Abstract
The use of transgenic mouse models has revolutionized the study of many human diseases. However, murine models are limited in their representation of spontaneously arising tumors and often lack key clinical signs and pathological changes. Thus, a closer representation of complex human diseases is of high therapeutic relevance. Given the high failure rate of drugs at the clinical trial phase (i.e., around 90%), there is a critical need for additional clinically relevant animal models. Companion animals like cats and dogs display chronic inflammatory or neoplastic diseases that closely resemble the human counterpart. Cat and dog patients can also be treated with clinically approved inhibitors or, if ethics and drug safety studies allow, pilot studies can be conducted using, e.g., inhibitors of the evolutionary conserved JAK-STAT pathway. The incidence by which different types of cancers occur in companion animals as well as mechanisms of disease are unique between humans and companion animals, where one can learn from each other. Taking advantage of this situation, existing inhibitors of known oncogenic STAT3/5 or JAK kinase signaling pathways can be studied in the context of rare human diseases, benefitting both, the development of drugs for human use and their application in veterinary medicine.
1. Introduction
Almost half of all households in the United States have at least one companion animal. This means that approximately 77 million dogs and 58 million cats share a common environment with their human owners and are largely exposed to the same health risk factors [1]. In the absence of significant cardiovascular disease, cancer is the number one cause of death for dogs, killing between 40% and 50% of individuals older than 10 years, and between 20% and 25% regardless of age [2,3,4]. Numbers are less detailed for cats, but the overall tumor incidence ranges between 30% and 35% [5]. The prevalence of cancer in companion animals has increased in the last decades, which may be the result of a real increase in cancer incidence, an increase in the population of companion animals at risk or the awareness and willingness of the animal owners to pursue diagnostic and treatment options [6]. While the full spectrum of tumor types seen in humans also occurs in cats and dogs, the rates for individual tumor types are often different. Canine osteosarcoma, soft tissue sarcoma and feline non-Hodgkin’s lymphoma for example are significantly higher than in humans (Table 1), whereas other tumor entities like lung, prostate and colon tumors are rare in companion animals. Cancer in companion animals, particularly in dogs, resembles cancer in humans in many ways, including spontaneous disease occurring without an isogenic background or genetic engineering and shared environmental and societal status with owners. Further similarities in chronology of the disease adapted to lifespan, organization into various well-characterized breeds that show different incidences of tumor types, and shared environmental and societal status with owners makes them attractive objects for comparison.

Table 1.
Incidence rates of various tumor types from human, dog and cat.
The mouse has been an extremely useful tool to gain cellular and mechanistic understanding into the development of cancer [17]. The concept of oncogenes and tumor suppressors balancing cellular proliferation of multicellular organisms has been proven in vivo in genetically modified mice [18,19]. The use of mice deleted for a single gene has allowed us to determine the involvement of signaling pathways, genetic regulators like transcription factors, epigenetic factors, etc. in the development and sustained growth of cancer. Overall, experiments in mouse models have been invaluable in understanding the mechanistic basis of cancer biology. However, while offering critical insights into basic concepts, murine models underrepresent the heterogeneity and complex interplay between human immune and cancer cells [20,21].
The Janus kinase (JAK)—signal transducer and activator of transcription (STAT) signaling pathway—provides a fast and efficient way for relaying signals from the extracellular space to the nucleus and modifying gene expression [22]. Main targets of this pathway represent regulators of cell division and apoptosis as broadly discussed in several publications in this special issue [23,24,25]. As these are central aspects in the development of cancer, it is not surprising that members of this pathway are over-activated in many cancers [26,27]. The kinases of this pathway, JAK 1-3 and TYK2 are constantly activated in several different tumor types and are subject to pharmacological inhibition. Recently, also STAT proteins have come into focus of cancer researchers. Particularly STAT3 and STAT5 are activated in over 70% of all human cancer types and constitute a critical node in the signaling networks of tumor cells [28].
This review will highlight, why companion animals and particularly the dog represent an attractive link between murine models, addressing basic mechanistic aspects and human diseases specifically in the context of JAK-STAT signaling.
2. Preclinical Models
The vast majority of cancer models currently are represented by mice, and their fundamental importance for preclinical research is clearly established [20,29]. These animals come as various strains that have been inbred over many generations and thereby are genetically highly homogenous [30]. When used for experiments, they are matched for age, sex and size, receive the same sterile diets and are housed under specific pathogen-free (SPF) conditions. All of these factors are controlled for in order to generate standardized conditions that allow us to draw scientifically sound conclusions based on the variation of one single parameter [31]. Furthermore, this reduction of variables and thereby “noise” allows us to reduce the number of animals necessary to reach statistically significant results. While this concept has proven its scientific merits and is logical within itself, it is questionable, whether young, sex-matched and inbred mice on sterile nutrition are a good representation of the typically older, obese and genetically diverse human cancer patients [32].
The controlled environment also affects the outcome of cancer-related experiments. For example, it has become clear recently, that the microbiome influences the response to cancer treatment [33,34]. Accordingly, mice raised under SPF conditions in various research institutions show differences in the composition of their gut microbiome, affecting tumor growth rates [35]. Again, while differences in the gut microbiome are likely to exist in human cancer patients, the single standardized composition under which these experiments are carried out in laboratory mice are very likely no close representation of the much broader spectrum in humans. Additionally, rodents have adopted to most ecosystems metabolically, but in regard to drug metabolism, due to growth-hormone regulated p450 cytochrome components, pharmacodynamic and pharmacokinetic properties, canines are by far superior and therefore used as key models for FDA-drug approved testing [36].
Another important aspect is the genetic background and its modification particularly with respect to the immune system. Mice used for tumor experiments usually are highly inbred, reducing genetic variability [37]. Experiments regarding the consequences of a narrow genetic background have demonstrated potential phenotypic tilting, which may result in unrepresentative biased phenotypes [38]. Furthermore, the buffering of genetic variation, including disease-causing mutations, is impaired under these circumstances [39]. One effort to overcome limitations due to a narrow genetic background is to establish new reference populations derived from the crossing of several different mouse strains, as exemplified by the Collaborative Cross project [40]. By broadening and defining the genetic basis, it offers the perspective of enhancing genetic stability and reproducibility, thereby also representing a new and potentially better resource of murine models for human diseases.
Additionally, and probably of higher practical relevance is the fact that development and proliferation of tumor cells happens in a complex interplay with cells of the immune system [41]. However, cancer-studies in mice are often performed in the absence of a fully functional immune system, using immunocompromised mice as hosts for transplantations of human cell lines, patient-derived xenografts or human tumor-derived immune cell xenografts [42]. The immune system constitutes the major player in the counter-selection against tumor cells, thereby necessitating evasion or adaption strategies on the side of the tumor cells. Since this aspect is missing in such mouse models, the results from experiments thereof likely reflect only partial aspects of tumor biology and challenge their biological relevance.
One way to overcome these problems are humanized mice, which express human instead of murine components of the immune system like major histocompatibility complex, allowing the transplantation of tumor cells in an immunologically at least partially competent environment [43]. These models clearly represent a step forward, however, they are costly, technically complicated and still do not represent all components of a functional and homogenous immune system [44]. Moreover, human patients that have developed cancer are not living under special pathogen-free conditions and are very likely to have chronic viral infections, like the Epstein–Barr virus, cytomegalovirus or herpes simplex, which are present in up to 90% of the total population [45,46,47]. Such chronic infections exert a constant pressure and shape the immune system, which is not the case in mice under SPF conditions. Co-housing of “dirty” outbred immunocompetent mice could be a way for improvement, still mammalian viral species barriers exist, that make companion animals superior in these infectious aspects.
Finally, genetically engineered mice harboring the deletion of a tumor suppressor gene or the ectopic expression of an oncogene or combinations thereof can be used [48]. In this case, the tumors develop in the presence of a competent immune system and problems related to cross-species compatibility do not arise. This setting also enables the introduction of defined mutations that occur in human tumors. However, this comes at the price of costly and time-consuming development, often requiring years of work before availability. Furthermore, whereas the targeted insertion of defined mutations reflecting the human situation is a clear advantage, cancer development is a multi-step process, and the heterogeneity of these further steps is often different between engineered mice and human patients [49,50].
