Mechanistic Signatures of Human Papillomavirus Insertions in Anal Squamous Cell Carcinomas

 , , , , , , and
, , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Methods
2.2.1. DNA Extraction
2.2.2. HPV Genotyping
2.2.3. DNA Library Preparation
2.2.4. HPV Double Capture Method
2.2.5. HPV Viral Load
2.2.6. Bioinformatics Pipeline for Analysis of HPV Genotypes and Viral–Cellular Junctions
2.3. Statistical Analysis
3. Results
3.1. Patient Characteristics
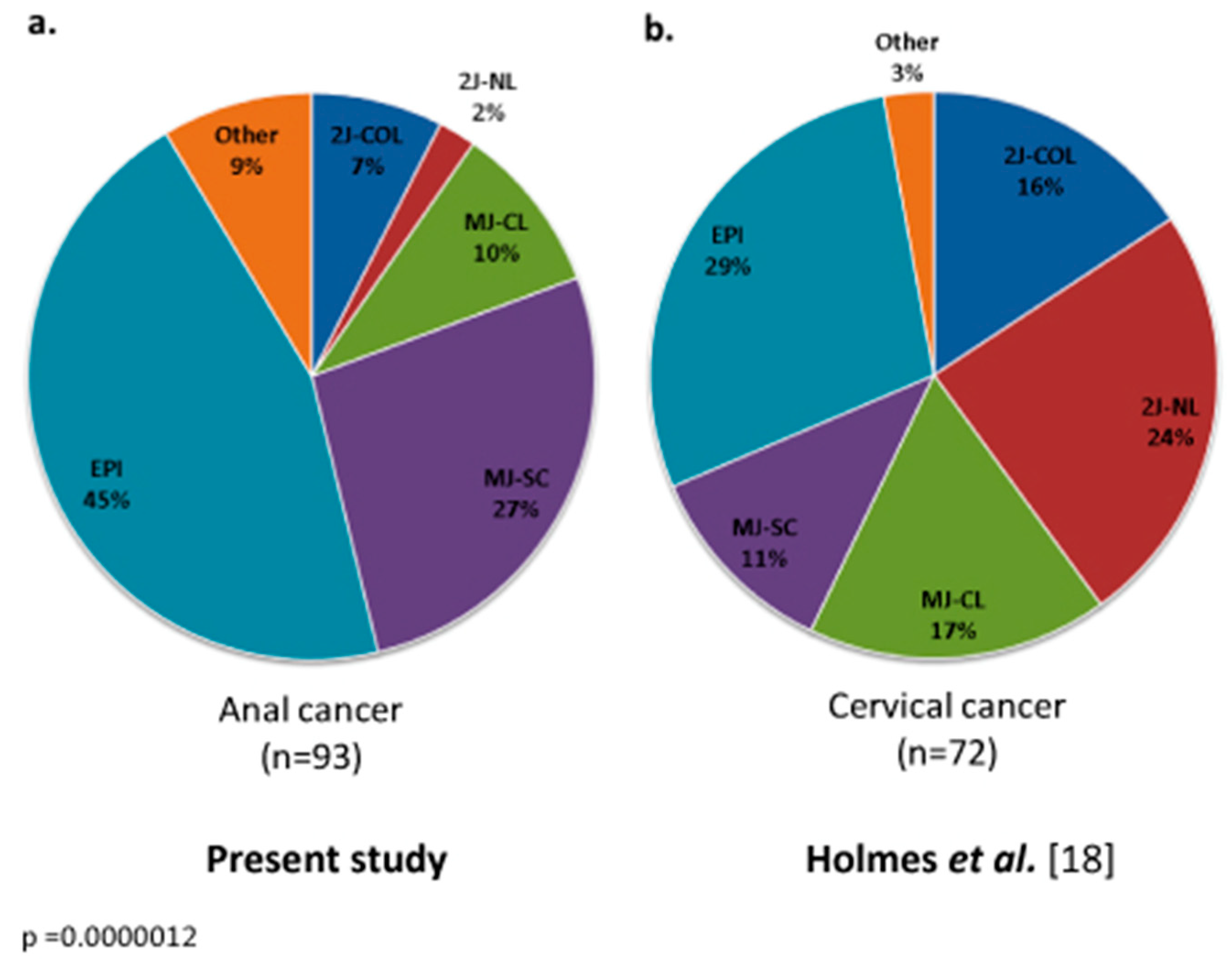
3.2. HPV Genotype and Mutational Analysis
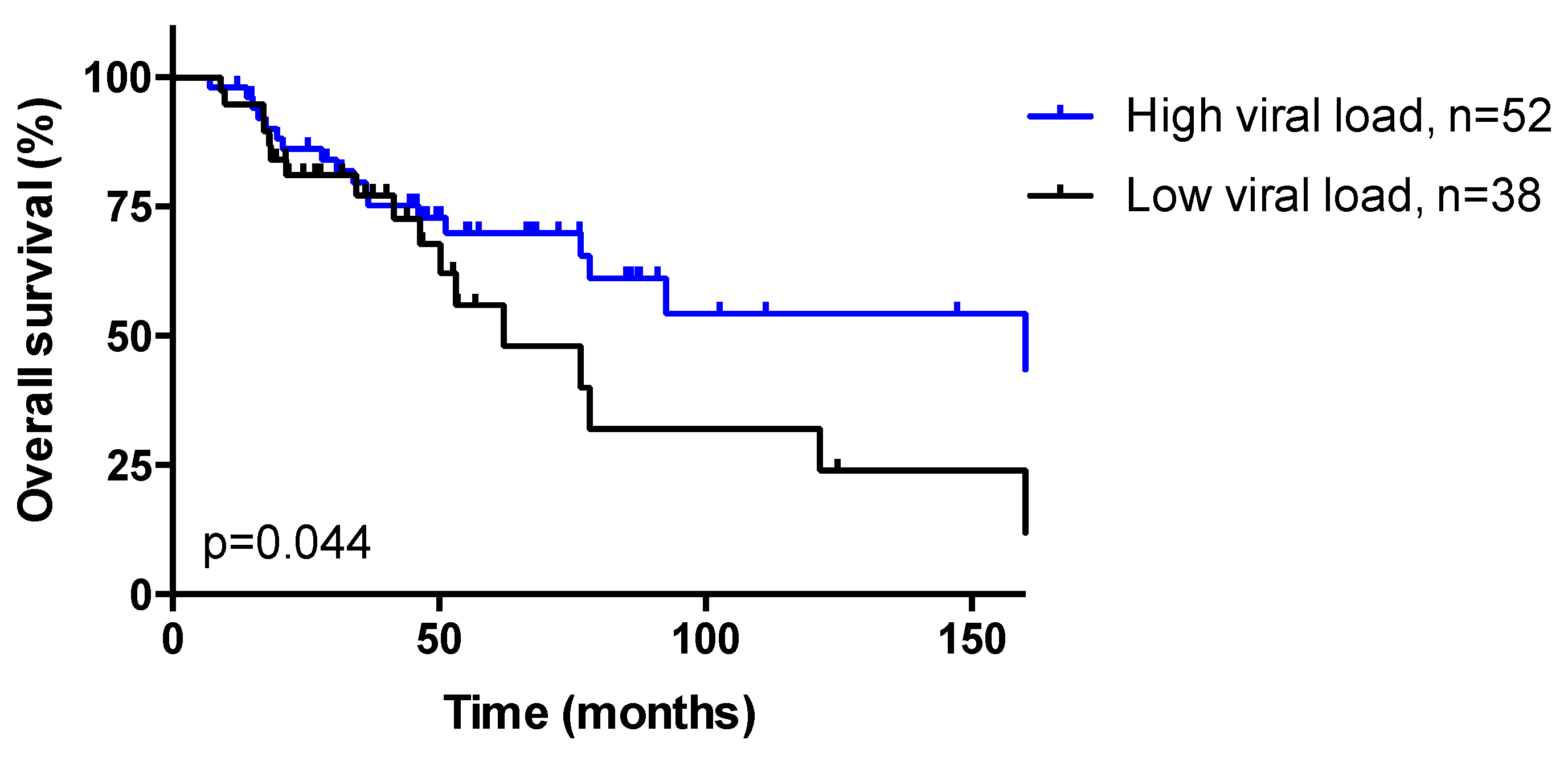
3.3. HPV Viral Load
3.4. Integration Mechanisms
3.5. Dynamic Evolution of Integration Status in Patients with Matched Samples
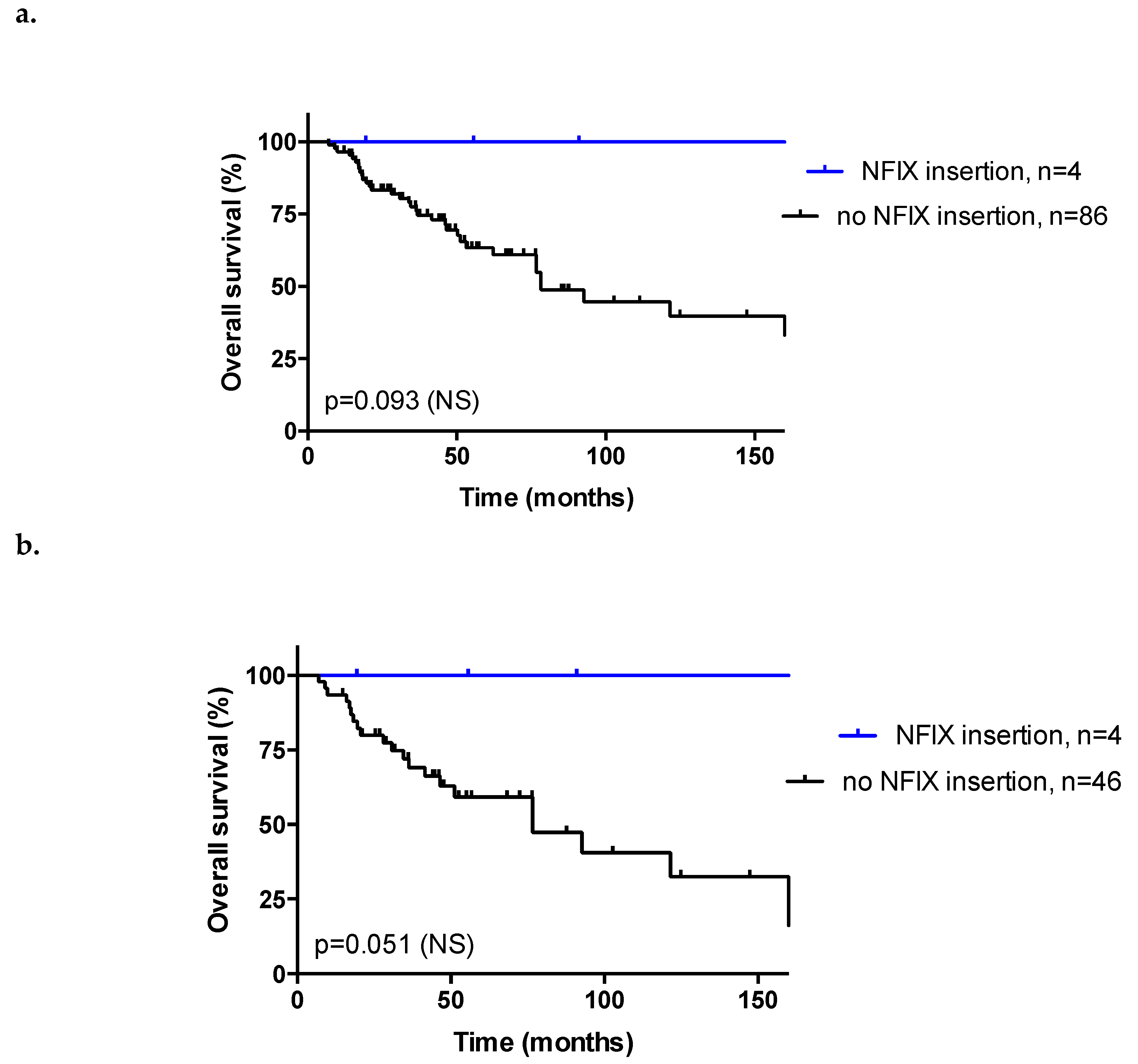
3.6. Insertion Sites
3.7. Association between Insertion Mechanisms and Patient Clinicopathological Data
3.8. Response to Treatment and Overall Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Access
