, 117, 3421–3429. [Google Scholar] [CrossRef] [PubMed]
- Mistry, H.; Hsieh, G.; Buhrlage, S.J.; Huang, M.; Park, E.; Cuny, G.D.; Galinsky, I.; Stone, R.M.; Gray, N.S.; D’Andrea, A.D.; et al. Small-molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol. Cancer Ther. 2013, 12, 2651–2662. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.; Scuto, A.; Yang, F.; Chen, W.; Park, S.; Yoo, H.S.; Konig, H.; Bhatia, R.; Cheng, X.; Merz, K.H.; et al. Indirubin derivatives induce apoptosis of chronic myelogenous leukemia cells involving inhibition of Stat5 signaling. Mol. Oncol. 2012, 6, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Page, B.D.; Khoury, H.; Laister, R.C.; Fletcher, S.; Vellozo, M.; Manzoli, A.; Yue, P.; Turkson, J.; Minden, M.D.; Gunning, P.T. Small molecule STAT5-SH2 domain inhibitors exhibit potent antileukemia activity. J. Med. Chem. 2012, 55, 1047–1055. [Google Scholar] [CrossRef]
- Cumaraswamy, A.A.; Lewis, A.M.; Geletu, M.; Todic, A.; Diaz, D.B.; Cheng, X.R.; Brown, C.E.; Laister, R.C.; Muench, D.; Kerman, K.; et al. Nanomolar-potency small molecule inhibitor of STAT5 protein. Am. Chem. Soc. Med. Chem. Lett. 2014, 5, 1202–1206. [Google Scholar] [CrossRef]
- Elumalai, N.; Berg, A.; Rubner, S.; Blechschmidt, L.; Song, C.; Natarajan, K.; Matysik, J.; Berg, T. Rational development of Stafib-2: A selective, nanomolar inhibitor of the transcription factor STAT5b. Sci. Rep. 2017, 7, 819. [Google Scholar] [CrossRef]
- Wingelhofer, B.; Maurer, B.; Heyes, E.C.; Cumaraswamy, A.A.; Berger-Becvar, A.; de Araujo, E.D.; Orlova, A.; Freund, P.; Ruge, F.; Park, J.; et al. Pharmacologic inhibition of STAT5 in acute myeloid leukemia. Leukemia 2018, 32, 1135–1146. [Google Scholar] [CrossRef]
- Prost, S.; Le Dantec, M.; Augé, S.; Le Grand, R.; Derdouch, S.; Auregan, G.; Déglon, N.; Relouzat, F.; Aubertin, A.M.; Maillere, B.; et al. Human and simian immunodeficiency viruses deregulate early hematopoiesis through a Nef/PPARγ/STAT5 signaling pathway in macaques. J. Clin. Investig. 2008, 118, 1765–1775. [Google Scholar] [CrossRef]
- Liu, H.; Zang, C.; Fenner, M.H.; Liu, D.; Possinger, K.; Koeffler, H.P.; Elstner, E. Growth inhibition and apoptosis in human Philadelphia chromosome-positive lymphoblastic leukemia cell lines by treatment with the dual PPARα/γ ligand TZD18. Blood 2006, 107, 3683–3692. [Google Scholar] [CrossRef]
- Bertz, J.; Zang, C.; Liu, H.; Wächter, M.; Possinger, K.; Koeffler, H.P.; Elstner, E. Compound 48, a novel dual PPAR α/γ ligand, inhibits the growth of human CML cell lines and enhances the anticancer-effects of imatinib. Leuk. Res. 2009, 33, 686–692. [Google Scholar] [CrossRef]