Overall, the mouse has been highly instructive in determining genes involved and their mechanistic contribution to the origin and development of cancer [51]. However, the limitations of this model, in particular the differences in environment and microbiome, life span, tumor etiology and genetic status may be the reason why certain aspects are not reflected closely in this model, resulting in only 11% of oncology drugs that work in mice being approved for human use [52]. Therefore, it is desirable from a translational perspective to add another layer that closer reflects human biology and cancer development.
3. Advantages and Disadvantages of Canine Tumor Models
Roughly 4,000,000 dogs and a similar number of cats are diagnosed with cancer each year in the U.S. [53]. Although exact epidemiological data are not available for companion animals, this translates into approximately 5300 cases per 100,000 dogs, which is around ten times higher than in humans with 500 cases per 100,000 persons. This large number of pets provides the opportunity to study spontaneous cancers that are highly similar to those occurring in humans, especially since most pet owners are highly motivated to seek out novel treatments for their companion animals.
There are several advantages of using companion animals as models for human cancer. Among them is the fact that tumors arise spontaneously, just as in humans, and that tumor initiation and progression are influenced by similar factors like age, nutrition, sex, reproductive status and environmental exposure [54]. The risk for developing cancer of the nasal cavity for example is increased up to 60% in animals that are kept by smokers in comparison to pets of non-smoking owners [55,56]. Although there are differences in the diet of humans and companion animals, many components such as meat, vegetables and carbohydrates are derived from the same sources and are consumed non-sterile as opposed to mice under SPF conditions [57]. Furthermore, studies have shown that the contact between owner and pet leads to a large overlap in the microbiome, the importance of which for human tumor development has come into focus recently [58].
Pets, and in particular dogs are large and relatively outbred in comparison to laboratory mice. In fact, the genetic variation across dog breeds or in mixed breeds is similar to the variation in humans on the basis of single nucleotide polymorphisms [59]. In individual pure breeds however, the level of genetic diversity is more restricted [60]. The canine genome has been sequenced with a coverage of 99%, revealing that the approximately 19,000 genes identified in the dog match to homologous or orthologous genes in humans [59]. Actually, for many gene families, particularly for those associated with cancer, the homologies are significantly closer than the relationship between human and mouse [61]. Accordingly, most oncogenes and tumor suppressors that are known from human cancers have been shown to contribute to canine cancers [62].
Dogs and cats of all breeds develop cancer, and the spectrum of cancers seen in companion animals is as diverse as that seen in human patients [63]. The dog is the species in which comparative oncology has shown the most growth, and where it is best characterized [64]. Interestingly, there are breed-specific differences as to the cancer subtypes, reflecting the underlying genetics of the various breeds (Table 2). Mast cell tumors and gliomas for example are over-represented in Boxers, Staffordshire, Weimaraner and Golden Retriever, osteosarcoma in Rottweilers, Greyhounds and Golden Retrievers, bladder cancer in Scottish Terriers, histiocytic sarcomas in Flat-Coated Retrievers and Bernese Mountain Dogs and melanoma and gastric carcinoma in Chow-Chows [65,66,67].
Table 2.
Oncological disposition of various dog breeds.
The global expression pattern of canine and human osteosarcoma for example shows a strong similarity, and cluster analysis of orthologous gene signatures does not segregate human and canine tumors [73]. Finally, many chemotherapy protocols used for the treatment of canine cancers have been adopted from human medicine. The same chemotherapeutics used in human lymphoma for example are also active in canine lymphoma (e.g., vincristine, cyclophosphamide, doxorubicin, mitoxantrone, cytarabine and methotrexate), and drugs that are ineffective in human lymphomas are also inactive in canines (i.e., gemcitabine, cisplatin and carboplatin) [5].
One of the biggest advantages of companion animals as models for human cancer is the spontaneous development of tumors in the presence of an intact immune system. Immune cells pose a significant barrier to the development of cancer, and undergo changes themselves, as cancer cells co-opt the immune response [76,77]. As a result, the tumor influences innate as well as adaptive immune cells to become regulatory, rather than tumoricidal [78,79]. This interplay results in the selection of tumor cells that are invisible to anti-tumor T-cell-mediated destruction, and is central for tumor editing and immune evasion. Additionally, similar to humans, dogs with advanced cancer exhibit intrinsic T-cell defects as well as T-cell exhaustion [80]. Immune cell interplay with cancer cells is a JAK-STAT3/5 affair, as detailed in the special issue in several articles [24,25,81].
Like all other model systems, companion animals have strengths as well as weaknesses, both practically and conceptually. The biggest hurdle, when working with cats and dogs in research is the paucity of investigational tools. Many antibodies and recombinant products that are available for humans and mice do not show cross-species reactivity. However, the sequencing of the canine genome and the development of genome editing via the CRISPR/Cas9 technology has relieved many of the restrictions [82]. It is now possible to introduced genetic alterations with high efficiency into any desired locus, facilitating for example the visualization of specific cell types via the expression of marker proteins, or targeted deletion of single genes [83,84]. Conceptually, the lack of standardized housing conditions, making it difficult to control for variables, has been held against non-rodent models. However, as pointed out here, this represents an advantage when it comes to closely mimicking the translational aspects in tumor biology.
The potential of companion animals as biological models between the basic mechanistic work that is possible in the mouse and translation to humans is demonstrated by 1. the high conservation at the genomic level, 2. the involvement of similar genetic and environmental risk factors, 3. the successful use of canine cancer as biological models for the early development of bone marrow transplantation protocols and 4. canine trials for the development of drug level and exposure durations. This sets an ideal stage to combine new perspectives of targeted therapies and specific molecular inhibitors in the field of comparative oncology for the benefit of human as well as veterinary medicine (Figure 1). Furthermore, this approach is not limited to cancer but applicable to any comparative condition including infectious or inflammatory diseases like inflammatory bowel disease or pre-malignant conditions like adenoma formation or clonal hematopoiesis.
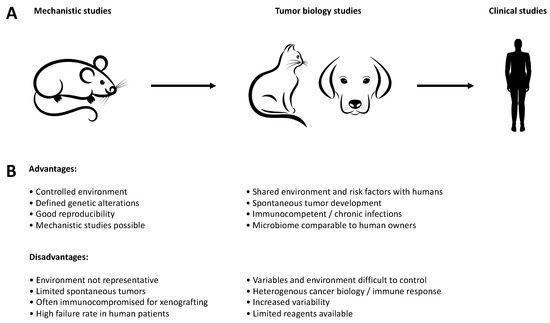
Figure 1.
Advantages and disadvantages of different models during drug discovery. (A) Companion animals can be used as an intermediate step between the mechanistic work in murine models and clinical studies in humans, particularly with regard to comparative aspects of tumor biology. (B) Advantages and disadvantages of the individual models for translation into human clinical studies.
4. Relevance and Conservation of the JAK-STAT Signaling Pathway
JAK-STAT proteins constitute an evolutionary conserved signaling pathway [85]. Ligand binding of receptors leads to the activation of JAK kinases and STAT proteins, inducing transcription. The family of JAK kinases consists of four members, JAK1-3 and TYK2, and there are seven highly homologous STAT proteins, STAT1–4, STAT5a and STAT5b and STAT6 [22,86]. For details on the mechanism see also other reviews in this issue [87,88]. As such, this pathway provides a remarkably elegant and straightforward mechanism to transduce signals from receptors to the nucleus.