Abbreviations
| 2J | Two hybrid junctions |
| 2J-COL | Two hybrid co-linear junctions |
| 2J-NL | Two hybrid non-linear junctions |
| 5FU-CDDP | 5-Fluorouracil-Cisplatin |
| 5FU-MMC | 5-Fluorouracil-Mitomycin C |
| AJCC | American Joint Committee on Cancer |
| ASCC | Anal squamous cell carcinoma |
| CR | Complete response |
| CRT | Chemo-radiotherapy |
| EPI | Episomal |
| HIV | Human immunodeficiency virus |
| HNSCC | Head and neck squamous cell carcinoma |
| HPV | Human papillomavirus |
| MJ | Multiple hybrid junction |
| MJ-CL | Multiple hybrid junctions clustered in one locus |
| MJ-SC | Multiple hybrid junctions scattered at distinct loci |
| NGS | Next-Generation Sequencing |
| NS | Not significant |
| OS | Overall survival |
| RT | Radiation therapy |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Simard, E.P.; Dorell, C.; Noone, A.M.; Markowitz, L.E.; Kohler, B.; Eheman, C.; Saraiya, M.; Bandi, P.; Saslow, D.; et al. Annual Report to the Nation on the Status of Cancer, 1975–2009, featuring the burden and trends in human papillomavirus(HPV)-associated cancers and HPV vaccination coverage levels. J. Natl. Cancer Inst. 2013, 105, 175–201. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.A.; Levine, A.M.; Bernstein, L.; Smith, D.D.; Lai, L.L. Changing patterns of anal canal carcinoma in the United States. J. Clin. Oncol. 2013, 31, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Alemany, L.; Saunier, M.; Alvarado-Cabrero, I.; Quiros, B.; Salmeron, J.; Shin, H.R.; Pirog, E.C.; Guimera, N.; Hernandez-Suarez, G.; Felix, A.; et al. Human papillomavirus DNA prevalence and type distribution in anal carcinomas worldwide. Int. J. Cancer 2015, 136, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Ouhoummane, N.; Steben, M.; Coutlee, F.; Vuong, T.; Forest, P.; Rodier, C.; Louchini, R.; Duarte, E.; Brassard, P. Squamous anal cancer: Patient characteristics and HPV type distribution. Cancer Epidemiol. 2013, 37, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Cacheux, W.; Rouleau, E.; Briaux, A.; Tsantoulis, P.; Mariani, P.; Richard-Molard, M.; Buecher, B.; Dangles-Marie, V.; Richon, S.; Lazartigues, J.; et al. Mutational analysis of anal cancers demonstrates frequent PIK3CA mutations associated with poor outcome after salvage abdominoperineal resection. Br. J. Cancer 2016, 114, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Glynne-Jones, R.; Nilsson, P.J.; Aschele, C.; Goh, V.; Peiffert, D.; Cervantes, A.; Arnold, D. Anal cancer: ESMO-ESSO-ESTRO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 5 (Suppl. 3), iii10–iii20. [Google Scholar] [CrossRef]
- NCCN Guidelines Version 1.2019—Anal Cancer 2019. Available online: www.nccn.org (accessed on 21 November 2019).