- Parmenon, C.; Guillard, J.; Caignard, D.-H.; Hennuyer, N.; Staels, B.; Audinot-Bouchez, V.; Boutin, J.A.; Dacquet, C.; Ktorza, A.; Viaud-Massuard, M.C. 4,4-Dimethyl-1,2,3,4-Tetrahydroquinoline-Based PPARα/γ agonists. Part I: Synthesis and pharmacological evaluation. Bioorg. Med. Chem. Lett. 2008, 18, 1617–1622. [Google Scholar] [CrossRef]
- Parmenon, C.; Guillard, J.; Caignard, D.-H.; Hennuyer, N.; Staels, B.; Audinot-Bouchez, V.; Boutin, J.A.; Dacquet, C.; Ktorza, A.; Viaud-Massuard, M.C. 4,4-Dimethyl-1,2,3,4-Tetrahydroquinoline-Based PPARα/γ agonists. Part II: Synthesis and pharmacological evaluation of oxime and acidic head group structural variations. Bioorg. Med. Chem. Lett. 2009, 19, 2683–2687. [Google Scholar] [CrossRef]
- Juen, L.; Brachet-Botineau, M.; Parmenon, C.; Bourgeais, J.; Hérault, O.; Gouilleux, F.; Viaud-Massuard, M.C.; Prié, G. New inhibitor targeting signal transducer and activator of transcription 5 (STAT5) signaling in myeloid leukemias. J. Med. Chem. 2017, 60, 6119–6136. [Google Scholar] [CrossRef]
- Kontro, M.; Kuusanmäki, H.; Eldfors, S.; Burmeister, T.; Andersson, E.I.; Bruserud, O.; Brümmendorf, T.H.; Edgren, H.; Gjertsen, B.T.; Itälä-Remes, M.; et al. Novel activating STAT5B mutations as putative drivers of T-cell acute lymphoblastic leukemia. Leukemia 2014, 28, 1738–1742. [Google Scholar] [CrossRef]
- Bandapalli, O.R.; Schuessele, S.; Kunz, J.B.; Rausch, T.; Stütz, A.M.; Tal, N.; Geron, I.; Gershman, N.; Izraeli, S.; Eilers, J.; et al. The activating STAT5B N642H mutation is a common abnormality in pediatric T-cell acute lymphoblastic leukemia and confers a higher risk of relapse. Haematologica 2014, 99, 188–192. [Google Scholar] [CrossRef]
- Nicolae, A.; Xi, L.; Pittaluga, S.; Abdullaev, Z.; Pack, S.D.; Chen, J.; Waldmann, T.A.; Jaffe, E.S.; Raffeld, M. Frequent STAT5B mutations in γδ hepatosplenic T-cell lymphomas. Leukemia 2014, 28, 2244–2248. [Google Scholar] [CrossRef]
- Pham, H.T.T.; Maurer, B.; Prchal-Murphy, M.; Grausenburger, R.; Grundschober, E.; Javaheri, T.; Nivarthi, H.; Boersma, A.; Kolbe, T.; Elabd, M.; et al. STAT5BN642H is a driver mutation for T cell neoplasia. J. Clin. Investig. 2018, 128, 387–401. [Google Scholar] [CrossRef]
- Liao, Z.; Nevalainen, M.T. Targeting transcription factor Stat5a/b as a therapeutic strategy for prostate cancer. Am. J. Transl. Res. 2011, 3, 133–138. [Google Scholar]
- Ko, Y.C.; Hu, C.Y.; Liu, Z.H.; Tien, H.F.; Ou, D.L.; Chien, H.F.; Lin, L.I. Cytarabine-Resistant FLT3-ITD Leukemia Cells are Associated with TP53 Mutation and Multiple Pathway Alterations—Possible Therapeutic Efficacy of Cabozantinib. Int. J. Mol. Sci. 2019, 20, 1230. [Google Scholar] [CrossRef]
- Goh, E.L.; Zhu, T.; Leong, W.Y.; Lobie, P.E. c-Cbl is a negative regulator of GH-stimulated STAT5-mediated transcription. Endocrinology 2002, 143, 3590–3603. [Google Scholar] [CrossRef][Green Version]