All family members show the same structural organization, i.e., an N-terminal domain required for oligomerization of dimers into tetramers, a coiled-coil domain, a DNA-binding domain, a linker domain, a Src homology 2 (Sh2) domain for dimerization and a C-terminal transactivation domain (Figure 2). Functionally, STAT2, STAT4 and STAT6 regulate specific immune cell responses, whereas STAT1, STAT3 and STAT5 have diverse physiological roles. STAT1 is mostly involved in immunity, host defense against pathogens and cell death, stimulating the transcription of pro-inflammatory and anti-proliferative genes like caspases, NOS2, MDM2, CDKN1A and CDKN1B. On the contrary, STAT3 and STAT5 are mostly involved in cell proliferation and prevention of apoptosis, activating the transcription of genes like CCND1, BIRC5, c-MYC, VEGF, MCL1, BCL2L1 and BCL2. Additionally, STAT3 can also be found in mitochondria, where it supports RAS-dependent malignant transformation via sustained altered glycolytic and oxidative phosphorylation [89,90]. Given their roles in the stimulation of cellular proliferation, the prevention of apoptosis and the stimulation of metabolism, STAT5, and even more so STAT3, are activated in nearly 70% of solid and hematological human tumors [91,92,93].

Figure 2.
Cross-species conservation of STAT protein domains. (A) STAT1, STAT3, STAT5a and STAT5b from dog, cat and mouse are analyzed for their overall homology compared to the respective human protein (grey boxes, left). In the schematic representation of STAT protein domains, the amino acid positions are indicated above. All proteins share the same domain positions, except for murine STAT1, which has a five amino acid insertion in the DNA binding domain (numbers below the scheme indicate the aa position in this case). Percentages in the domain boxes of dog, cat and mouse STAT proteins show the homology of each domain to the human counterpart. Analyses were carried out using ClustalX. (B) Comparison of key phosphorylation sites in the transactivation domain of STAT1, STAT3, STAT5a and STAT5b from dog, cat and mouse to the human sequence. Amino acid sequence is shown, with phosphorylation sites in green and position indicated; positive amino acid exchanges (conserving protein function) are indicated in yellow, other exchanges in red. (STAT1: human NP_009330.1, dog XP_848353.1, cat XP_006935505.1, mouse NP_001192242.1; STAT3: human NP_644805.1, dog XP_005624514.1, cat XP_003996930.1, mouse NP_998824.1; STAT5a: human NP_001275647.1, dog XP_548091.2, cat XP_023099834.1, mouse NP_001157534.1; STAT5b: human NP_036580.2, dog XP_548092.1, cat XP_023100377.1, mouse NP_035619.3).
Silencing or inhibition of STAT3 or STAT5 signaling impairs tumor growth and survival in murine and human studies, while only slightly affecting normal differentiated cells [94,95,96,97]. These findings lead to the concept of STAT3 and STAT5 constituting a “signaling bottleneck” situation for tumor cells, making them attractive targets for inhibition [98]. However, caution has to be exerted with regard to tissue-specificity, as tumor-suppressive functions have been ascribed to STAT3 in neuronal, hepatic and colorectal tumors and to STAT5 in breast cancer [99,100].
Several different ways of inhibiting STAT signaling are possible. Upstream of STAT proteins, JAK kinases are mutated in a broad range of diseases from severe combined immunodeficiency to various forms of cancer, including JAK1 in acute myeloid leukemia, JAK2 in myeloproliferative diseases and JAK3 in different leukemias and lymphomas, and inhibitors against JAK kinases are already approved by the US Food and Drug Administration (FDA) for clinical use [27]. Interestingly, different layers of negative regulators of JAK-STAT signaling are present such as suppressor of cytokine signaling (SOCS), protein inhibitor of activated STAT (PIAS) and protein tyrosine phosphatases, arguing for the necessity of a tightly controlled down-regulation of this signaling pathway [101].
Due to the broad activation, minor side-effects and the overall importance, major efforts by many laboratories and pharmaceutical companies are ongoing to develop inhibitors against STAT3 and STAT5. In both cases, all current inhibitors target one of three STAT motifs: the SH2 domain necessary for the interaction of phosphorylated monomers to form dimers, the N-terminal domain mediating the formation of tetramers from activated STAT dimers and the DNA-binding domain [102]. STAT3 and STAT5 from companion animals show more than 96% homology at the overall protein level to their human counterparts, with a particular high level of conservation of 98% to complete alignment in these three domains (Figure 2). This high level of conservation opens up the possibility to use pet animals as models for diseases in which the JAK-STAT signaling pathway is over-activated.
A good example for such a successful application is already established. Cytokine dysregulation has been implicated in allergic skin disease, particularly in atopic dermatitis in humans. T-helper cells type 2 (Th2) produce increased levels of IL4, IL5, IL10, IL13 and IL31, in addition to elevated production of IFNγ by T-helper cells type 1 (Th1), signals that all converge on the JAK-STAT signaling pathway [27,103,104,105]. Dermatological problems are the second most common reason for dogs to present to veterinary practices, frequently including allergic skin diseases like atopic dermatitis [106,107]. In the skin of atopic dogs, a cytokine profile can be found that resembles the human condition, and in an experimental model of canine allergic dermatitis, elevated transcripts of IL6, IL13 and IL18 and IFNγ were detected, supporting the idea that cytokine dysregulation plays a role in allergic skin disease [108,109,110]. The novel JAK inhibitor oclacitinib is most potent against JAK1, but also affects JAK2 and JAK3 at reduced efficiency, inhibiting the function of JAK1-dependent inflammatory cytokines [111]. Treatment of dogs suffering from atopic dermatitis results in a reduction of associated skin lesions and oclacitinib recently has been approved in the US and Europe for the treatment of allergic/atopic dermatitis [112,113,114]. Overall, inhibition of JAK1-dependent cytokines is an effective and novel way to treat canine allergic skin disease, proving the high similarity and cross-species conservation of JAK-STAT signaling.
5. Inhibition of STAT3 and STAT5 in Companion Animals: Current Status/Future Perspectives
Currently, STAT3 and STAT5 are studied in several cancer types of companion animals. Canine mammary cancer cells and diffuse large B cell lymphoma as well as feline oral squamous cell carcinoma and mammary tumors show activation of STAT3 or STAT5. First results indicate reduced proliferation and increased apoptosis upon inhibition of JAK1/2 or STAT3 respectively [115,116,117,118,119,120,121]. Two areas exemplify the potential of research and development concerning STAT proteins and human—canine comparative oncology best [122,123]. Osteosarcoma is the most frequent form of malignant bone disease in dogs, however, it is relatively rare in humans. The estimated incidence rate is at least 13.9/100,000 in canines and 1.02/100,000 in humans, affecting primarily children [124,125]. Such a high incidence rate provides a good opportunity to study a rare human disease using dogs as a preclinical model. Moreover, canine and human osteosarcoma share many key features like tumor location, early metastasis, development of chemotherapy-resistant metastases and altered expression or activation of several proteins [126].
Consistent activation of STAT3 occurs in a large subset of human and canine osteosarcoma and osteosarcoma cell lines, but not in normal osteoblasts. Down-regulation of STAT3 expression or activity reduces proliferation and induces apoptosis in human and canine cell lines [127,128,129,130]. Additionally, human and canine osteosarcoma possess overlapping transcriptional profiles, further supporting the concept that these diseases are similar at the molecular level [131]. Spontaneous canine osteosarcoma has been used for the development of novel therapeutics such as muramyl tripeptide, IGF-1R inhibitors and rapamycin [132]. However, despite aggressive treatment, 30–40% of children and >90% of dogs are still dying from disease, demonstrating the need for new therapeutic options. The similarities between human and canine osteosarcoma, together with the dependency on JAK-STAT signaling, particularly STAT3, make osteosarcoma one of the prime areas for comparative oncology studies, especially since a large number of STAT3 inhibitors are currently being developed.
Mast cell tumors arise from the uncontrolled proliferation of transformed mast cells mostly in skin, spreading primarily to spleen, liver and bone marrow [133]. While mast cell tumors are relatively rare in humans, they are frequent in dogs, accounting for 7–21% of cutaneous tumors [134,135]. Metastasized tumors in humans as well as in canines have a poor prognosis and short survival times [133,134,136]. Stem cell factor and its receptor c-KIT are essential for mast cell survival and inhibition of apoptosis, and gain-of-function mutations are present in human and canine mast cell tumors [137,138,139,140,141,142]. Accordingly, two inhibitors of c-KIT, masitinib and toceranib have been approved for use in c-KIT driven mast cell tumors in dogs. Both drugs are able to suppress tumor growth temporarily, however, relapses are high, indicating the need for the identification of further targets and therapeutic options [143,144].