- Cacheux, W.; Lievre, A.; De La Rochefordiere, A.; Dieumegard, B.; Cvitkovic, F.; Labib, A.; Mitry, E.; Buecher, B. Chemotherapy in the treatment of anal canal carcinoma. Dig. Liver Dis. 2012, 44, 803–811. [Google Scholar] [CrossRef]
- Meulendijks, D.; Tomasoa, N.B.; Dewit, L.; Smits, P.H.; Bakker, R.; van Velthuysen, M.L.; Rosenberg, E.H.; Beijnen, J.H.; Schellens, J.H.; Cats, A. HPV-negative squamous cell carcinoma of the anal canal is unresponsive to standard treatment and frequently carries disruptive mutations in TP53. Br. J. Cancer 2015, 112, 1358–1366. [Google Scholar] [CrossRef]
- Serup-Hansen, E.; Linnemann, D.; Skovrider-Ruminski, W.; Hogdall, E.; Geertsen, P.F.; Havsteen, H. Human papillomavirus genotyping and p16 expression as prognostic factors for patients with American Joint Committee on Cancer stages I to III carcinoma of the anal canal. J. Clin. Oncol. 2014, 32, 1812–1817. [Google Scholar] [CrossRef]
- Koerber, S.A.; Schoneweg, C.; Slynko, A.; Krug, D.; Haefner, M.F.; Herfarth, K.; Debus, J.; Sterzing, F.; von Knebel Doeberitz, M.; Prigge, E.S.; et al. Influence of human papillomavirus and p16(INK4a) on treatment outcome of patients with anal cancer. Radiother. Oncol. 2014, 113, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Forman, D.; de Martel, C.; Lacey, C.J.; Soerjomataram, I.; Lortet-Tieulent, J.; Bruni, L.; Vignat, J.; Ferlay, J.; Bray, F.; Plummer, M.; et al. Global burden of human papillomavirus and related diseases. Vaccine 2012, 30 (Suppl. 5), F12–F23. [Google Scholar] [CrossRef] [PubMed]
- Harden, M.E.; Munger, K. Human papillomavirus molecular biology. Mutat. Res. Rev. Mutat. Res. 2017, 772, 3–12. [Google Scholar] [CrossRef]
- Jeannot, E.; Harle, A.; Holmes, A.; Sastre-Garau, X. Nuclear factor I X is a recurrent target for HPV16 insertions in anal carcinomas. Genes Chromosomes Cancer 2018, 57, 638–644. [Google Scholar] [CrossRef] [PubMed]
- zur Hausen, H. Papillomaviruses in anogenital cancer as a model to understand the role of viruses in human cancers. Cancer Res. 1989, 49, 4677–4681. [Google Scholar] [PubMed]
- Ziegert, C.; Wentzensen, N.; Vinokurova, S.; Kisseljov, F.; Einenkel, J.; Hoeckel, M.; von Knebel Doeberitz, M. A comprehensive analysis of HPV integration loci in anogenital lesions combining transcript and genome-based amplification techniques. Oncogene 2003, 22, 3977–3984. [Google Scholar] [CrossRef]
- Holmes, A.; Lameiras, S.; Jeannot, E.; Marie, Y.; Castera, L.; Sastre-Garau, X.; Nicolas, A. Mechanistic signatures of HPV insertions in cervical carcinomas. NPJ Genom. Med. 2016, 1, 16004. [Google Scholar] [CrossRef]
- Ferrari, A.; Vincent-Salomon, A.; Pivot, X.; Sertier, A.S.; Thomas, E.; Tonon, L.; Boyault, S.; Mulugeta, E.; Treilleux, I.; MacGrogan, G.; et al. A whole-genome sequence and transcriptome perspective on HER2-positive breast cancers. Nat. Commun. 2016, 7, 12222. [Google Scholar] [CrossRef]
- Lombard, I.; Vincent-Salomon, A.; Validire, P.; Zafrani, B.; de la Rochefordiere, A.; Clough, K.; Favre, M.; Pouillart, P.; Sastre-Garau, X. Human papillomavirus genotype as a major determinant of the course of cervical cancer. J. Clin. Oncol. 1998, 16, 2613–2619. [Google Scholar] [CrossRef]
