Structure and Function of the Nuclear Receptor Superfamily and Current Targeted Therapies of Prostate Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Class I: Overview of Steroid Hormone Receptors, Structure and Function
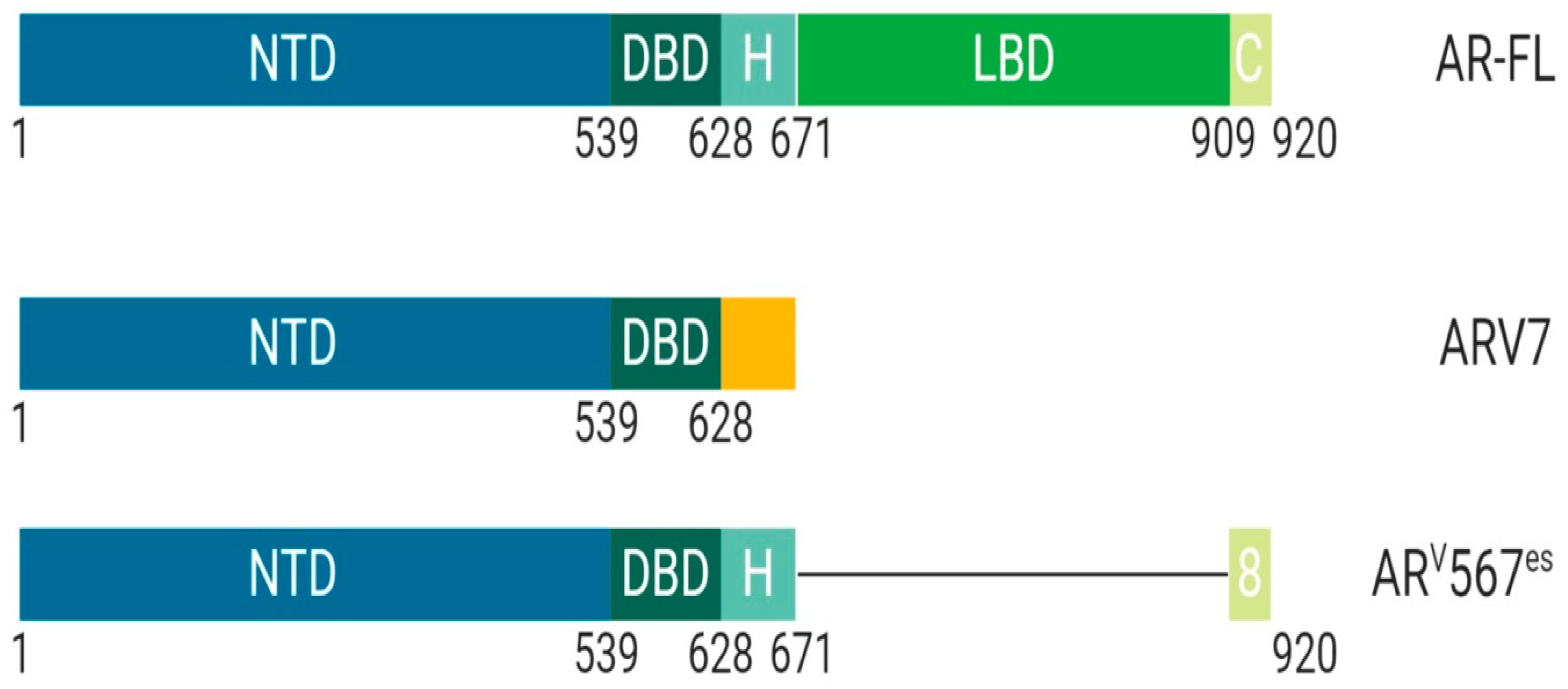
1.1.1. Class I: The Androgen Receptor, Structural and Functional Differences
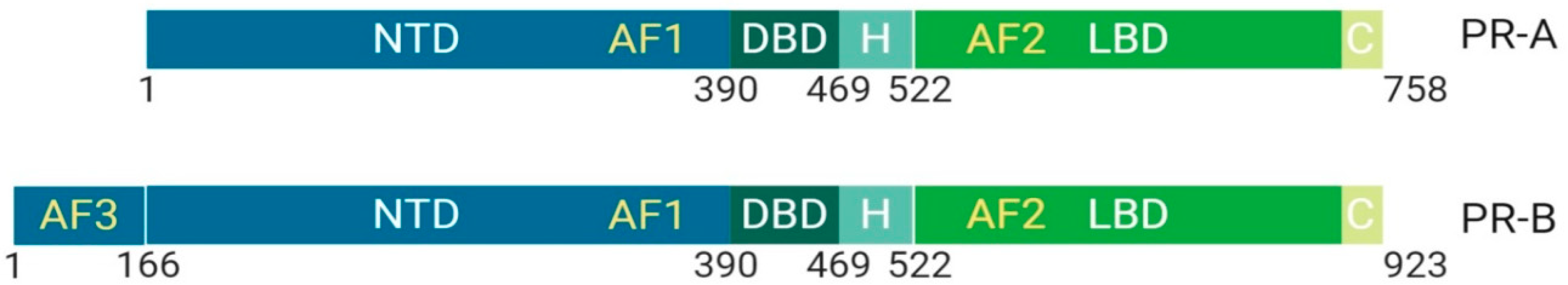
1.1.2. Class I: The Progesterone Receptor, Structural and Functional Differences
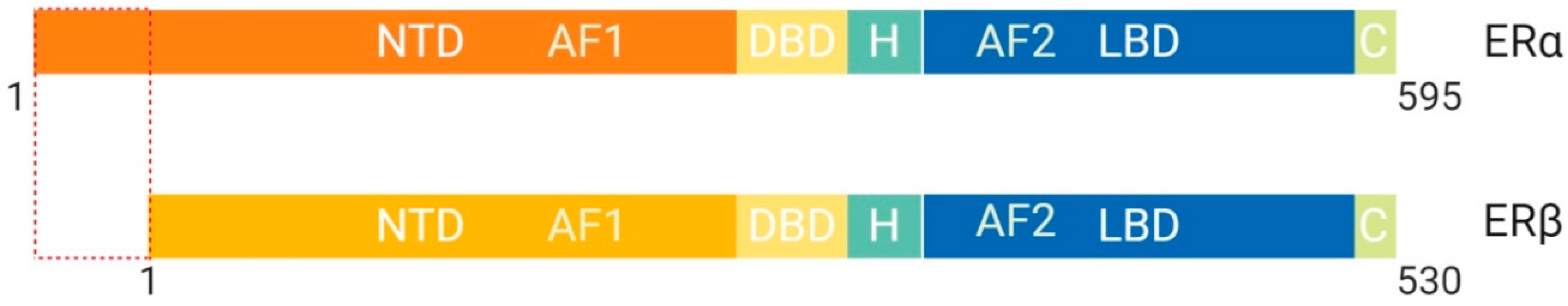
1.1.3. Class I: The Estrogen Receptor, Structural and Functional Differences
1.1.4. Class I: The Glucocorticoid Receptor, Structural and Functional Differences
1.2. Class II: RXR Heterodimers, Structure and Function
1.3. Class III: Homodimeric Orphan Receptors, Structure and Function
1.4. Class IV: Monomeric Orphan Receptors, Structure and Function
2. Overview of Targeting Transcription Factors in Prostate Cancer
2.1. Currently Targeted Transcription Factor in Primary Prostate Cancer
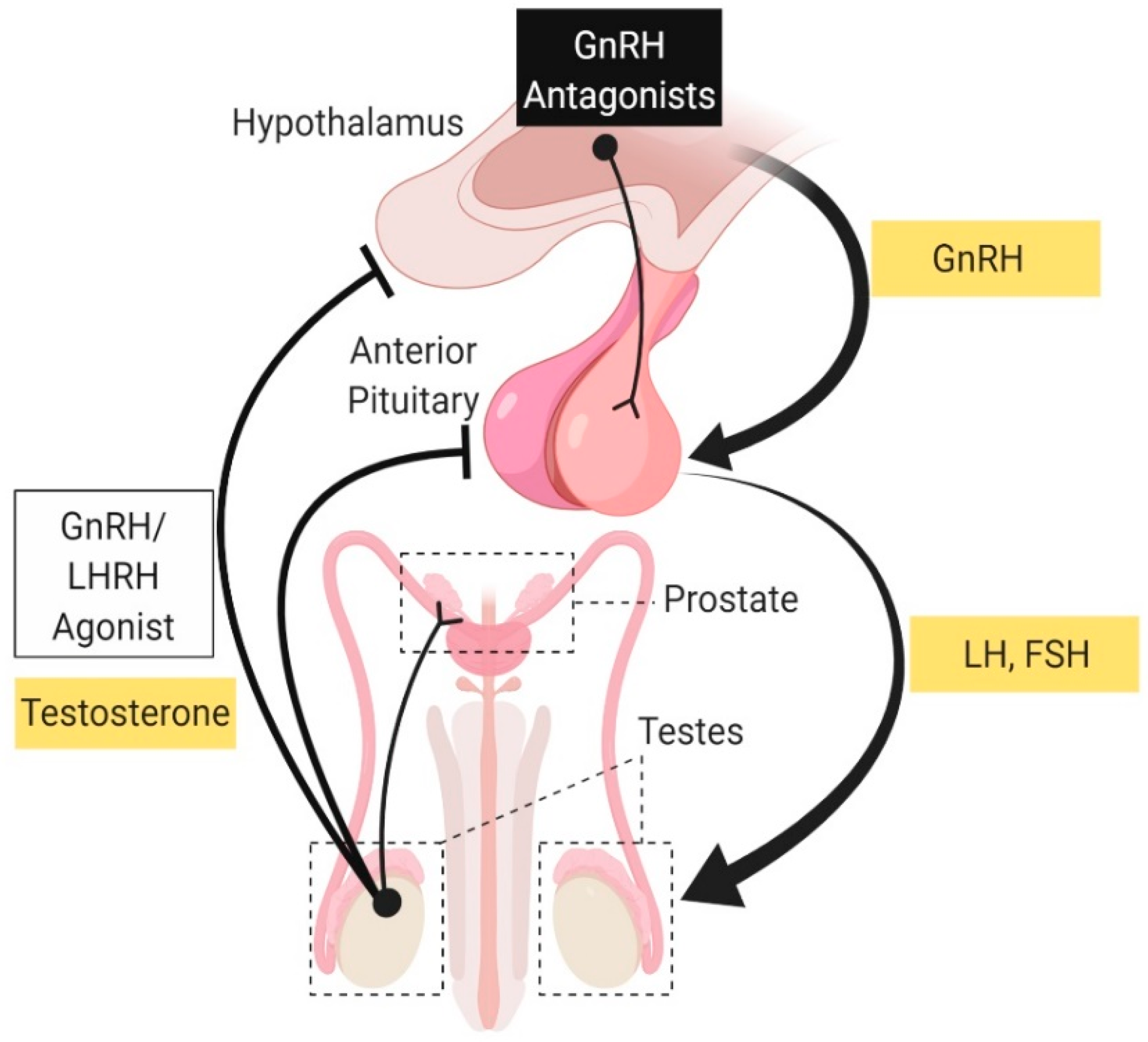
2.1.1. Androgen Deprivation Therapy: Luteinizing Hormone-Releasing Hormone (LHRH) Agonist and Antagonist
2.1.2. Anti-Androgen Therapy: Bicalutamide
2.2. Currently Targeted Transcription Factors in Castrate Resistant Prostate Cancer (CRPC)
2.2.1. Androgen Biosynthesis Inhibitor: Abiraterone
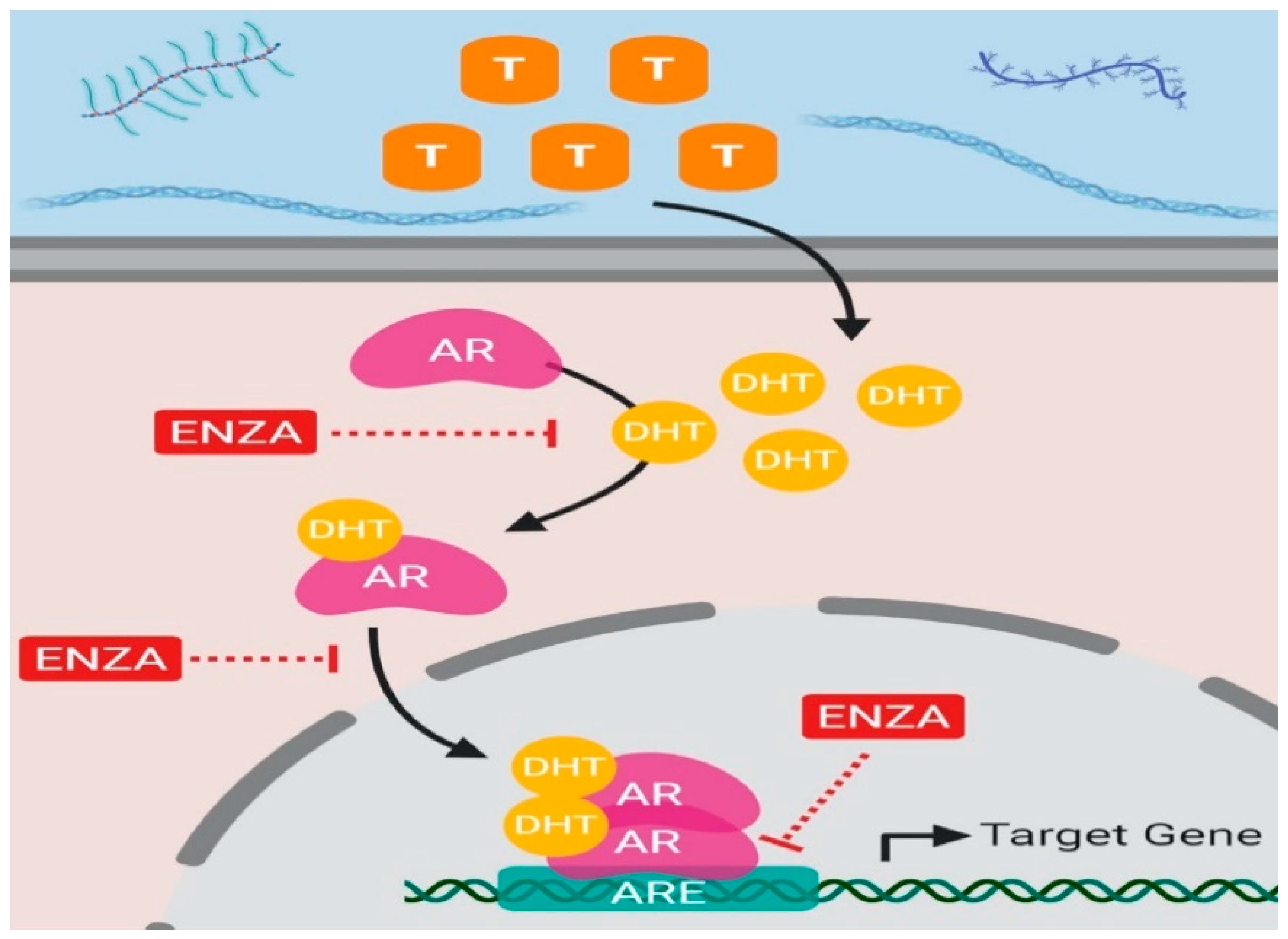
2.2.2. Androgen Receptor Antagonists: Enzalutamide, Darolutamide
2.3. Targeting STAT3 in Androgen-Independent Prostate Cancer
2.4. Use of Antisense Oligonucleotides (ASO) in Androgen-Independent Prostate Cancer
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schütz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef]
- Umesono, K.; Evans, R.M. Determinants of target gene specificity for steroid/thyroid hormone receptors. Cell 1989, 57, 1139–1146. [Google Scholar] [CrossRef]
- Mangelsdorf, D.J.; Evans, R.M. The RXR heterodimers and orphan receptors. Cell 1995, 83, 841–850. [Google Scholar] [CrossRef]
- Hall, J.M.; McDonnell, D.P.; Korach, K.S. Allosteric regulation of estrogen receptor structure, function, and coactivator recruitment by different estrogen response elements. Mol. Endocrinol. 2002, 16, 469–486. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.H.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, P.L.; Jivan, A.; Dollins, D.E.; Claessens, F.; Gewirth, D.T. Structural basis of androgen receptor binding to selective androgen response elements. Proc. Natl. Acad. Sci. USA 2004, 101, 4758–4763. [Google Scholar] [CrossRef]
