Targeting Angiogenesis by Blocking the ATM–SerRS–VEGFA Pathway for UV-Induced Skin Photodamage and Melanoma Growth


Abstract
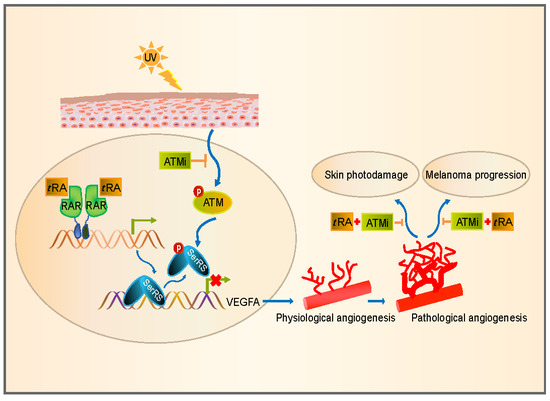
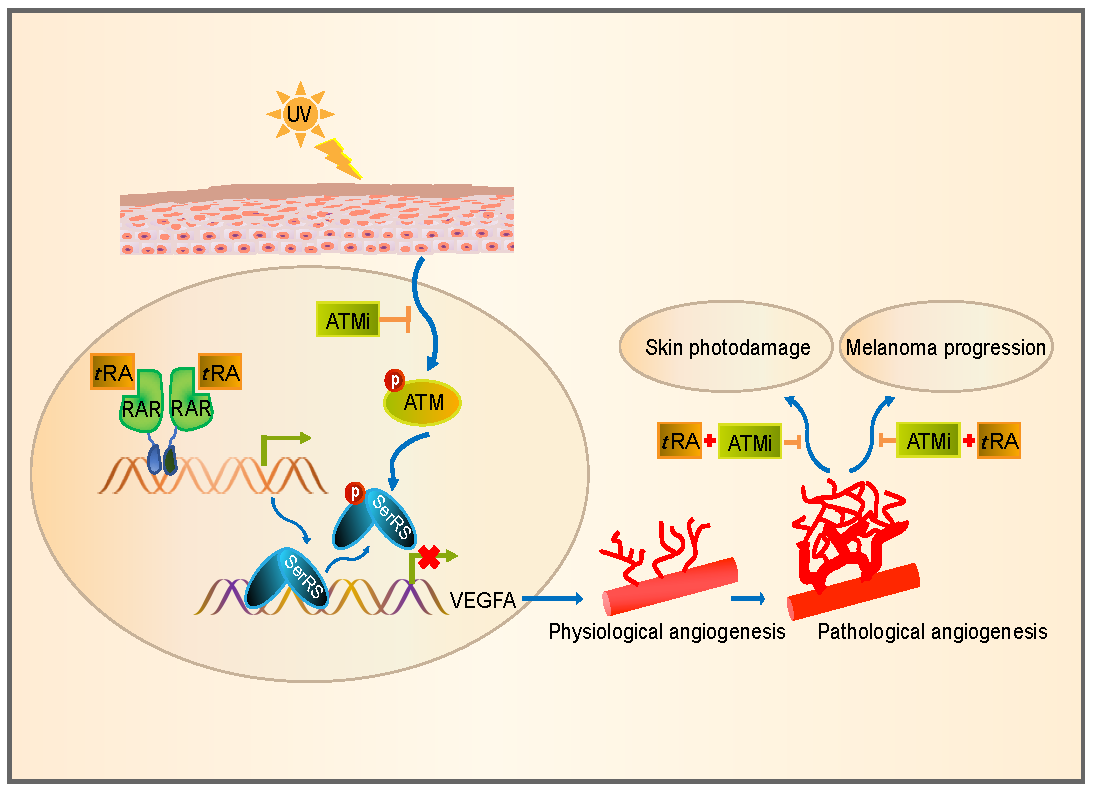
1. Introduction
2. Results
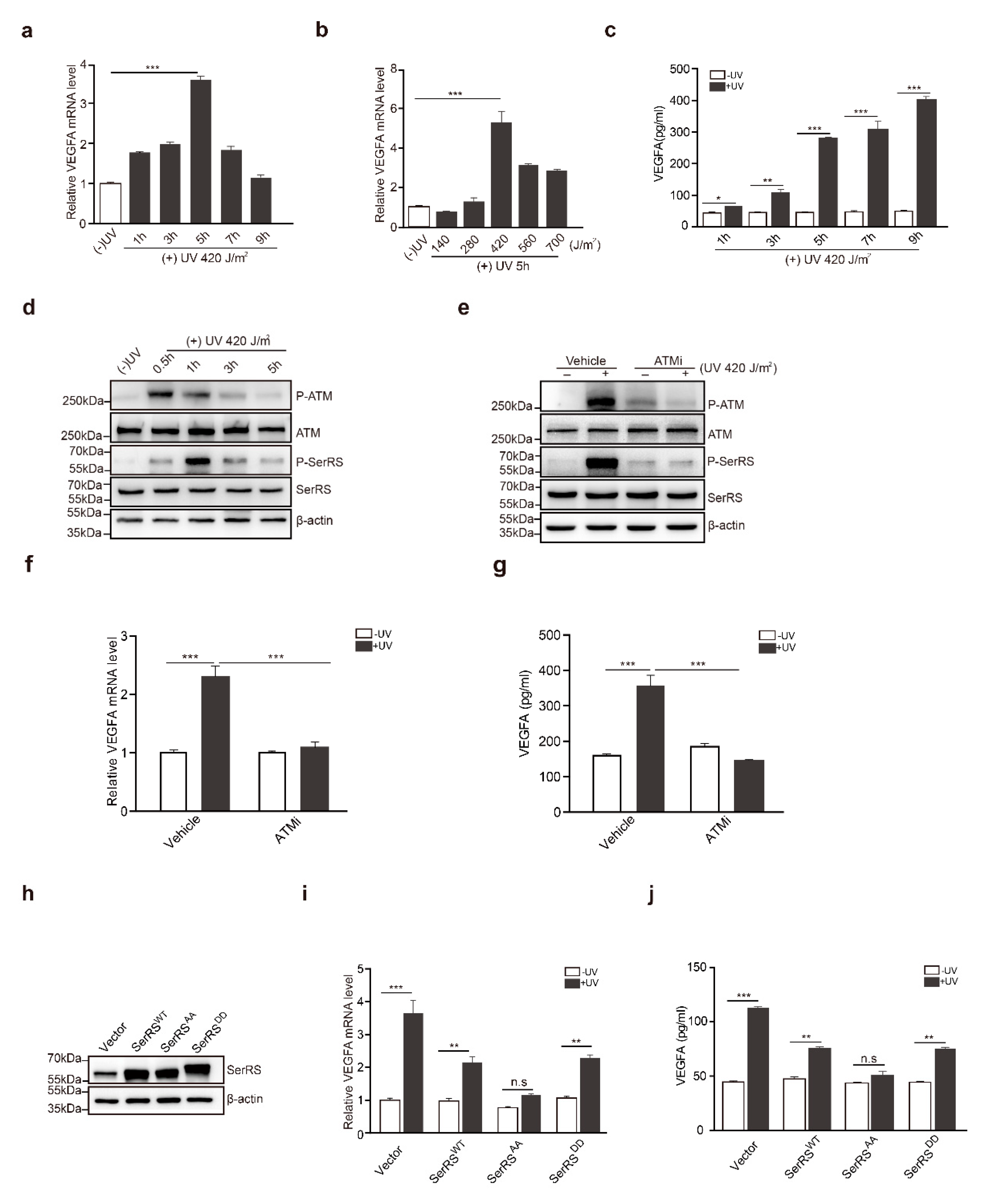
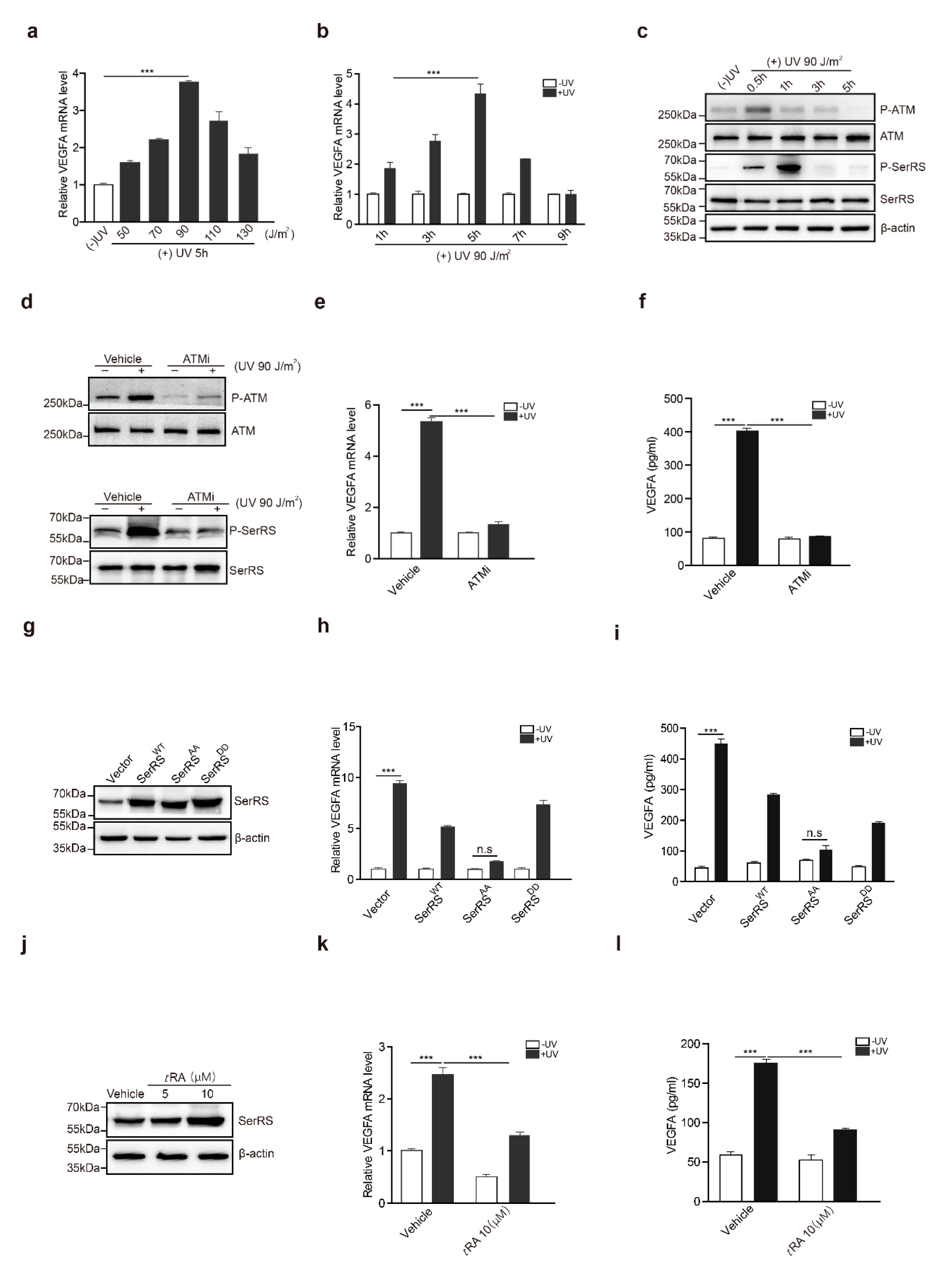