- Hanley, J.A.; McNeil, B.J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982, 143, 29–36. [Google Scholar] [CrossRef]
- Olthof, N.C.; Speel, E.J.; Kolligs, J.; Haesevoets, A.; Henfling, M.; Ramaekers, F.C.; Preuss, S.F.; Drebber, U.; Wieland, U.; Silling, S.; et al. Comprehensive analysis of HPV16 integration in OSCC reveals no significant impact of physical status on viral oncogene and virally disrupted human gene expression. PLoS ONE 2014, 9, e88718. [Google Scholar] [CrossRef]
- Parfenov, M.; Pedamallu, C.S.; Gehlenborg, N.; Freeman, S.S.; Danilova, L.; Bristow, C.A.; Lee, S.; Hadjipanayis, A.G.; Ivanova, E.V.; Wilkerson, M.D.; et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 15544–15549. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Huang, Y.; An, J.; Yang, X.; Li, N.; Huang, M.; Wu, L. Nomograms based on HPV load for predicting survival in cervical squamous cell carcinoma: An observational study with a long-term follow-up. Chin. J. Cancer Res. 2019, 31, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Park, S.; Nam, B.H.; Roh, J.W.; Lee, C.H.; Kim, Y.H.; Shin, H.J.; Lee, S.K.; Kong, S.Y.; Seong, M.W.; et al. Low initial human papilloma viral load implicates worse prognosis in patients with uterine cervical cancer treated with radiotherapy. J. Clin. Oncol. 2009, 27, 5088–5093. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Feng, Y.; Zheng, J.; Huang, Q.; Liu, J. Low initial human papillomavirus viral load may indicate worse prognosis in patients with cervical carcinoma treated with surgery. J. Gynecol. Oncol. 2015, 26, 111–117. [Google Scholar] [CrossRef]
- Hampson, L.; El Hady, E.S.; Moore, J.V.; Kitchener, H.; Hampson, I.N. The HPV16 E6 and E7 proteins and the radiation resistance of cervical carcinoma. FASEB J. 2001, 15, 1445–1447. [Google Scholar] [CrossRef]
- Valmary-Degano, S.; Jacquin, E.; Pretet, J.L.; Monnien, F.; Girardo, B.; Arbez-Gindre, F.; Joly, M.; Bosset, J.F.; Kantelip, B.; Mougin, C. Signature patterns of human papillomavirus type 16 in invasive anal carcinoma. Hum. Pathol. 2013, 44, 992–1002. [Google Scholar] [CrossRef]
- Petrelli, F.; Sarti, E.; Barni, S. Predictive value of human papillomavirus in oropharyngeal carcinoma treated with radiotherapy: An updated systematic review and meta-analysis of 30 trials. Head Neck 2014, 36, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.; Chakravarthy, A.; Su, X.; Boshoff, C.; Fenton, T.R. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep. 2014, 7, 1833–1841. [Google Scholar] [CrossRef]
- Zhang, Y.; Koneva, L.A.; Virani, S.; Arthur, A.E.; Virani, A.; Hall, P.B.; Warden, C.D.; Carey, T.E.; Chepeha, D.B.; Prince, M.E.; et al. Subtypes of HPV-Positive Head and Neck Cancers Are Associated with HPV Characteristics, Copy Number Alterations, PIK3CA Mutation, and Pathway Signatures. Clin. Cancer Res. 2016, 22, 4735–4745. [Google Scholar] [CrossRef]