- Huang, P.; Chandra, V.; Rastinejad, F. Structural overview of the nuclear receptor superfamily: Insights into physiology and therapeutics. Annu. Rev. Physiol. 2010, 72, 247–272. [Google Scholar] [CrossRef]
- Simons, S.S., Jr.; Edwards, D.P.; Kumar, R. Minireview: Dynamic Structures of Nuclear Hormone Receptors: New Promises and Challenges. Mol. Endocrinol. 2014, 28, 173–182. [Google Scholar] [CrossRef]
- Askew, E.B.; Minges, J.T.; Hnat, A.T.; Wilson, E.M. Structural features discriminate androgen receptor N/C terminal and coactivator interactions. Mol. Cell. Endocrinol. 2012, 348, 403–410. [Google Scholar] [CrossRef]
- Hill, K.K.; Roemer, S.C.; Churchill, M.E.; Edwards, D.P. Structural and functional analysis of domains of the progesterone receptor. Mol. Cell. Endocrinol. 2012, 348, 418–429. [Google Scholar] [CrossRef]
- Kumar, R.; Thompson, E.B. Transactivation Functions of the N-Terminal Domains of Nuclear Hormone Receptors: Protein Folding and Coactivator Interactions. Mol. Endocrinol. 2003, 17, 1–10. [Google Scholar] [CrossRef]
- McEwan, I.J.; Lavery, D.; Fischer, K.; Watt, K. Natural disordered sequences in the amino terminal domain of nuclear receptors: Lessons from the androgen and glucocorticoid receptors. Nucl. Recept. Signal. 2007, 5, e001. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.A.; Grossmann, M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin. Biochem. Rev. 2016, 37, 3–15. [Google Scholar] [PubMed]
- Davies, P.; Watt, K.; Kelly, S.M.; Clark, C.; Price, N.C.; McEwan, I.J. Consequences of poly-glutamine repeat length for the conformation and folding of the androgen receptor amino-terminal domain. J. Mol. Endocrinol. 2008, 41, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Cato, L.; de Tribolet-Hardy, J.; Lee, I.; Rottenberg, J.T.; Coleman, I.; Melchers, D.; Houtman, R.; Xiao, T.; Li, W.; Uo, T.; et al. ARv7 Represses Tumor-Suppressor Genes in Castration-Resistant Prostate Cancer. Cancer Cell 2019, 35, 401–413.e6. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Sprenger, C.C.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Investig. 2010, 120, 2715–2730. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yang, O.; Fazli, L.; Rennie, P.S.; Gleave, M.E.; Dong, X. Progesterone receptor expression during prostate cancer progression suggests a role of this receptor in stromal cell differentiation. Prostate 2015, 75, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Hopp, T.A.; Weiss, H.L.; Hilsenbeck, S.G.; Cui, Y.; Allred, D.C.; Horwitz, K.B.; Fuqua, S.A. Breast cancer patients with progesterone receptor PR-A-rich tumors have poorer disease-free survival rates. Clin. Cancer Res. 2004, 10, 2751–2760. [Google Scholar] [CrossRef] [PubMed]
- Jonak, C.R.; Lainez, N.M.; Roybal, L.L.; Williamson, A.D.; Coss, D. c-JUN Dimerization Protein 2 (JDP2) Is a Transcriptional Repressor of Follicle-stimulating Hormone β (FSHβ) and Is Required for Preventing Premature Reproductive Senescence in Female Mice. J. Biol. Chem. 2017, 292, 2646–2659. [Google Scholar] [CrossRef]
- Wardell, S.E.; Boonyaratanakornkit, V.; Adelman, J.S.; Aronheim, A.; Edwards, D.P. Jun dimerization protein 2 functions as a progesterone receptor N-terminal domain coactivator. Mol. Cell. Biol. 2002, 22, 5451–5466. [Google Scholar] [CrossRef]
- Kato, J.; Svensson, C.I. Chapter Nine—Role of Extracellular Damage-Associated Molecular Pattern Molecules (DAMPs) as Mediators of Persistent Pain. In Progress in Molecular Biology and Translational Science; Price, T.J., Dussor, G., Eds.; Academic Press: Elsevier, MA, USA, 2015; pp. 251–279. [Google Scholar]
- Parker, K.H.; Beury, D.W.; Ostrand-Rosenberg, S. Chapter Three—Myeloid-Derived Suppressor Cells: Critical Cells Driving Immune Suppression in the Tumor Microenvironment. In Advances in Cancer Research; Wang, X.Y., Fisher, P.B., Eds.; Academic Press: Elsevier, MA, USA, 2015; pp. 95–139. [Google Scholar]
- Amornsupak, K.; Jamjuntra, P.; Warnnissorn, M.; O-Charoenrat, P.; Sa-Nguanraksa, D.; Thuwajit, P.; Eccles, S.A.; Thuwajit, C. High ASMA (+) Fibroblasts and Low Cytoplasmic HMGB1(+) Breast Cancer Cells Predict Poor Prognosis. Clin. Breast Cancer 2017, 17, 441–452.e2. [Google Scholar] [CrossRef]
- Yu, Y.; Lee, J.S.; Xie, N.; Li, E.; Hurtado-Coll, A.; Fazli, L.; Cox, M.; Plymate, S.; Gleave, M.; Dong, X. Prostate stromal cells express the progesterone receptor to control cancer cell mobility. PLoS ONE 2014, 9, e92714. [Google Scholar] [CrossRef] [PubMed]