2.1. The ATM–SerRS–VEGFA Pathway Played an Essential Role in UV-Induced VEGFA Expression in HaCaT Cells
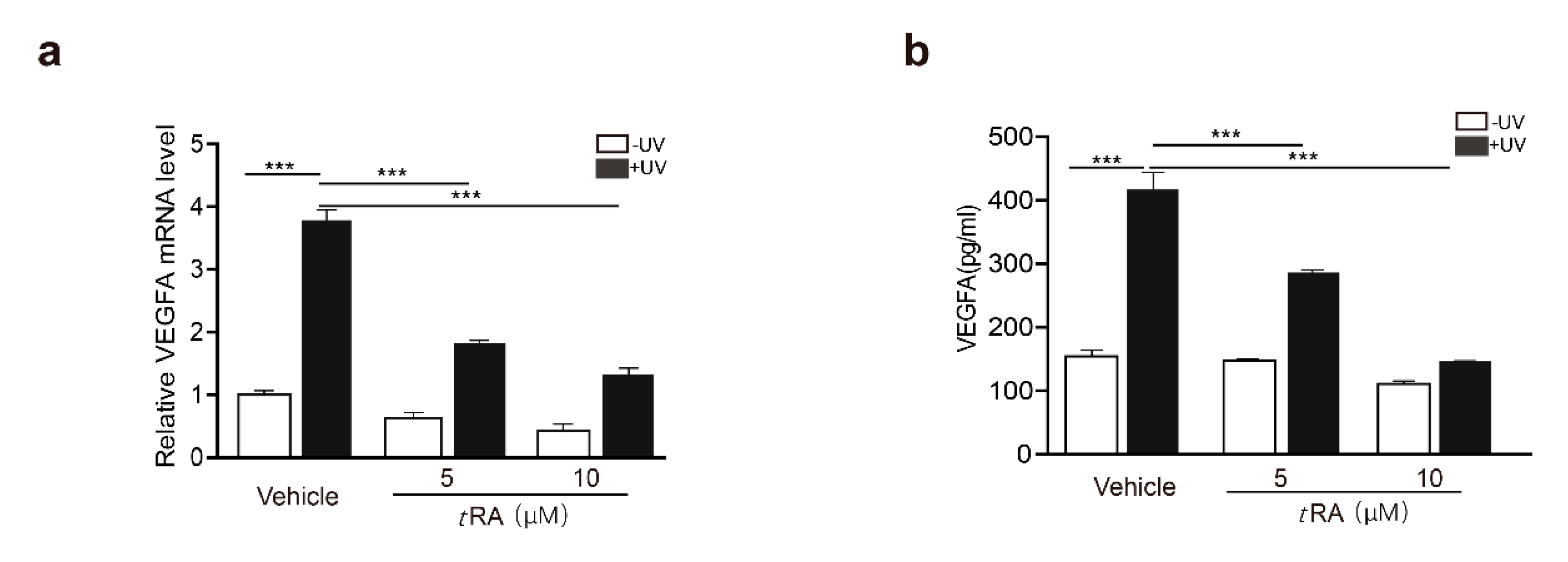
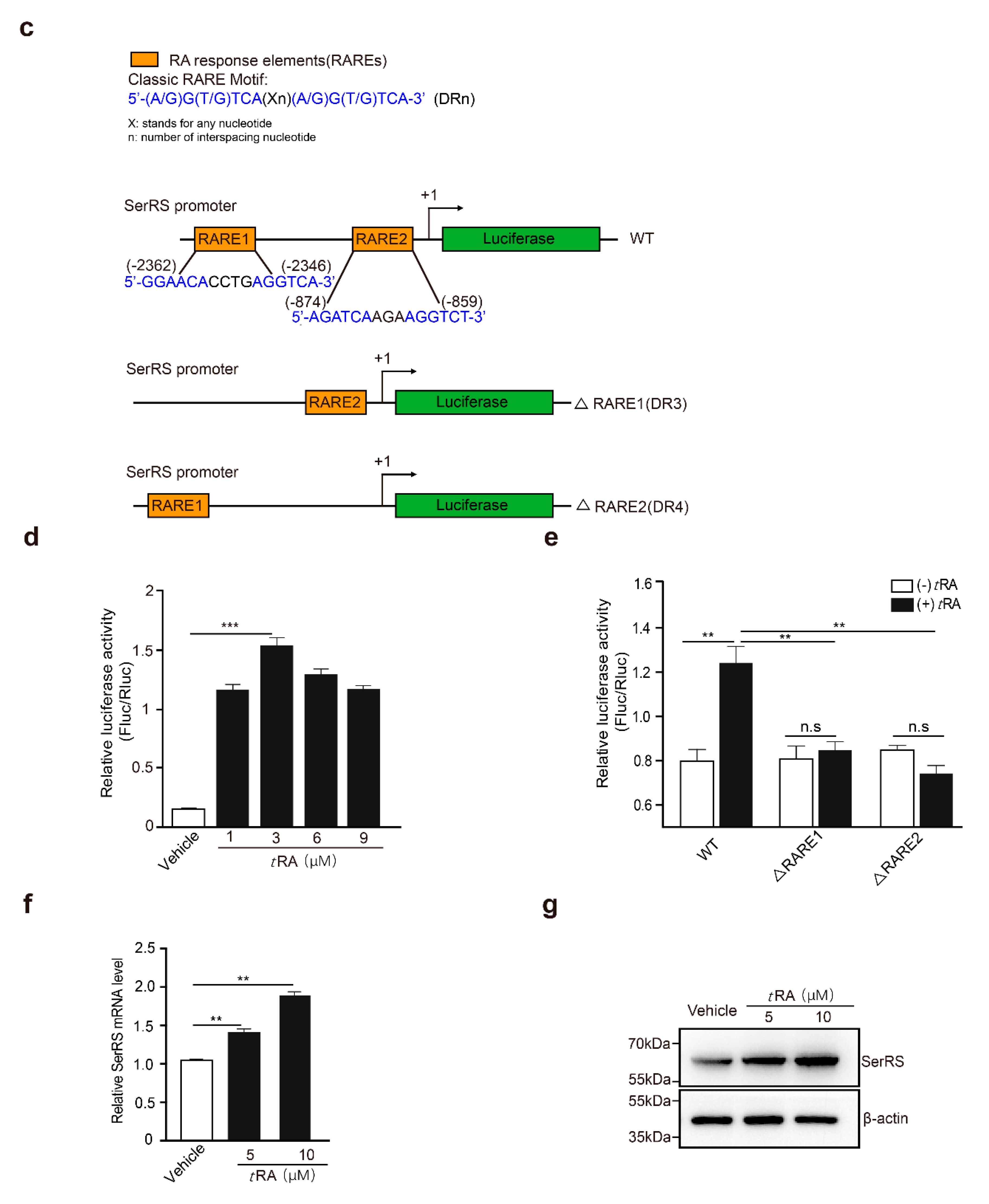