- Chen, Y.; Dai, X.; Haas, A.L.; Wen, R.; Wang, D. Proteasome-dependent down-regulation of activated Stat5A in the nucleus. Blood 2006, 108, 566–574. [Google Scholar] [CrossRef]
- Rathinam, C.; Thien, C.B.; Langdon, W.Y.; Gu, H.; Flavell, R.A. The E3 ubiquitin ligase c-Cbl restricts development and functions of hematopoietic stem cells. Genes Dev. 2008, 22, 992–997. [Google Scholar] [CrossRef]
- Moulick, K.; Ahn, J.H.; Zong, H.; Rodina, A.; Cerchietti, L.; Gomes DaGama, E.M.; Caldas-Lopes, E.; Beebe, K.; Perna, F.; Hatzi, K.; et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat. Chem. Biol. 2011, 7, 818–826. [Google Scholar] [CrossRef]
- Guo, F.; Sigua, C.; Bali, P.; George, P.; Fiskus, W.; Scuto, A.; Annavarapu, S.; Mouttaki, A.; Sondarva, G.; Wei, S.; et al. Mechanistic role of heat shock protein 70 in Bcr-Abl-mediated resistance to apoptosis in human acute leukemia cells. Blood 2005, 105, 1246–1255. [Google Scholar] [CrossRef]
- Wu, L.; Yu, J.; Chen, R.; Liu, Y.; Lou, L.; Wu, Y.; Huang, L.; Fan, Y.; Gao, P.; Huang, M.; et al. Dual inhibition of Bcr-Abl and Hsp90 by C086 potently inhibits the proliferation of imatinib-resistant CML cells. Clin. Cancer Res. 2015, 21, 833–843. [Google Scholar] [CrossRef]
- Yao, Q.; Nishiuchi, R.; Kitamura, T.; Kersey, J.H. Human leukemias with mutated FLT3 kinase are synergistically sensitive to FLT3 and Hsp90 inhibitors: The key role of the STAT5 signal transduction pathway. Leukemia 2005, 19, 1605–1612. [Google Scholar] [CrossRef]
- Katayama, K.; Noguchi, K.; Sugimoto, Y. Heat shock protein 90 inhibitors overcome the resistance to Fms-like tyrosine kinase 3 inhibitors in acute myeloid leukemia. Oncotarget 2018, 9, 34240–34258. [Google Scholar] [CrossRef]
- Pocaly, M.; Lagarde, V.; Etienne, G.; Ribeil, J.A.; Claverol, S.; Bonneu, M.; Moreau-Gaudry, F.; Guyonnet-Duperat, V.; Hermine, O.; Melo, J.V.; et al. Overexpression of the heat-shock protein 70 is associated to imatinib resistance in chronic myeloid leukemia. Leukemia 2007, 21, 93–101. [Google Scholar] [CrossRef]
- Dhennin-Duthille, I.; Nyga, R.; Yahiaoui, S.; Gouilleux-Gruart, V.; Régnier, A.; Lassoued, K.; Gouilleux, F. The tumor suppressor hTid1 inhibits STAT5b activity via functional interaction. J. Biol. Chem. 2011, 286, 5034–5042. [Google Scholar] [CrossRef]
- Bae, M.K.; Jeong, J.W.; Kim, S.H.; Kim, S.Y.; Kang, H.J.; Kim, D.M.; Bae, S.K.; Yun, I.; Trentin, G.A.; Rozakis-Adcock, M.; et al. Tid-1 interacts with the von Hippel-Lindau protein and modulates angiogenesis by destabilization of HIF-1α. Cancer Res. 2005, 65, 2520–2525. [Google Scholar] [CrossRef]
- Schaller-Schönitz, M.; Barzan, D.; Williamson, A.J.; Griffiths, J.R.; Dallmann, I.; Battmer, K.; Ganser, A.; Whetton, A.D.; Scherr, M.; Eder, M. BCR-ABL affects STAT5A and STAT5B differentially. PLoS ONE 2014, 9, e97243. [Google Scholar] [CrossRef]