- Le Romancer, M.; Treilleux, I.; Bouchekioua-Bouzaghou, K.; Sentis, S.; Corbo, L. Methylation, a key step for nongenomic estrogen signaling in breast tumors. Steroids 2010, 75, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Woo, E.M.; Chong, Y.T.; Homenko, D.R.; Kraus, W.L. Acetylation of estrogen receptor alpha by p300 at lysines 266 and 268 enhances the deoxyribonucleic acid binding and transactivation activities of the receptor. Mol. Endocrinol. 2006, 20, 1479–1493. [Google Scholar] [CrossRef] [PubMed]
- Christoforou, P.; Christopoulos, P.F.; Koutsilieris, M. The role of estrogen receptor β in prostate cancer. Mol. Med. Camb. Mass 2014, 20, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Ström, A.; Treuter, E.; Warner, M.; et al. Estrogen Receptors: How Do They Signal and What Are Their Targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar] [CrossRef]
- Chanal, M.; Chevallier, P.; Raverot, V.; Fonteneau, G.; Lucia, K.; Monteserin Garcia, J.L.; Rachwan, A.; Jouanneau, E.; Trouillas, J.; Honnorat, J.; et al. Differential Effects of PI3K and Dual PI3K/mTOR Inhibition in Rat Prolactin-Secreting Pituitary Tumors. Mol. Cancer Ther. 2016, 15, 1261–1270. [Google Scholar] [CrossRef]
- Tremont, A.; Lu, J.; Cole, J.T. Endocrine Therapy for Early Breast Cancer: Updated Review. Ochsner J. 2017, 17, 405–411. [Google Scholar]
- Meijsing, S.H.; Pufall, M.A.; So, A.Y.; Bates, D.L.; Chen, L.; Yamamoto, K.R. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science 2009, 324, 407–410. [Google Scholar] [CrossRef]
- Wärnmark, A.; Gustafsson, J.Å.; Wright, A.P.H. Architectural Principles for the Structure and Function of the Glucocorticoid Receptor τ1 Core Activation Domain. J. Biol. Chem. 2000, 275, 15014–15018. [Google Scholar] [CrossRef]
- Khan, S.H.; Awasthi, S.; Guo, C.; Goswami, D.; Ling, J.; Griffin, P.R.; Simons, S.S., Jr.; Kumar, R. Binding of the N-terminal region of coactivator TIF2 to the intrinsically disordered AF1 domain of the glucocorticoid receptor is accompanied by conformational reorganizations. J. Biol. Chem. 2012, 287, 44546–44560. [Google Scholar] [CrossRef] [PubMed]
- Webb, P.; Nguyen, P.; Shinsako, J.; Anderson, C.; Feng, W.; Nguyen, M.P.; Chen, D.; Huang, S.M.; Subramanian, S.; McKinerney, E.; et al. Estrogen receptor activation function 1 works by binding p160 coactivator proteins. Mol. Endocrinol. 1998, 12, 1605–1618. [Google Scholar] [CrossRef] [PubMed]