2.2. tRA Enhanced the Transcription of SerRS to Suppress VEGFA Expression in HaCaT Cells
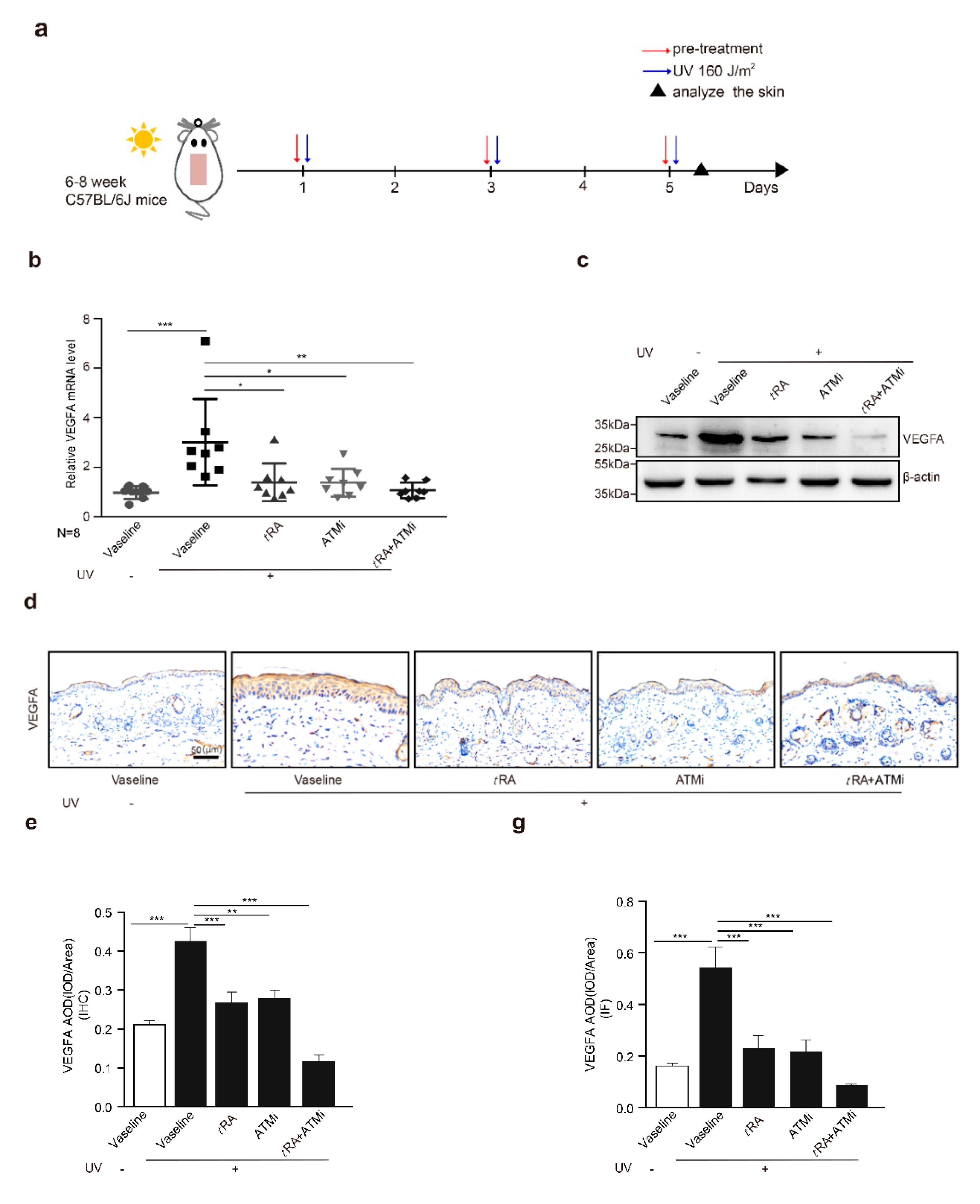
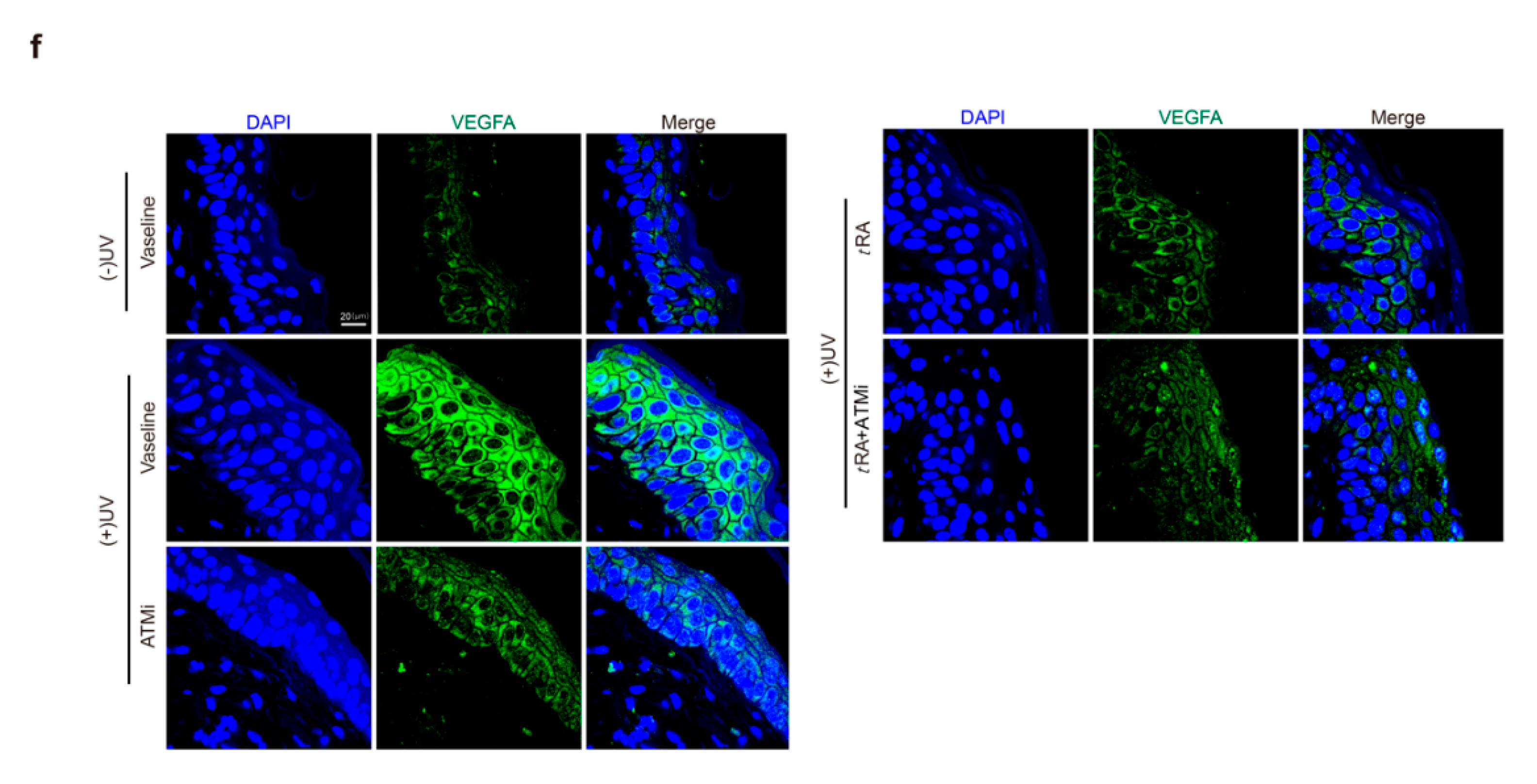