- Chen, K.S.; Lim, J.W.C.; Richards, L.J.; Bunt, J. The convergent roles of the nuclear factor I transcription factors in development and cancer. Cancer Lett. 2017, 410, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N.I.A.; Abdul Murad, N.A.; Mollah, M.M.; Jamal, R.; Harun, R. NFIX as a Master Regulator for Lung Cancer Progression. Front. Pharmacol. 2017, 8, 540. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Cai, G.; Xu, Y.; Cai, S. The Plasma microRNA miR-1914* and -1915 Suppresses Chemoresistant in Colorectal Cancer Patients by Down-regulating NFIX. Curr. Mol. Med. 2016, 16, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Wu, S.N.; Gao, C.C.; Yang, X.Z.; Wang, H.G.; Zhang, J.L.; Yan, W.; Ma, T.H. Prognostic value of combined and individual expression of microRNA-1290 and its target gene nuclear factor I/X in human esophageal squamous cell carcinoma. Cancer Biomark. 2017, 20, 325–331. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Heading | Patients (%) | Univariable Analysis | Multivariable Analysis | |||
|---|---|---|---|---|---|---|
| Patients (%) | Events (%) a | OS (p-Value) a, b | HR f | 95% CI h | p-Value i | |
| Total | 93 (100) | 37 (40.9) | ||||
| Age (years) <65 ≥65 | 60 (64.5) 33 (35.5) | 24 (40.0) 13 (39.4) | 0.32 (NS) | |||
| Gender Male Female | 18 (19.4) 75 (80.6) | 6 (33.3) 31 (41.3) | 0.83 (NS) | |||
| Tumor differentiation Well/Moderate Poor | 83 (89.2) 10 (10.8) | 31 (37.3) 6 (60.0) | 0.014 | 0.40 1 | 0.14–1.16 | 0.092 (NS) |
| Basaloid contingent Present Absent | 9 (9.7) 84 (90.3) | 2 (22.2) 35 (41.7) | 0.23 (NS) | |||
| HPV status Genotype 16 Other genotypes | 85 (91.4) 8 (8.6) | 33 (38.8) 4 (50.0) | 0.005 | 1 6.78 | 1.66–27.8 | 0.008 |
| Concomitant HIV infection c Yes No | 8 (8.8) 83 (91.2) | 5 (62.5) 31 (37.3) | 0.099 (NS) | 1.87 1 | 0.57–6.16 | 0.31 (NS) |
| Tumor stage (AJCC 2010) a I II IIIA IIIB IV | 8 (8.9) 37 (41.1) 21 (23.3) 20 (22.2) 4 (4.4) | 3 (37.5) 16 (43.2) 4 (19.0) 9 (45.0) 2 (50.0) | 0.088 (NS) | 1 2.49 2.82 8.04 9.07 | 0.49–12.7 0.18–43.5 0.44–146 0.55–148 | 0.048 |
| Lymph node invasion c Yes No | 40 (44.0) 51 (56.0) | 15 (37.5) 20 (39.2) | 0.094 (NS) | 0.51 1 | 0.15–1.80 | 0.30 (NS) |
| Synchronous metastasis d Yes No | 4 (4.3) 88 (95.7) | 2 (50.0) 34 (38.6) | 0.44 (NS) | |||
| Sample status Treatment-naive tumor Tumor recurrence | 54 (58.1) 39 (41.9) | 16 (29.6) 21 (53.8) | 0.45 (NS) | |||
| Complete response after RT/CRT e Yes No | 72 (86.7) 11 (13.3) | 25 (34.7) 7 (63.6) | <0.0001 | 0.16 1 | 0.00–0.46 | 0.001 |
| PIK3CA mutational status Wild-type Mutated | 71 (76.3) 22 (23.7) | 27 (38.0) 10 (45.5) | 0.57 (NS) | |||
| Patients (%) | Number of Patients (%) | p-Value a | |||
|---|---|---|---|---|---|
| Heading | EPI | 2J | MJ | ||
| Total | 93 (100) | 42 (45.2) | 15 (16.1) | 36 (38.7) | |
| Age (years) <65 ≥65 | 60 (64.5) 33 (35.5) | 28 (66.7) 14 (33.3) | 10 (66.7) 5 (33.3) | 22 (61.1) 14 (38.9) | 0.86 (NS) |
| Gender Male Female | 18 (19.4) 75 (80.6) | 7 (16.7) 35 (83.3) | 3 (20.0) 12 (80.0) | 8 (22.2) 28 (77.8) | 0.83 (NS) |
| Tumor differentiation Well/Moderate Poor | 83 (89.2) 10 (10.8) | 37 (88.1) 5 (11.9) | 14 (93.3) 1 (6.7) | 32 (88.9) 4 (11.1) | 0.85 (NS) |
| Basaloid contingent Present Absent | 9 (9.7) 84 (90.3) | 6 (14.3) 36 (85.7) | 0 (0.0) 15 (100.0) | 3 (8.3) 33 (91.7) | 0.26 (NS) |