- Morel, L. 13—Immunometabolism. In Dubois’ Lupus Erythematosus and Related Syndromes (Ninth Edition); Wallace, D.J., Hahn, B.H., Eds.; Content Repository Only: London, UK, 2019; pp. 153–163. [Google Scholar]
- Chen, Y.E.; Fu, M.; Zhang, J.; Lin, Y.; Akinbami, M.A.; Song, Q. Peroxisome Proliferator-Activated Receptors and the Cardiovascular System. In Vitamins & Hormones; Academic Press: Elsevier, MA, USA, 2003; pp. 157–188. [Google Scholar]
- Szanto, A.; Narkar, V.; Shen, Q.; Uray, I.P.; Davies, P.J.; Nagy, L. Retinoid X receptors: X-ploring their (patho)physiological functions. Cell Death Differ. 2004, 11 (Suppl. 2), S126–S143. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, J.; Robertson, C.L.; Rajasekaran, D.; Gredler, R.; Siddiq, A.; Emdad, L.; Mukhopadhyay, N.D.; Ghosh, S.; Hylemon, P.B.; Gil, G.; et al. AEG-1 Regulates Retinoid X Receptor and Inhibits Retinoid Signaling. Cancer Res. 2014, 74, 4364–4377. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Lazar, M.A. The monomer-binding orphan receptor Rev-Erb represses transcription as a dimer on a novel direct repeat. Mol. Cell. Biol. 1995, 15, 4791–4802. [Google Scholar] [CrossRef] [PubMed]
- Dussault, I.; Fawcett, D.; Matthyssen, A.; Bader, J.A.; Giguere, V. Orphan nuclear receptor ROR alpha-deficient mice display the cerebellar defects of staggerer. Mech. Dev. 1998, 70, 147–153. [Google Scholar] [CrossRef]
- Moraitis, A.N.; Giguѐre, V. Transition from Monomeric to Homodimeric DNA Binding by Nuclear Receptors: Identification of RevErbAα Determinants Required for RORα Homodimer Complex Formation. Mol. Endocrinol. 1999, 13, 431–439. [Google Scholar] [CrossRef][Green Version]
- Lala, D.S.; Rice, D.A.; Parker, K.L. Steroidogenic factor I, a key regulator of steroidogenic enzyme expression, is the mouse homolog of fushi tarazu-factor I. Mol. Endocrinol. 1992, 6, 1249–1258. [Google Scholar]
- Lin, L.; Achermann, J.C. Steroidogenic factor-1 (SF-1, Ad4BP, NR5A1) and disorders of testis development. Sex. Dev. 2008, 2, 200–209. [Google Scholar] [CrossRef]
- Lambert, M.; Jambon, S.; Depauw, S.; David-Cordonnier, M.H. Targeting Transcription Factors for Cancer Treatment. Molecules 2018, 23, 1479. [Google Scholar] [CrossRef]