2.3. ATM Inhibitor Enhanced the Effect of tRA on the Protection of Mouse Skin from UV-Induced VEGFA Expression
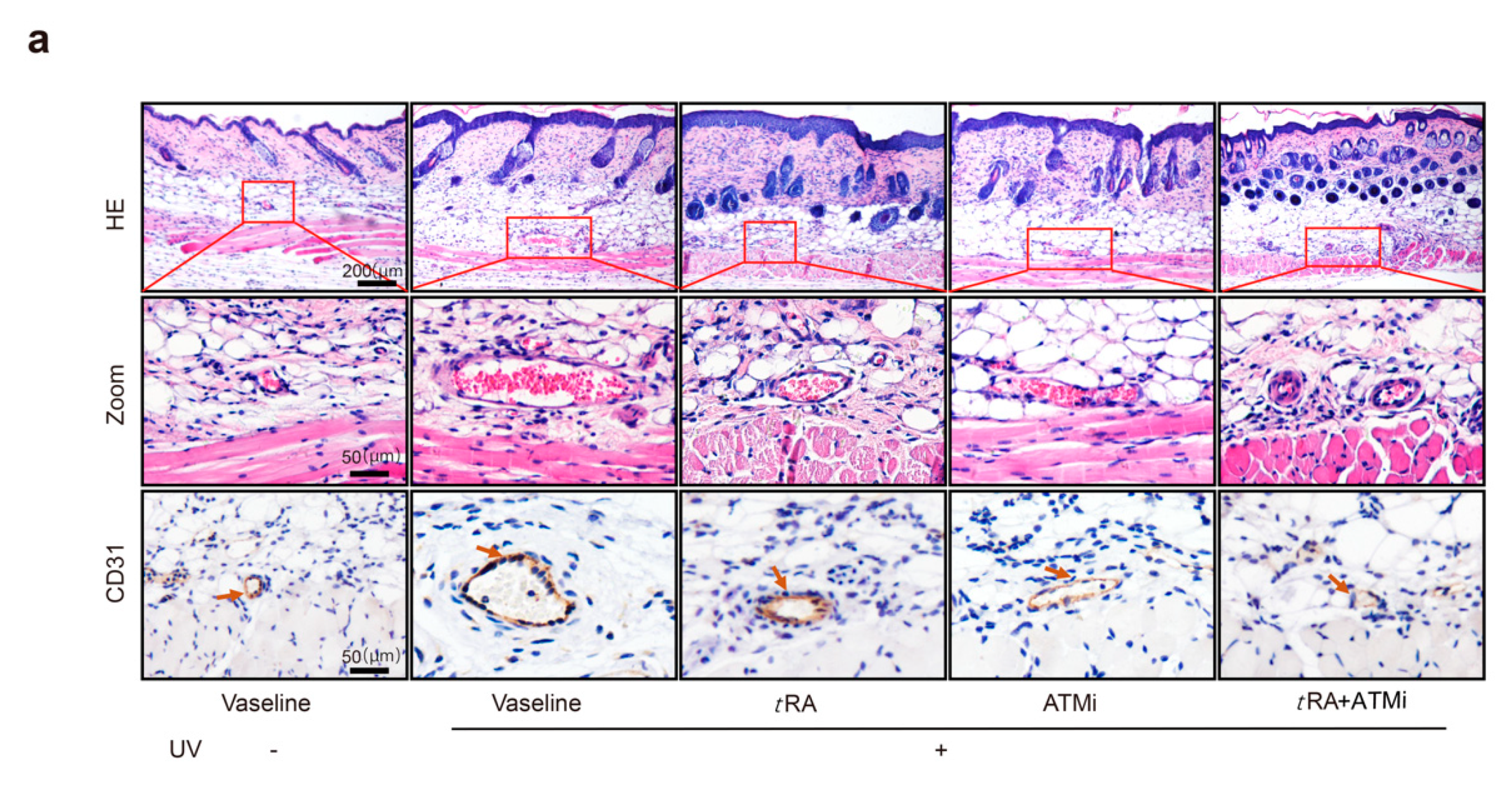