STAT1, STAT3 and STAT5 are activated down-stream of mutant c-KIT, but only STAT5 is transcriptionally active in neoplastic mast cells [145,146]. Inhibition of JAK2 and STAT5 was recently discovered to inhibit proliferation and survival of canine mastocytoma cell lines, identifying JAK2/STAT5 signaling as a new potential target in mast cell tumors [147]. Therefore, an attractive option is to use the frequent occurrence of canine mast cell tumors to determine if inhibitors of STAT5 can be used alone or in combination with inhibitors of c-kit or other kinases as a new therapeutic option. Indeed, the potential of STAT5 inhibitors to overcome resistance to a multi-kinase inhibitor in neoplastic mast cells has been demonstrated [148]. Consequently, the next step is to determine the potential of new STAT5 inhibitors in vivo [149]. Both of these examples, osteosarcoma as well as mast cell tumors offer thereby the possibility of using new pharmaceutical in companion animals as a closer biological mimic of the human situation.
6. Conclusions
Taken together, cancer in dogs resembles cancer in humans in many ways, like its latency, clinical manifestation and metastatic behavior, its pathobiological characteristics like tumor heterogeneity, its genomic instability and pharmacogenomic signatures including chemoresistance and last but not least its multifactorial nature, including genetic and environmental risk factors [150]. The inability of murine cancer models to recapitulate certain aspects of human tumors is increasingly recognized and illuminates the huge potential of spontaneous canine and, to a lesser extent, feline cancer [151,152,153]. The JAK-STAT pathway is activated in the vast majority of solid and hematological tumors, and is necessary for tumor growth and prevention of apoptosis. The major efforts that are ongoing to develop inhibitors specifically for STAT3 and STAT5 can be extended to studies in companion animals, an option that is particularly attractive for rare human diseases occurring more frequently in dogs and/or cats.
Funding
This research received no external funding.
Acknowledgments
Open Access Funding by the University of Veterinary Medicine Vienna.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vail, D.M.; MacEwen, E.G. Spontaneously occurring tumors of companion animals as models for human cancer. Cancer Invest. 2000, 18, 781–792. [Google Scholar] [CrossRef]
- Bonnett, B.N.; Egenvall, A.; Hedhammar, A.; Olson, P. Mortality in over 350,000 insured Swedish dogs from 1995-2000: I. Breed-, gender-, age- and cause-specific rates. Acta Vet. Scand. 2005, 46, 105–120. [Google Scholar] [CrossRef]
- Egenvall, A.; Bonnett, B.N.; Hedhammar, A.; Olson, P. Mortality in over 350,000 insured Swedish dogs from 1995-2000: II. Breed-specific age and survival patterns and relative risk for causes of death. Acta Vet. Scand. 2005, 46, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Vascellari, M.; Baioni, E.; Ru, G.; Carminato, A.; Mutinelli, F. Animal tumour registry of two provinces in northern Italy: Incidence of spontaneous tumours in dogs and cats. BMC Vet. Res. 2009, 5, 39. [Google Scholar] [CrossRef] [PubMed]
- Withrow, S.J.; Vail, D.M. Withrow & MacEwen’s Small Animal Clinical Oncology, 5th ed.; Saunders Elsevier: Philadelphia, PA, USA, 2013. [Google Scholar]
- Paoloni, M.; Khanna, C. Translation of new cancer treatments from pet dogs to humans. Nat. Rev. Cancer 2008, 8, 147–156. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Surveillance, Epidemiology and End Results (SEER) Program 2012–2016 Data. Available online: www.seer.cancer.gov/statfacts (accessed on 6 November 2019).
- Vascellari, M.; Capello, K.; Carminato, A.; Zanardello, C.; Baioni, E.; Mutinelli, F. Incidence of mammary tumors in the canine population living in the Veneto region (Northeastern Italy): Risk factors and similarities to human breast cancer. Prev. Vet. Med. 2016, 126, 183–189. [Google Scholar] [CrossRef]
- Hassan, B.B.; Elshafae, S.M.; Supsavhad, W.; Simmons, J.K.; Dirksen, W.P.; Sokkar, S.M.; Rosol, T.J. Feline Mammary Cancer. Vet. Pathol. 2017, 54, 32–43. [Google Scholar] [CrossRef]
- Graf, R.; Pospischil, A.; Guscetti, F.; Meier, D.; Welle, M.; Dettwiler, M. Cutaneous tumors in Swiss dogs: Retrospective data from the Swiss canine cancer registry, 2008–2013. Vet. Pathol. 2018, 55, 809–820. [Google Scholar] [CrossRef]
- Merlo, D.F.; Rossi, L.; Pellegrino, C.; Ceppi, M.; Cardellino, U.; Capurro, C.; Ratto, A.; Sambucco, P.L.; Sestito, V.; Tanara, G.; et al. Cancer incidence in pet dogs: Findings of the animal tumor registry of Genoa, Italy. J. Vet. Intern. Med. 2008, 22, 976–984. [Google Scholar] [CrossRef]
- Lomas, A.; Leonardi-Bee, J.; Bath-Hextall, F. A systematic review of worldwide incidence of nonmelanoma skin cancer. Br. J. Dermatol. 2012, 166, 1069–1080. [Google Scholar] [CrossRef]
- Pinello, K.C.; Niza-Ribeiro, J.; Fonseca, L.; de Matos, A.J. Incidence, characteristics and geographical distributions of canine and human non-Hodgkin’s lymphoma in the Porto region (North West Portugal). Vet. J. 2019, 245, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Grover, S. Gastronintestinal lymphoma in cats. Oncol. Compend. 2005, 27, 741–751. [Google Scholar]
- Egenvall, A.; Nodtvedt, A.; von Euler, H. Bone tumors in a population of 400 000 insured Swedish dogs up to 10 y of age: Incidence and survival. Can. J. Vet. Res. 2007, 71, 292–299. [Google Scholar] [PubMed]
- Dimopoulou, M.; Kirpensteijn, J.; Moens, H.; Kik, M. Histologic prognosticators in feline osteosarcoma: A comparison with phenotypically similar canine osteosarcoma. Vet. Surg. 2008, 37, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Cheon, D.J.; Orsulic, S. Mouse models of cancer. Annu. Rev. Pathol 2011, 6, 95–119. [Google Scholar] [CrossRef] [PubMed]
- Ghebranious, N.; Donehower, L.A. Mouse models in tumor suppression. Oncogene 1998, 17, 3385–3400. [Google Scholar] [CrossRef] [PubMed]
- Fowlis, D.J.; Balmain, A. Oncogenes and tumour suppressor genes in transgenic mouse models of neoplasia. Eur. J. Cancer 1993, 29A, 638–645. [Google Scholar] [CrossRef]
- Murphy, W.J. Of mice and men. Biol. Blood Marrow Transplant. 2013, 19, 1140–1141. [Google Scholar] [CrossRef][Green Version]
- Cekanova, M.; Rathore, K. Animal models and therapeutic molecular targets of cancer: Utility and limitations. Drug Des. Devel. Ther. 2014, 8, 1911–1921. [Google Scholar] [CrossRef]
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef]
- Igelmann, S.; Neubauer, H.A.; Ferbeyre, G. STAT3 and STAT5 Activation in Solid Cancers. Cancers 2019, 11, 1428. [Google Scholar] [CrossRef] [PubMed]