- Wade, C.A.; Kyprianou, N. Profiling Prostate Cancer Therapeutic Resistance. Int. J. Mol. Sci. 2018, 19, 904. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Solo, K.; Valant, J.; Todd, M.B.; Mehra, M. Prevalence of Prostate Cancer Clinical States and Mortality in the United States: Estimates Using a Dynamic Progression Model. PLoS ONE 2015, 10, e0139440. [Google Scholar] [CrossRef] [PubMed]
- Schally, A.V.; Comaru-Schally, A.M. Mode of Action of LHRH Analogs. In Holland-Frei Cancer Medicine, 6th ed.; BC Decker: Hamilton, ON, USA, 2003. [Google Scholar]
- Furr, B.J.A.; Tucker, H. The preclinical development of bicalutamide: Pharmacodynamics and mechanism of action. Urology 1996, 47, 13–25. [Google Scholar] [CrossRef]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a Second-Generation Antiandrogen for Treatment of Advanced Prostate Cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef]
- Hodgson, M.C.; Astapova, I.; Hollenberg, A.N.; Balk, S.P. Activity of Androgen Receptor Antagonist Bicalutamide in Prostate Cancer Cells Is Independent of NCoR and SMRT Corepressors. Cancer Res. 2007, 67, 8388–8395. [Google Scholar] [CrossRef]
- Sartor, O.; de Bono, J.S. Metastatic Prostate Cancer. N. Engl. J. Med. 2018, 378, 645–657. [Google Scholar] [CrossRef]
- Karantanos, T.; Evans, C.P.; Tombal, B.; Thompson, T.C.; Montironi, R.; Isaacs, W.B. Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level. Eur. Urol. 2015, 67, 470–479. [Google Scholar] [CrossRef]
- Haapala, K.; Hyytinen, E.R.; Roiha, M.; Laurila, M.; Rantala, I.; Helin, H.J.; Koivisto, P.A. Androgen receptor alterations in prostate cancer relapsed during a combined androgen blockade by orchiectomy and bicalutamide. Lab. Investig. 2001, 81, 1647–1651. [Google Scholar] [CrossRef][Green Version]
- Titus, M.A.; Schell, M.J.; Lih, F.B.; Tomer, K.B.; Mohler, J.L. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin. Cancer Res. 2005, 11, 4653–4657. [Google Scholar] [CrossRef]
- Waltering, K.K.; Urbanucci, A.; Visakorpi, T. Androgen receptor (AR) aberrations in castration-resistant prostate cancer. Mol. Cell. Endocrinol. 2012, 360, 38–43. [Google Scholar] [CrossRef]
- Chen CDWelsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Rehman, Y.; Rosenberg, J.E. Abiraterone acetate: Oral androgen biosynthesis inhibitor for treatment of castration-resistant prostate cancer. Drug Des. Dev. Ther. 2012, 6, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Cancio, M.; Camats, N.; Flück, C.E.; Zalewski, A.; Dick, B.; Frey, B.M.; Monné, R.; Torán, N.; Audí, L.; Pandey, A.V. Mechanism