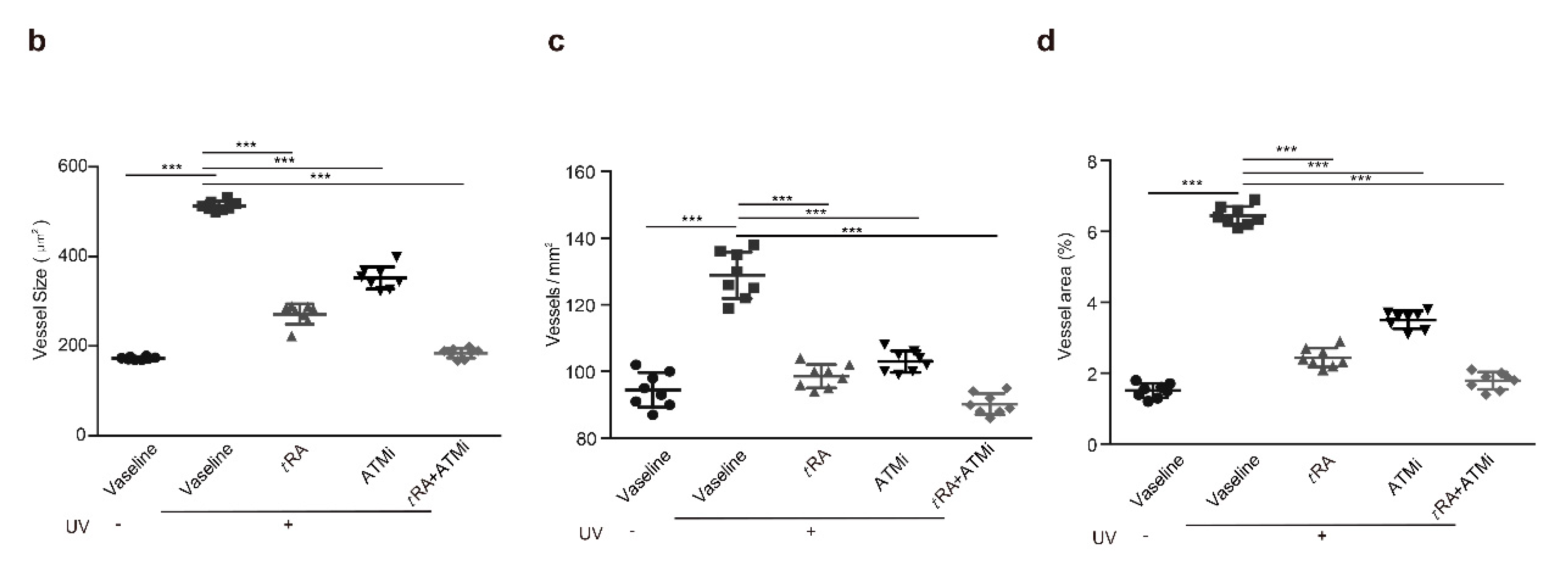
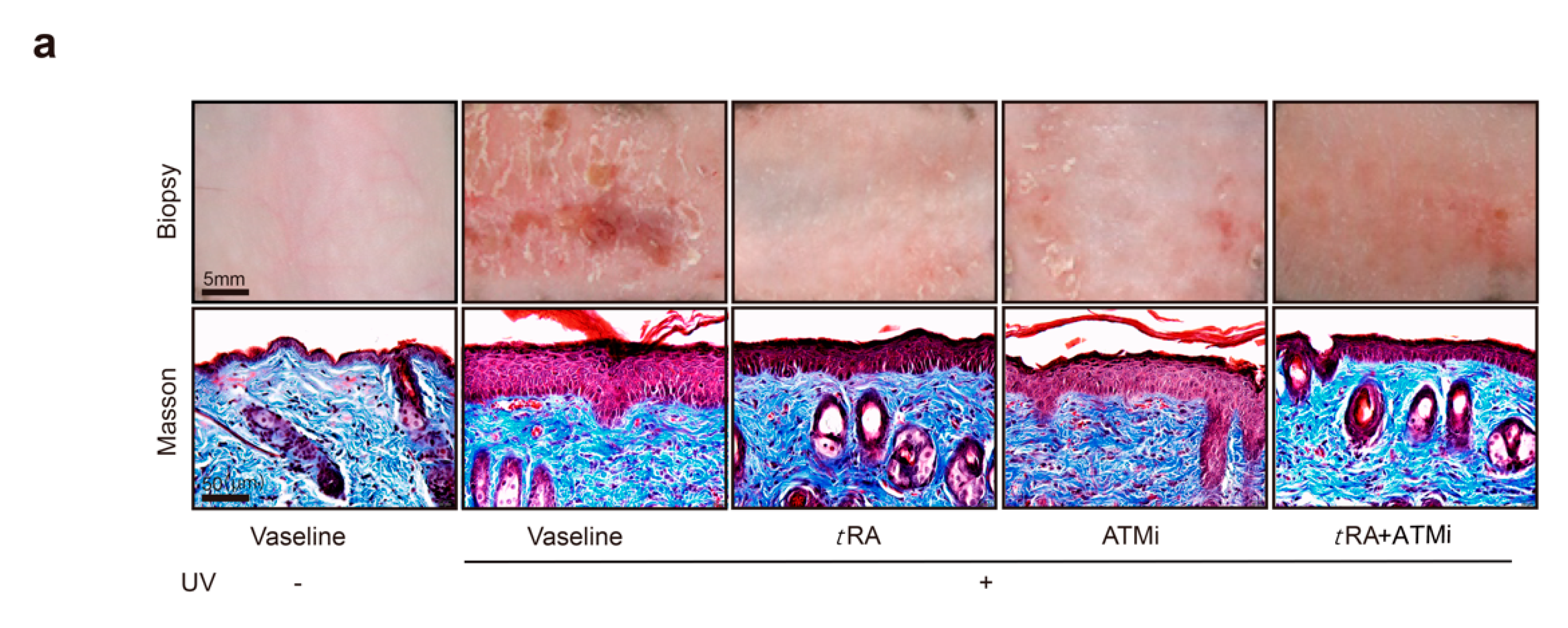
2.4. The ATM Inhibitor Improved the Efficiency of tRA Against Pathological Skin