- Logotheti, S.; Putzer, B.M. STAT3 and STAT5 targeting for simultaneous management of melanoma and autoimmune diseases. Cancers 2019, 11, 1448. [Google Scholar] [CrossRef] [PubMed]
- Rebe, C.; Ghiringhelli, F. STAT3, a master regulator of anti-tumor immune response. Cancers 2019, 11, 1280. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J. Stat proteins and oncogenesis. J. Clin. Invest. 2002, 109, 1139–1142. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef]
- Fagard, R.; Metelev, V.; Souissi, I.; Baran-Marszak, F. STAT3 inhibitors for cancer therapy: Have all roads been explored? JAKSTAT 2013, 2, e22882. [Google Scholar] [CrossRef]
- Rangarajan, A.; Weinberg, R.A. Opinion: Comparative biology of mouse versus human cells: Modelling human cancer in mice. Nat. Rev. Cancer 2003, 3, 952–959. [Google Scholar] [CrossRef]
- Dobrowolski, P.; Fischer, M.; Naumann, R. Novel insights into the genetic background of genetically modified mice. Transgenic Res. 2018, 27, 265–275. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]
- Bouchlaka, M.N.; Murphy, W.J. Impact of aging in cancer immunotherapy: The importance of using accurate preclinical models. Oncoimmunology 2013, 2, e27186. [Google Scholar] [CrossRef]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Honda, K. Intestinal commensal microbes as immune modulators. Cell Host Microbe 2012, 12, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Antonovic, L.; Martinez, M. Role of the cytochrome P450 enzyme system in veterinary pharmacokinetics: Where are we now? Where are we going? Future Med. Chem. 2011, 3, 855–879. [Google Scholar] [CrossRef] [PubMed]
- Brekke, T.D.; Steele, K.A.; Mulley, J.F. Inbred or outbred? Genetic diversity in laboratory rodent colonies. G3 (Bethesda) 2018, 8, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Cutler, G.; Kassner, P.D. Copy number variation in the mouse genome: Implications for the mouse as a model organism for human disease. Cytogenet. Genome Res. 2008, 123, 297–306. [Google Scholar] [CrossRef]
- Hartman, J.L.t.; Garvik, B.; Hartwell, L. Principles for the buffering of genetic variation. Science 2001, 291, 1001–1004. [Google Scholar] [CrossRef]
- Srivastava, A.; Morgan, A.P.; Najarian, M.L.; Sarsani, V.K.; Sigmon, J.S.; Shorter, J.R.; Kashfeen, A.; McMullan, R.C.; Williams, L.H.; Giusti-Rodriguez, P.; et al. Genomes of the mouse collaborative cross. Genetics 2017, 206, 537–556. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Cespedes, M.V.; Casanova, I.; Parreno, M.; Mangues, R. Mouse models in oncogenesis and cancer therapy. Clin. Transl. Oncol. 2006, 8, 318–329. [Google Scholar] [CrossRef]
- Holzapfel, B.M.; Wagner, F.; Thibaudeau, L.; Levesque, J.P.; Hutmacher, D.W. Concise review: Humanized models of tumor immunology in the 21st century: Convergence of cancer Research and tissue engineering. Stem Cells 2015, 33, 1696–1704. [Google Scholar] [CrossRef] [PubMed]
- Richmond, A.; Su, Y. Mouse xenograft models vs. GEM models for human cancer therapeutics. Dis Model. Mech. 2008, 1, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Wald, A.; Corey, L. Persistence in the population: Epidemiology, transmission. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Smatti, M.K.; Al-Sadeq, D.W.; Ali, N.H.; Pintus, G.; Abou-Saleh, H.; Nasrallah, G.K. Epstein-Barr virus epidemiology, serology, and genetic variability of LMP-1 oncogene among healthy population: An update. Front. Oncol. 2018, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Colugnati, F.A.; Staras, S.A.; Dollard, S.C.; Cannon, M.J. Incidence of cytomegalovirus infection among the general population and pregnant women in the United States. BMC Infect. Dis 2007, 7, 71. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; Merlino, G.; Van Dyke, T. Preclinical mouse cancer models: A maze of opportunities and challenges. Cell 2015, 163, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Willyard, C. The mice with human tumours: Growing pains for a popular cancer model. Nature 2018, 560, 156–157. [Google Scholar] [CrossRef]
- Ireson, C.R.; Alavijeh, M.S.; Palmer, A.M.; Fowler, E.R.; Jones, H.J. The role of mouse tumour models in the discovery and development of anticancer drugs. Br. J. Cancer 2019, 121, 101–108. [Google Scholar] [CrossRef]
- Ciociola, A.A.; Cohen, L.B.; Kulkarni, P.; Gastroenterology, F.D.-R.M.C.o.t.A.C.o. How drugs are developed and approved by the FDA: Current process and future directions. Am. J. Gastroenterol. 2014, 109, 620–623. [Google Scholar] [CrossRef]
- Schiffman, J.D.; Breen, M. Comparative oncology: What dogs and other species can teach us about humans with cancer. Philos. Trans. R Soc. Lond. B Biol. Sci. 2015, 370. [Google Scholar] [CrossRef]
- Hahn, K.A.; Bravo, L.; Adams, W.H.; Frazier, D.L. Naturally occurring tumors in dogs as comparative models for cancer therapy research. In Vivo 1994, 8, 133–143. [Google Scholar] [PubMed]
- Reif, J.S.; Bruns, C.; Lower, K.S. Cancer of the nasal cavity and paranasal sinuses and exposure to environmental tobacco smoke in pet dogs. Am. J. Epidemiol. 1998, 147, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Reif, J.S.; Dunn, K.; Ogilvie, G.K.; Harris, C.K. Passive smoking and canine lung cancer risk. Am. J. Epidemiol. 1992, 135, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, D.; Izquierdo, O.; Eirmann, L.; Binder, S. Myths and misperceptions about ingredients used in commercial pet foods. Vet. Clin. North. Am. Small Anim. Pract. 2014, 44, 689–698. [Google Scholar] [CrossRef]
- Song, S.J.; Lauber, C.; Costello, E.K.; Lozupone, C.A.; Humphrey, G.; Berg-Lyons, D.; Caporaso, J.G.; Knights, D.; Clemente, J.C.; Nakielny, S.; et al. Cohabiting family members share microbiota with one another and with their dogs. Elife 2013, 2, e00458. [Google Scholar] [CrossRef]
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J., 3rd; Zody, M.C.; et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef]
- Vonholdt, B.M.; Pollinger, J.P.; Lohmueller, K.E.; Han, E.; Parker, H.G.; Quignon, P.; Degenhardt, J.D.; Boyko, A.R.; Earl, D.A.; Auton, A.; et al. Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature 2010, 464, 898–902. [Google Scholar] [CrossRef]
- Hoffman, M.M.; Birney, E. Estimating the neutral rate of nucleotide substitution using introns. Mol. Biol Evol 2007, 24, 522–531. [Google Scholar] [CrossRef]
- Paoloni, M.C.; Khanna, C. Comparative oncology today. Vet. Clin. North. Am. Small Anim Pract 2007, 37, 1023–1032. [Google Scholar] [CrossRef]
- Khanna, C.; Lindblad-Toh, K.; Vail, D.; London, C.; Bergman, P.; Barber, L.; Breen, M.; Kitchell, B.; McNeil, E.; Modiano, J.F.; et al. The dog as a cancer model. Nat. Biotechnol. 2006, 24, 1065–1066. [Google Scholar] [CrossRef]