- Kollmann, S.; Grundschober, E.; Maurer, B.; Warsch, W.; Grausenburger, R.; Edlinger, L.; Huuhtanen, J.; Lagger, S.; Hennighausen, L.; Valent, P.; et al. Twins with different personalities: STAT5B-but not STAT5A-has a key role in BCR/ABL-induced leukemia. Leukemia 2019, 33, 1583–1597. [Google Scholar] [CrossRef]
- Park, H.J.; Li, J.; Hannah, R.; Biddie, S.; Leal-Cervantes, A.I.; Kirschner, K.; Flores Santa Cruz, D.; Sexl, V.; Göttgens, B.; Green, A.R. Cytokine-induced megakaryocytic differentiation is regulated by genome-wide loss of a uSTAT transcriptional program. Eur. Mol. Biol. Organ. J. 2016, 35, 580–594. [Google Scholar] [CrossRef]
- Nagy, Z.S.; Rui, H.; Stepkowski, S.M.; Karras, J.; Kirken, R.A. A preferential role for STAT5, not constitutively active STAT3, in promoting survival of a human lymphoid tumor. J. Immunol. 2006, 177, 5032–5040. [Google Scholar] [CrossRef]
- Vignon, C.; Debeissat, C.; Georget, M.-T.; Bouscary, D.; Gyan, E.; Rosset, P.; Herault, O. Flow cytometric quantification of all phases of the cell cycle and apoptosis in a two-color fluorescence plot. PLoS ONE 2013, 7, e68425. [Google Scholar] [CrossRef]
- Moriggl, R.; Gouilleux-Gruart, V.; Jähne, R.; Berchtold, S.; Gartmann, C.; Liu, X.; Hennighausen, L.; Sotiropoulos, A.; Groner, B.; Gouilleux, F. Deletion of the carboxyl-terminal transactivation domain of MGF-Stat5 results in sustained DNA binding and a dominant negative phenotype. Mol. Cell. Biol. 1996, 16, 5691–5700. [Google Scholar] [CrossRef][Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brachet-Botineau, M.; Deynoux, M.; Vallet, N.; Polomski, M.; Juen, L.; Hérault, O.; Mazurier, F.; Viaud-Massuard, M.-C.; Prié, G.; Gouilleux, F. A Novel Inhibitor of STAT5 Signaling Overcomes Chemotherapy Resistance in Myeloid Leukemia Cells. Cancers 2019, 11, 2043. https://doi.org/10.3390/cancers11122043
Brachet-Botineau M, Deynoux M, Vallet N, Polomski M, Juen L, Hérault O, Mazurier F, Viaud-Massuard M-C, Prié G, Gouilleux F. A Novel Inhibitor of STAT5 Signaling Overcomes Chemotherapy Resistance in Myeloid Leukemia Cells. Cancers. 2019; 11(12):2043. https://doi.org/10.3390/cancers11122043
Chicago/Turabian StyleBrachet-Botineau, Marie, Margaux Deynoux, Nicolas Vallet, Marion Polomski, Ludovic Juen, Olivier Hérault, Frédéric Mazurier, Marie-Claude Viaud-Massuard, Gildas Prié, and Fabrice Gouilleux. 2019. "A Novel Inhibitor of STAT5 Signaling Overcomes Chemotherapy Resistance in Myeloid Leukemia Cells" Cancers 11, no. 12: 2043. https://doi.org/10.3390/cancers11122043
APA StyleBrachet-Botineau, M., Deynoux, M., Vallet, N., Polomski, M., Juen, L., Hérault, O., Mazurier, F., Viaud-Massuard, M.-C., Prié, G., & Gouilleux, F. (2019). A Novel Inhibitor of STAT5 Signaling Overcomes Chemotherapy Resistance in Myeloid Leukemia Cells. Cancers, 11(12), 2043. https://doi.org/10.3390/cancers11122043