Angiogenesis Caused by UV Irradiation in Mice
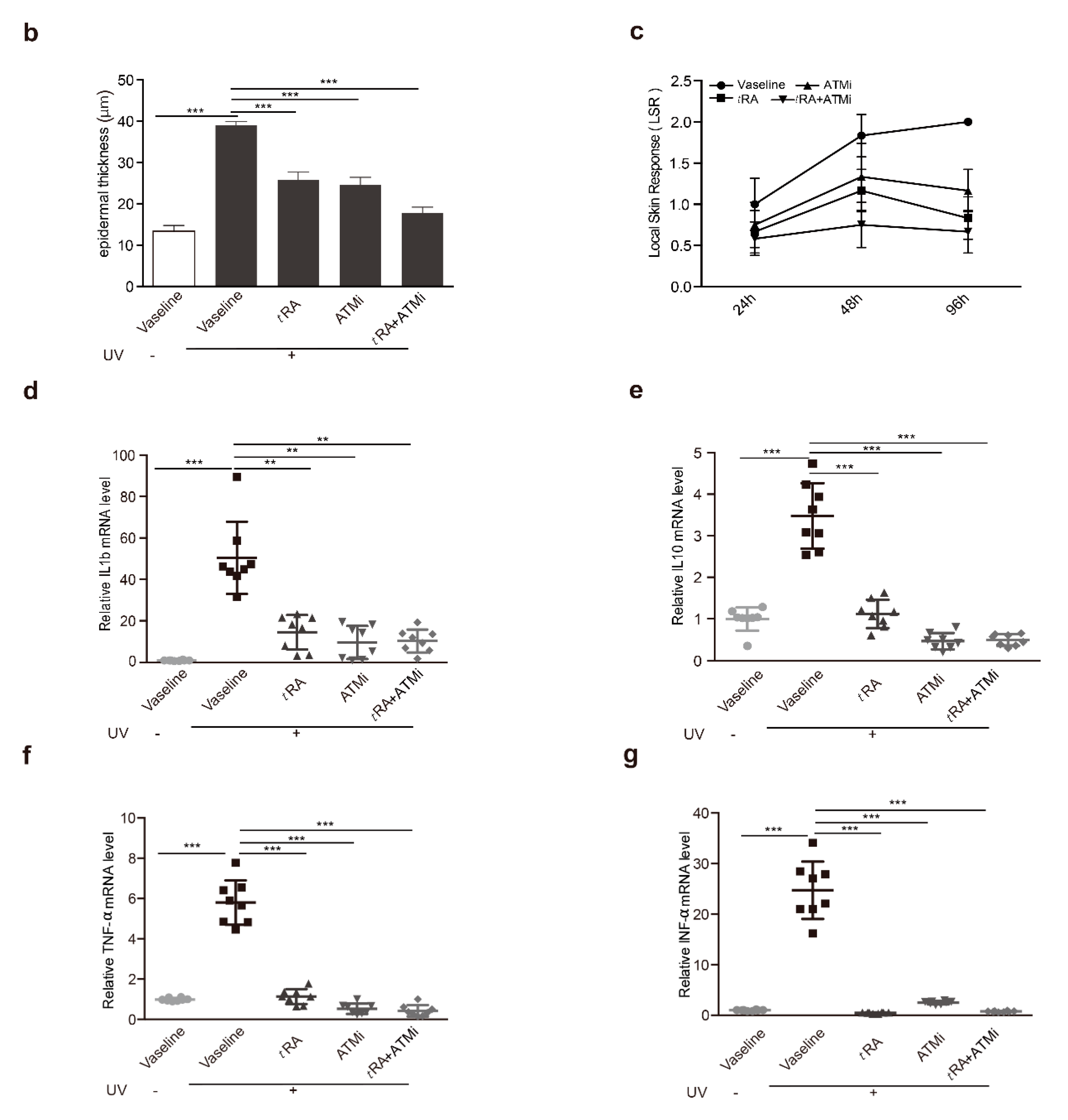
2.5. ATM Inhibitor Improved the Effects of tRA on Alleviating UVB-Induced Thickening