| HPV status Genotype 16 Other genotypes | 85 (91.4) 8 (8.6) | 36 (85.7) 6 (14.3) | 14 (93.3) 1 (6.7) | 35 (97.2) 1 (2.8) | 0.44 (NS) |
| Concomitant HIV infection b Yes No | 8 (8.8) 83 (91.2) | 3 (7.3) 38 (92.7) | 2 (13.3) 13 (86.7) | 3 (8.6) 32 (91.4) | 0.92 (NS) |
| Tumor stage (AJCC 2010) c I II IIIA IIIB IV | 8 (8.9) 37 (41.1) 21 (23.3) 20 (22.2) 4 (4.4) | 5 (11.9) 18 (42.9) 9 (21.4) 8 (19.0) 2 (4.8) | 1 (6.7) 6 (40.0) 3 (20.0) 4 (26.7) 1 (6.7) | 2 (5.6) 13 (36.1) 9 (25.0) 8 (22.2) 1 (2.8) | 0.98 (NS) |
| Lymph node invasion b Yes No | 40 (44.0) 51 (56.0) | 14 (33.3) 28 (66.7) | 8 (53.3) 7 (46.7) | 18 (52.9) 16 (47.1) | 0.17 (NS) |
| Synchronous metastasis d Yes No | 4 (4.3) 88 (95.7) | 2 (4.8) 40 (95.2) | 1 (6.7) 14 (93.3) | 1 (2.9) 34 (97.1) | 0.82 (NS) |
| Sample status Treatment-naive tumor Tumor recurrence | 54 (58.1) 39 (41.9) | 25 (59.5) 17 (40.5) | 9 (60.0) 6 (40.0) | 20 (55.6) 16 (44.4) | 0.93 (NS) |
| Initial therapy Upfront surgery d No Yes RT or CRT b RT CRT With concomitant 5FU-MMC With concomitant 5FU-CDDP With other concomitant CT No RT/CRT | 77 (83.7) 15 (16.3) 22 (24.2) 61 (67.0) 11 (18.0) 44 (77.0) 3 (4.9) 8 (8.8) | 36 (85.7) 6 (14.3) 10 (24.4) 27 (65.9) 5 (18.5) 21 (77.8) 1 (3.7) 4 (9.8) | 12 (80.0) 3 (20.0) 2 (13.3) 11 (73.3) 1 (9.1) 10 (90.9) 0 (0.0) 2 (13.3) | 29 (82.9) 6 (17.1) 10 (27.8) 23 (63.9) 5 (21.7) 16 (69.6) 2 (8.7) 2 (5.6) | 0.86 (NS) 0.76 (NS) |
| Complete Response after RT/CRT e Yes No | 72 (86.7) 11 (13.3) | 34 (91.9) 3 (7.1) | 10 (76.9) 3 (23.1) | 28 (84.8) 5 (15.2) | 0.36 (NS) |
| PIK3CA mutational status Wild-type Mutated | 71 (76.3) 22 (23.7) | 38 (90.5) 4 (9.5) | 8 (53.3) 7 (46.7) | 25 (69.4) 11 (30.6) | 0.0069 |
| HPV viral load Low (<12) High (≥12) | 41 (44.1) 52 (55.9) | 21 (50.0) 21 (50.0) | 10 (66.7) 5 (33.3) | 10 (27.8) 26 (72.2) | 0.022 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morel, A.; Neuzillet, C.; Wack, M.; Lameiras, S.; Vacher, S.; Deloger, M.; Servant, N.; Veyer, D.; Péré, H.; Mariani, O.; et al. Mechanistic Signatures of Human Papillomavirus Insertions in Anal Squamous Cell Carcinomas. Cancers 2019, 11, 1846. https://doi.org/10.3390/cancers11121846
Morel A, Neuzillet C, Wack M, Lameiras S, Vacher S, Deloger M, Servant N, Veyer D, Péré H, Mariani O, et al. Mechanistic Signatures of Human Papillomavirus Insertions in Anal Squamous Cell Carcinomas. Cancers. 2019; 11(12):1846. https://doi.org/10.3390/cancers11121846
Chicago/Turabian StyleMorel, Adeline, Cindy Neuzillet, Maxime Wack, Sonia Lameiras, Sophie Vacher, Marc Deloger, Nicolas Servant, David Veyer, Hélène Péré, Odette Mariani, and et al. 2019. "Mechanistic Signatures of Human Papillomavirus Insertions in Anal Squamous Cell Carcinomas" Cancers 11, no. 12: 1846. https://doi.org/10.3390/cancers11121846
APA StyleMorel, A., Neuzillet, C., Wack, M., Lameiras, S., Vacher, S., Deloger, M., Servant, N., Veyer, D., Péré, H., Mariani, O., Baulande, S., Rouzier, R., Kamal, M., El Alam, E., Jeannot, E., Nicolas, A., Bièche, I., & Cacheux, W. (2019). Mechanistic Signatures of Human Papillomavirus Insertions in Anal Squamous Cell Carcinomas. Cancers, 11(12), 1846. https://doi.org/10.3390/cancers11121846