- Di Cerbo, A.; Palmieri, B.; De Vico, G.; Iannitti, T. Onco-epidemiology of domestic animals and targeted therapeutic attempts: Perspectives on human oncology. J. Cancer Res. Clin. Oncol 2014, 140, 1807–1814. [Google Scholar] [CrossRef] [PubMed]
- Modiano, J.F.; Breen, M.; Burnett, R.C.; Parker, H.G.; Inusah, S.; Thomas, R.; Avery, P.R.; Lindblad-Toh, K.; Ostrander, E.A.; Cutter, G.C.; et al. Distinct B-cell and T-cell lymphoproliferative disease prevalence among dog breeds indicates heritable risk. Cancer Res. 2005, 65, 5654–5661. [Google Scholar] [CrossRef] [PubMed]
- Nishiya, A.T.; Massoco, C.O.; Felizzola, C.R.; Perlmann, E.; Batschinski, K.; Tedardi, M.V.; Garcia, J.S.; Mendonca, P.P.; Teixeira, T.F.; Zaidan Dagli, M.L. Comparative aspects of canine melanoma. Vet. Sci. 2016, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Dobson, J.M. Breed-predispositions to cancer in pedigree dogs. ISRN Vet. Sci 2013, 2013, 941275. [Google Scholar] [CrossRef] [PubMed]
- Klopfenstein, M.; Howard, J.; Rossetti, M.; Geissbuhler, U. Life expectancy and causes of death in Bernese mountain dogs in Switzerland. BMC Vet. Res. 2016, 12, 153. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.W.; Wiles, B.M.; Llewellyn-Zaidi, A.M.; Evans, K.M.; O’Neill, D.G. Longevity and mortality in Kennel Club registered dog breeds in the UK in 2014. Canine Genet. Epidemiol. 2018, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Dobson, J.; Hoather, T.; McKinley, T.J.; Wood, J.L. Mortality in a cohort of flat-coated retrievers in the UK. Vet. Comp. Oncol. 2009, 7, 115–121. [Google Scholar] [CrossRef]
- Zink, M.C.; Farhoody, P.; Elser, S.E.; Ruffini, L.D.; Gibbons, T.A.; Rieger, R.H. Evaluation of the risk and age of onset of cancer and behavioral disorders in gonadectomized Vizslas. J. Am. Vet. Med. Assoc. 2014, 244, 309–319. [Google Scholar] [CrossRef]
- Henry, C.J.; Higginbotham, H.M. Cancer Management in Small Animal Practice; Saunders Elsevier: Philadelphia, PA, USA, 2009. [Google Scholar]
- Fan, T.M.; Khanna, C. Comparative Aspects of Osteosarcoma Pathogenesis in Humans and Dogs. Vet. Sci 2015, 2, 210–230. [Google Scholar] [CrossRef]
- Urfer, S.R.; Gaillard, C.; Steiger, A. Lifespan and disease predispositions in the Irish Wolfhound: A review. Vet. Q. 2007, 29, 102–111. [Google Scholar] [CrossRef]
- Zandvliet, M. Canine lymphoma: A review. Vet. Q. 2016, 36, 76–104. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A. Adaptive immune resistance: How cancer protects from immune attack. Cancer Discov. 2015, 5, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Goulart, M.R.; Pluhar, G.E.; Ohlfest, J.R. Identification of myeloid derived suppressor cells in dogs with naturally occurring cancer. PLoS ONE 2012, 7, e33274. [Google Scholar] [CrossRef]
- Pinheiro, D.; Chang, Y.M.; Bryant, H.; Szladovits, B.; Dalessandri, T.; Davison, L.J.; Yallop, E.; Mills, E.; Leo, C.; Lara, A.; et al. Dissecting the regulatory microenvironment of a large animal model of non-Hodgkin lymphoma: Evidence of a negative prognostic impact of FOXP3+ T cells in canine B cell lymphoma. PLoS ONE 2014, 9, e105027. [Google Scholar] [CrossRef]
- Coy, J.; Caldwell, A.; Chow, L.; Guth, A.; Dow, S. PD-1 expression by canine T cells and functional effects of PD-1 blockade. Vet. Comp. Oncol. 2017, 15, 1487–1502. [Google Scholar] [CrossRef]
- Zerdes, I.; Wallerius, M.; Sifakis, E.G.; Wallmann, T.; Betts, S.; Bartish, M.; Tsesmetzis, N.; Tobin, N.P.; Coucoravas, C.; Bergh, J.; et al. STAT3 activity promotes programmed-death ligand 1 expression and suppresses immune responses in breast cancer. Cancers 2019, 11, 1479. [Google Scholar] [CrossRef]
- Reardon, S. CRISPR gene-editing creates wave of exotic model organisms. Nature 2019, 568, 441–442. [Google Scholar] [CrossRef]
- Simoff, I.; Karlgren, M.; Backlund, M.; Lindstrom, A.C.; Gaugaz, F.Z.; Matsson, P.; Artursson, P. Complete knockout of endogenous Mdr1 (Abcb1) in MDCK cells by CRISPR-Cas9. J. Pharm. Sci. 2016, 105, 1017–1021. [Google Scholar] [CrossRef]
- Amoasii, L.; Hildyard, J.C.W.; Li, H.; Sanchez-Ortiz, E.; Mireault, A.; Caballero, D.; Harron, R.; Stathopoulou, T.R.; Massey, C.; Shelton, J.M.; et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 2018, 362, 86–91. [Google Scholar] [CrossRef]
- Liongue, C.; Ward, A.C. Evolution of the JAK-STAT pathway. JAKSTAT 2013, 2, e22756. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [PubMed]
- Polak, K.L.; Chernosky, N.M.; Smigiel, J.M.; Tamagno, I.; Jackson, M.W. Balancing STAT Activity as a Therapeutic Strategy. Cancers 2019, 11, 1716. [Google Scholar] [CrossRef] [PubMed]
- Yue-Ting, K.L.; Ramaiyer, M.; E. Johnson, D.; R. Grandis, J. Targeting STAT3 in cancer with nucleotide therapeutics. Cancers 2019, 11, 1681. [Google Scholar] [CrossRef]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef]
- Meier, J.A.; Larner, A.C. Toward a new STATe: The role of STATs in mitochondrial function. Semin. Immunol. 2014, 26, 20–28. [Google Scholar] [CrossRef]
- Frank, D.A. STAT3 as a central mediator of neoplastic cellular transformation. Cancer Lett. 2007, 251, 199–210. [Google Scholar] [CrossRef]
- Bar-Natan, M.; Nelson, E.A.; Xiang, M.; Frank, D.A. STAT signaling in the pathogenesis and treatment of myeloid malignancies. JAKSTAT 2012, 1, 55–64. [Google Scholar] [CrossRef]
- Roeser, J.C.; Leach, S.D.; McAllister, F. Emerging strategies for cancer immunoprevention. Oncogene 2015, 34, 6029–6039. [Google Scholar] [CrossRef]
- Yu, H.; Jove, R. The STATs of cancer--new molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef]
- Sen, M.; Thomas, S.M.; Kim, S.; Yeh, J.I.; Ferris, R.L.; Johnson, J.T.; Duvvuri, U.; Lee, J.; Sahu, N.; Joyce, S.; et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: Implications for cancer therapy. Cancer Discov. 2012, 2, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kwok, S.K.; Lim, M.A.; Kim, E.K.; Ryu, J.G.; Kim, S.M.; Oh, H.J.; Ju, J.H.; Park, S.H.; Kim, H.Y.; et al. STA-21, a promising STAT-3 inhibitor that reciprocally regulates Th17 and Treg cells, inhibits osteoclastogenesis in mice and humans and alleviates autoimmune inflammation in an experimental model of rheumatoid arthritis. Arthritis Rheumatol. 2014, 66, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Kunigal, S.; Lakka, S.S.; Sodadasu, P.K.; Estes, N.; Rao, J.S. Stat3-siRNA induces Fas-mediated apoptosis in vitro and in vivo in breast cancer. Int. J. Oncol. 2009, 34, 1209–1220. [Google Scholar] [PubMed]
- Darnell, J.E., Jr. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Ferbeyre, G.; Moriggl, R. The role of Stat5 transcription factors as tumor suppressors or oncogenes. Biochim. Biophys. Acta 2011, 1815, 104–114. [Google Scholar] [CrossRef]