of the Dual Activities of Human CYP17A1 and Binding to Anti-Prostate Cancer Drug Abiraterone Revealed by a Novel V366M Mutation Causing 17,20 Lyase Deficiency. Pharmaceuticals 2018, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B.; Saad, F.; et al. Abiraterone and Increased Survival in Metastatic Prostate Cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Mostaghel, E.A.; Marck, B.T.; Plymate, S.R.; Vessella, R.L.; Balk, S.; Matsumoto, A.M.; Nelson, P.S.; Montgomery, R.B. Resistance to CYP17A1 Inhibition with Abiraterone in Castration-Resistant Prostate Cancer: Induction of Steroidogenesis and Androgen Receptor Splice Variants. Clin. Cancer Res. 2011, 17, 5913. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Lu, C.; Mostaghel, E.A.; Yegnasubramanian, S.; Gurel, M.; Tannahil, C.; Edwards, J.; Isaascs, W.B.; Nelson, P.S.; Bluemn, E.; et al. Distinct Transcriptional Programs Mediated by the Ligand-Dependent Full-Length Androgen Receptor and Its Splice Variants in Castration-Resistant Prostate Cancer. Cancer Res. 2012, 72, 3457–3462. [Google Scholar] [CrossRef]
- Schalken, J.; Fitzpatrick, J.M. Enzalutamide: Targeting the androgen signalling pathway in metastatic castration-resistant prostate cancer. BJU Int. 2016, 117, 215–225. [Google Scholar] [CrossRef]
- Balbas, M.D.; Evans, M.J.; Hosfield, D.J.; Wongvipat, J.; Arora, V.K.; Watson, P.A.; Chen, Y.; Greene, G.L.; Shen, Y.; Sawyers, C.L. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife 2013, 2, e00499. [Google Scholar] [CrossRef]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef]
- Yang, Y.C.; Banuelos, C.A.; Mawji, N.R.; Wang, J.; Kato, M.; Haile, S.; McEwan, I.J.; Plymate, S.; Sadar, M.D. Targeting Androgen Receptor Activation Function-1 with EPI to Overcome Resistance Mechanisms in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 4466–4477. [Google Scholar] [CrossRef]
- Fizazi, K.; Shore, N.; Tammela, T.L.; Ulys, A.; Vjaters, E.; Polyakov, S.; Jievaltas, M.; Luz, M.; Alekseev, B.; Kuss, I.; et al. Darolutamide in Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2019, 380, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.M.; Howard, L.E.; Sourbeer, K.N.; Amarasekara, H.S.; Chow, L.C.; Cockrell, D.C.; Pratson, C.L.; Hanyok, B.T.; Aronson, W.J.; Kane, C.J.; et al. Predicting Time From Metastasis to Overall Survival in Castration-Resistant Prostate Cancer: Results From SEARCH. Clin. Genitourin. Cancer 2017, 15, 60–66.e2. [Google Scholar] [CrossRef] [PubMed]