- Zhang, H.F.; Lai, R. STAT3 in Cancer-Friend or Foe? Cancers 2014, 6, 1408–1440. [Google Scholar] [CrossRef]
- Starr, R.; Hilton, D.J. Negative regulation of the JAK/STAT pathway. Bioessays 1999, 21, 47–52. [Google Scholar] [CrossRef]
- Wake, M.S.; Watson, C.J. STAT3 the oncogene - still eluding therapy? FEBS J. 2015, 282, 2600–2611. [Google Scholar] [CrossRef]
- Carmi-Levy, I.; Homey, B.; Soumelis, V. A modular view of cytokine networks in atopic dermatitis. Clin. Rev. Allergy Immunol. 2011, 41, 245–253. [Google Scholar] [CrossRef]
- Brandt, E.B.; Sivaprasad, U. Th2 Cytokines and Atopic Dermatitis. J. Clin. Cell Immunol 2011, 2. [Google Scholar] [CrossRef]
- Ong, P.Y.; Leung, D.Y. Immune dysregulation in atopic dermatitis. Curr. Allergy Asthma Rep. 2006, 6, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Hill, P.B.; Lo, A.; Eden, C.A.; Huntley, S.; Morey, V.; Ramsey, S.; Richardson, C.; Smith, D.J.; Sutton, C.; Taylor, M.D.; et al. Survey of the prevalence, diagnosis and treatment of dermatological conditions in small animals in general practice. Vet. Rec. 2006, 158, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Olivry, T.; Bizikova, P. A systematic review of randomized controlled trials for prevention or treatment of atopic dermatitis in dogs: 2008-2011 update. Vet. Dermatol. 2013, 24, 97–117, e25–e26. [Google Scholar] [CrossRef] [PubMed]
- Marsella, R.; Olivry, T.; Maeda, S. Cellular and cytokine kinetics after epicutaneous allergen challenge (atopy patch testing) with house dust mites in high-IgE beagles. Vet. Dermatol. 2006, 17, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Nuttall, T.J.; Knight, P.A.; McAleese, S.M.; Lamb, J.R.; Hill, P.B. Expression of Th1, Th2 and immunosuppressive cytokine gene transcripts in canine atopic dermatitis. Clin. Exp. Allergy 2002, 32, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Schlotter, Y.M.; Rutten, V.P.; Riemers, F.M.; Knol, E.F.; Willemse, T. Lesional skin in atopic dogs shows a mixed Type-1 and Type-2 immune responsiveness. Vet. Immunol. Immunopathol. 2011, 143, 20–26. [Google Scholar] [CrossRef]
- Gonzales, A.J.; Bowman, J.W.; Fici, G.J.; Zhang, M.; Mann, D.W.; Mitton-Fry, M. Oclacitinib (APOQUEL((R))) is a novel Janus kinase inhibitor with activity against cytokines involved in allergy. J. Vet. Pharmacol. Ther. 2014, 37, 317–324. [Google Scholar] [CrossRef]
- Cosgrove, S.B.; Wren, J.A.; Cleaver, D.M.; Walsh, K.F.; Follis, S.I.; King, V.I.; Tena, J.K.; Stegemann, M.R. A blinded, randomized, placebo-controlled trial of the efficacy and safety of the Janus kinase inhibitor oclacitinib (Apoquel(R)) in client-owned dogs with atopic dermatitis. Vet. Dermatol. 2013, 24, 587–597, e141–e142. [Google Scholar] [CrossRef]
- Cosgrove, S.B.; Wren, J.A.; Cleaver, D.M.; Martin, D.D.; Walsh, K.F.; Harfst, J.A.; Follis, S.L.; King, V.L.; Boucher, J.F.; Stegemann, M.R. Efficacy and safety of oclacitinib for the control of pruritus and associated skin lesions in dogs with canine allergic dermatitis. Vet. Dermatol. 2013, 24, 479-e114. [Google Scholar] [CrossRef]
- Collard, W.T.; Hummel, B.D.; Fielder, A.F.; King, V.L.; Boucher, J.F.; Mullins, M.A.; Malpas, P.B.; Stegemann, M.R. The pharmacokinetics of oclacitinib maleate, a Janus kinase inhibitor, in the dog. J. Vet. Pharmacol Ther 2014, 37, 279–285. [Google Scholar] [CrossRef]
- Lu, Z.; Hong, C.C.; Jark, P.C.; Assumpcao, A.; Bollig, N.; Kong, G.; Pan, X. JAK1/2 Inhibitors AZD1480 and CYT387 inhibit canine B-Cell lymphoma growth by increasing apoptosis and disrupting cell proliferation. J. Vet. Intern. Med. 2017, 31, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- Assumpcao, A.; Jark, P.C.; Hong, C.C.; Lu, Z.; Ruetten, H.M.; Heaton, C.M.; Pinkerton, M.E.; Pan, X. STAT3 Expression and Activity are Up-Regulated in Diffuse Large B Cell Lymphoma of Dogs. J. Vet. Intern. Med. 2018, 32, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Krol, M.; Pawlowski, K.M.; Dolka, I.; Musielak, O.; Majchrzak, K.; Mucha, J.; Motyl, T. Density of Gr1-positive myeloid precursor cells, p-STAT3 expression and gene expression pattern in canine mammary cancer metastasis. Vet. Res. Commun. 2011, 35, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Van Garderen, E.; Swennenhuis, J.F.; Hellmen, E.; Schalken, J.A. Growth hormone induces tyrosyl phosphorylation of the transcription factors Stat5a and Stat5b in CMT-U335 canine mammary tumor cells. Domest. Anim. Endocrinol. 2001, 20, 123–135. [Google Scholar] [CrossRef]
- Brown, M.E.; Bear, M.D.; Rosol, T.J.; Premanandan, C.; Kisseberth, W.C.; London, C.A. Characterization of STAT3 expression, signaling and inhibition in feline oral squamous cell carcinoma. BMC Vet. Res. 2015, 11, 206. [Google Scholar] [CrossRef]
- Petterino, C.; Podesta, G.; Ratto, A.; Drigo, M.; Pellegrino, C. Immunohistochemical study of phospho-Stat3-ser727 expression in feline mammary gland tumours. Vet. Res. Commun. 2007, 31, 173–184. [Google Scholar] [CrossRef]
- Petterino, C.; Ratto, A.; Podesta, G.; Drigo, M.; Pellegrino, C. Immunohistochemical evaluation of STAT3-p-tyr705 expression in feline mammary gland tumours and correlation with histologic grade. Res. Vet. Sci. 2007, 82, 218–224. [Google Scholar] [CrossRef]
- Willmann, M.; Hadzijusufovic, E.; Hermine, O.; Dacasto, M.; Marconato, L.; Bauer, K.; Peter, B.; Gamperl, S.; Eisenwort, G.; Jensen-Jarolim, E.; et al. Comparative oncology: The paradigmatic example of canine and human mast cell neoplasms. Vet. Comp. Oncol. 2019, 17, 1–10. [Google Scholar] [CrossRef]
- Makielski, K.M.; Mills, L.J.; Sarver, A.L.; Henson, M.S.; Spector, L.G.; Naik, S.; Modiano, J.F. Risk factors for development of canine and human osteosarcoma: A comparative review. Vet. Sci. 2019, 6, 48. [Google Scholar] [CrossRef]
- Rowell, J.L.; McCarthy, D.O.; Alvarez, C.E. Dog models of naturally occurring cancer. Trends Mol. Med. 2011, 17, 380–388. [Google Scholar] [CrossRef]
- Mirabello, L.; Troisi, R.J.; Savage, S.A. Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the surveillance, epidemiology, and end results program. Cancer 2009, 115, 1531–1543. [Google Scholar] [CrossRef] [PubMed]
- Fenger, J.M.; London, C.A.; Kisseberth, W.C. Canine osteosarcoma: A naturally occurring disease to inform pediatric oncology. ILAR J. 2014, 55, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Fossey, S.L.; Liao, A.T.; McCleese, J.K.; Bear, M.D.; Lin, J.; Li, P.K.; Kisseberth, W.C.; London, C.A. Characterization of STAT3 activation and expression in canine and human osteosarcoma. BMC Cancer 2009, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Fossey, S.L.; Bear, M.D.; Lin, J.; Li, C.; Schwartz, E.B.; Li, P.K.; Fuchs, J.R.; Fenger, J.; Kisseberth, W.C.; London, C.A. The novel curcumin analog FLLL32 decreases STAT3 DNA binding activity and expression, and induces apoptosis in osteosarcoma cell lines. BMC Cancer 2011, 11, 112. [Google Scholar] [CrossRef] [PubMed]
- Couto, J.I.; Bear, M.D.; Lin, J.; Pennel, M.; Kulp, S.K.; Kisseberth, W.C.; London, C.A. Biologic activity of the novel small molecule STAT3 inhibitor LLL12 against canine osteosarcoma cell lines. BMC Vet. Res. 2012, 8, 244. [Google Scholar] [CrossRef] [PubMed]
- Onimoe, G.I.; Liu, A.; Lin, L.; Wei, C.C.; Schwartz, E.B.; Bhasin, D.; Li, C.; Fuchs, J.R.; Li, P.K.; Houghton, P.; et al. Small molecules, LLL12 and FLLL32, inhibit STAT3 and exhibit potent growth suppressive activity in osteosarcoma cells and tumor growth in mice. Invest. New Drugs 2012, 30, 916–926. [Google Scholar] [CrossRef]
- Paoloni, M.; Davis, S.; Lana, S.; Withrow, S.; Sangiorgi, L.; Picci, P.; Hewitt, S.; Triche, T.; Meltzer, P.; Khanna, C. Canine tumor cross-species genomics uncovers targets linked to osteosarcoma progression. BMC Genomics 2009, 10, 625. [Google Scholar] [CrossRef]
- Rodriguez, C.O., Jr. Using canine osteosarcoma as a model to assess efficacy of novel therapies: Can old dogs teach us new tricks? Adv. Exp. Med. Biol. 2014, 804, 237–256. [Google Scholar] [CrossRef]
- London, C.A.; Seguin, B. Mast cell tumors in the dog. Vet. Clin. North. Am. Small Anim. Pract. 2003, 33, 473–489. [Google Scholar] [CrossRef]
- Misdorp, W. Mast cells and canine mast cell tumours. A review. Vet. Q. 2004, 26, 156–169. [Google Scholar] [CrossRef]
- Shoop, S.J.; Marlow, S.; Church, D.B.; English, K.; McGreevy, P.D.; Stell, A.J.; Thomson, P.C.; O’Neill, D.G.; Brodbelt, D.C. Prevalence and risk factors for mast cell tumours in dogs in England. Canine Genet. Epidemiol. 2015, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Tefferi, A.; Lasho, T.L.; Finke, C.; Patnaik, M.; Butterfield, J.H.; McClure, R.F.; Li, C.Y.; Pardanani, A. Systemic mastocytosis in 342 consecutive adults: Survival studies and prognostic factors. Blood 2009, 113, 5727–5736. [Google Scholar] [CrossRef] [PubMed]
- Letard, S.; Yang, Y.; Hanssens, K.; Palmerini, F.; Leventhal, P.S.; Guery, S.; Moussy, A.; Kinet, J.P.; Hermine, O.; Dubreuil, P. Gain-of-function mutations in the extracellular domain of KIT are common in canine mast cell tumors. Mol. Cancer Res. 2008, 6, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- London, C.A.; Galli, S.J.; Yuuki, T.; Hu, Z.Q.; Helfand, S.C.; Geissler, E.N. Spontaneous canine mast cell tumors express tandem duplications in the proto-oncogene c-kit. Exp. Hematol. 1999, 27, 689–697. [Google Scholar] [CrossRef]
- Nagata, H.; Worobec, A.S.; Oh, C.K.; Chowdhury, B.A.; Tannenbaum, S.; Suzuki, Y.; Metcalfe, D.D. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc. Natl. Acad. Sci. USA 1995, 92, 10560–10564. [Google Scholar] [CrossRef]
- Metcalfe, D.D. Mast cells and mastocytosis. Blood 2008, 112, 946–956. [Google Scholar] [CrossRef]
- Iemura, A.; Tsai, M.; Ando, A.; Wershil, B.K.; Galli, S.J. The c-kit ligand, stem cell factor, promotes mast cell survival by suppressing apoptosis. Am. J. Pathol. 1994, 144, 321–328. [Google Scholar]
- Moller, C.; Alfredsson, J.; Engstrom, M.; Wootz, H.; Xiang, Z.; Lennartsson, J.; Jonsson, J.I.; Nilsson, G. Stem cell factor promotes mast cell survival via inactivation of FOXO3a-mediated transcriptional induction and MEK-regulated phosphorylation of the proapoptotic protein Bim. Blood 2005, 106, 1330–1336. [Google Scholar] [CrossRef]
- Hahn, K.A.; Ogilvie, G.; Rusk, T.; Devauchelle, P.; Leblanc, A.; Legendre, A.; Powers, B.; Leventhal, P.S.; Kinet, J.P.; Palmerini, F.; et al. Masitinib is safe and effective for the treatment of canine mast cell tumors. J. Vet. Intern. Med. 2008, 22, 1301–1309. [Google Scholar] [CrossRef]
- London, C.A.; Malpas, P.B.; Wood-Follis, S.L.; Boucher, J.F.; Rusk, A.W.; Rosenberg, M.P.; Henry, C.J.; Mitchener, K.L.; Klein, M.K.; Hintermeister, J.G.; et al. Multi-center, placebo-controlled, double-blind, randomized study of oral toceranib phosphate (SU11654), a receptor tyrosine kinase inhibitor, for the treatment of dogs with recurrent (either local or distant) mast cell tumor following surgical excision. Clin. Cancer Res. 2009, 15, 3856–3865. [Google Scholar] [CrossRef]
- Chaix, A.; Lopez, S.; Voisset, E.; Gros, L.; Dubreuil, P.; De Sepulveda, P. Mechanisms of STAT protein activation by oncogenic KIT mutants in neoplastic mast cells. J. Biol. Chem. 2011, 286, 5956–5966. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, C.; Cerny-Reiterer, S.; Sonneck, K.; Mayerhofer, M.; Gleixner, K.V.; Fritz, R.; Kerenyi, M.; Boudot, C.; Gouilleux, F.; Kornfeld, J.W.; et al. Expression of activated STAT5 in neoplastic mast cells in systemic mastocytosis: Subcellular distribution and role of the transforming oncoprotein KIT D816V. Am. J. Pathol. 2009, 175, 2416–2429. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Wingelhofer, B.; Peter, B.; Bauer, K.; Berger, D.; Gamperl, S.; Reifinger, M.; Cerny-Reiterer, S.; Moriggl, R.; Willmann, M.; et al. The JAK2/STAT5 signaling pathway as a potential therapeutic target in canine mastocytoma. Vet. Comp. Oncol. 2018, 16, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Peter, B.; Bibi, S.; Eisenwort, G.; Wingelhofer, B.; Berger, D.; Stefanzl, G.; Blatt, K.; Herrmann, H.; Hadzijusufovic, E.; Hoermann, G.; et al. Drug-induced inhibition of phosphorylation of STAT5 overrides drug resistance in neoplastic mast cells. Leukemia 2018, 32, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Page, B.D.; Khoury, H.; Laister, R.C.; Fletcher, S.; Vellozo, M.; Manzoli, A.; Yue, P.; Turkson, J.; Minden, M.D.; Gunning, P.T. Small molecule STAT5-SH2 domain inhibitors exhibit potent antileukemia activity. J. Med. Chem. 2012, 55, 1047–1055. [Google Scholar] [CrossRef]
- Park, J.S.; Withers, S.S.; Modiano, J.F.; Kent, M.S.; Chen, M.; Luna, J.I.; Culp, W.T.N.; Sparger, E.E.; Rebhun, R.B.; Monjazeb, A.M.; et al. Canine cancer immunotherapy studies: Linking mouse and human. J. Immunother. Cancer 2016, 4, 97. [Google Scholar] [CrossRef]
- Alvarez, C.E. Naturally occurring cancers in dogs: Insights for translational genetics and medicine. ILAR J. 2014, 55, 16–45. [Google Scholar] [CrossRef]
- Furdos, I.; Fazekas, J.; Singer, J.; Jensen-Jarolim, E. Translating clinical trials from human to veterinary oncology and back. J. Transl. Med. 2015, 13, 265. [Google Scholar] [CrossRef]
- Riccardo, F.; Aurisicchio, L.; Impellizeri, J.A.; Cavallo, F. The importance of comparative oncology in translational medicine. Cancer Immunol. Immunother. 2015, 64, 137–148. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).