- Abdulghani, J.; Gu, L.; Dagvadorj, A.; Lutz, J.; Leiby, B.; Bonuccelli, G.; Lisanti, M.P.; Zellweger, T.; Alanen, K.; Mirtti, T.; et al. Stat3 promotes metastatic progression of prostate cancer. Am. J. Pathol. 2008, 172, 1717–1728. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.; Adamus, T.; Zhao, X.; Su, Y.L.; Zhang, Z.; White, S.V.; Swideski, P.; Lu, X.; DePinho, R.A.; Pal, S.K.; et al. STAT3 Inhibition Combined with CpG Immunostimulation Activates Antitumor Immunity to Eradicate Genetically Distinct Castration-Resistant Prostate Cancers. Clin. Cancer Res. 2018, 24, 5948. [Google Scholar] [CrossRef]
- Levy, D.E.; Lee, C.K. What does Stat3 do? J. Clin. Investig. 2002, 109, 1143–1148. [Google Scholar] [CrossRef]
- Rothenfusser, S.; Tuma, E.; Endres, S.; Hartmann, G. Plasmacytoid dendritic cells: The key to CpG. The authors dedicate this article to Prof. K. Lennert, M.D., Department of Hematology and Oncology, University of Kiel, Germany, who was the first to describe plasmacytoid dendritic cells histologically in 1958. Hum. Immunol. 2002, 63, 1111–1119. [Google Scholar] [CrossRef]
- Talaro, K.P. Foundations in Microbiology, 4th ed.; McGraw-Hill: Boston, MA, USA, 2002. [Google Scholar]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porter, B.A.; Ortiz, M.A.; Bratslavsky, G.; Kotula, L. Structure and Function of the Nuclear Receptor Superfamily and Current Targeted Therapies of Prostate Cancer. Cancers 2019, 11, 1852. https://doi.org/10.3390/cancers11121852
Porter BA, Ortiz MA, Bratslavsky G, Kotula L. Structure and Function of the Nuclear Receptor Superfamily and Current Targeted Therapies of Prostate Cancer. Cancers. 2019; 11(12):1852. https://doi.org/10.3390/cancers11121852
Chicago/Turabian StylePorter, Baylee A., Maria A. Ortiz, Gennady Bratslavsky, and Leszek Kotula. 2019. "Structure and Function of the Nuclear Receptor Superfamily and Current Targeted Therapies of Prostate Cancer" Cancers 11, no. 12: 1852. https://doi.org/10.3390/cancers11121852
APA StylePorter, B. A., Ortiz, M. A., Bratslavsky, G., & Kotula, L. (2019). Structure and Function of the Nuclear Receptor Superfamily and Current Targeted Therapies of Prostate Cancer. Cancers, 11(12), 1852. https://doi.org/10.3390/cancers11121852