and the Inflammatory Response of the Epidermis
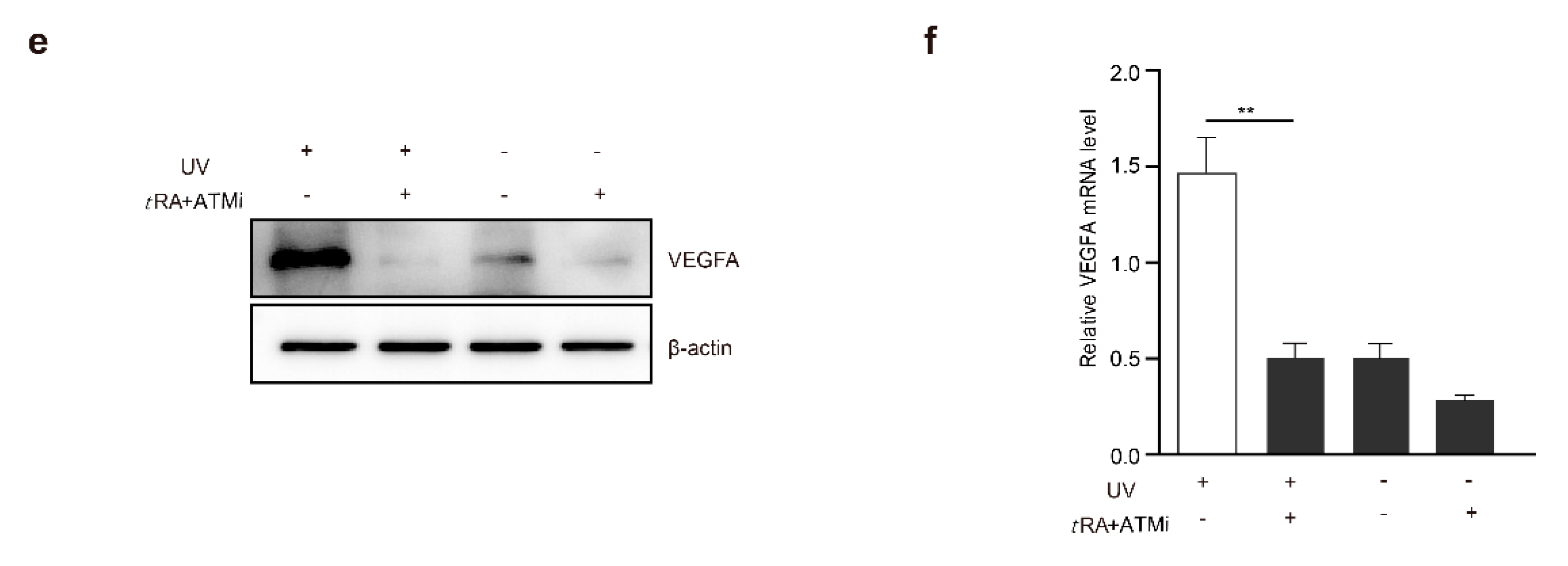
2.6. tRA and ATMi Could Greatly Inhibit UV-Induced VEGFA in Melanoma Cells
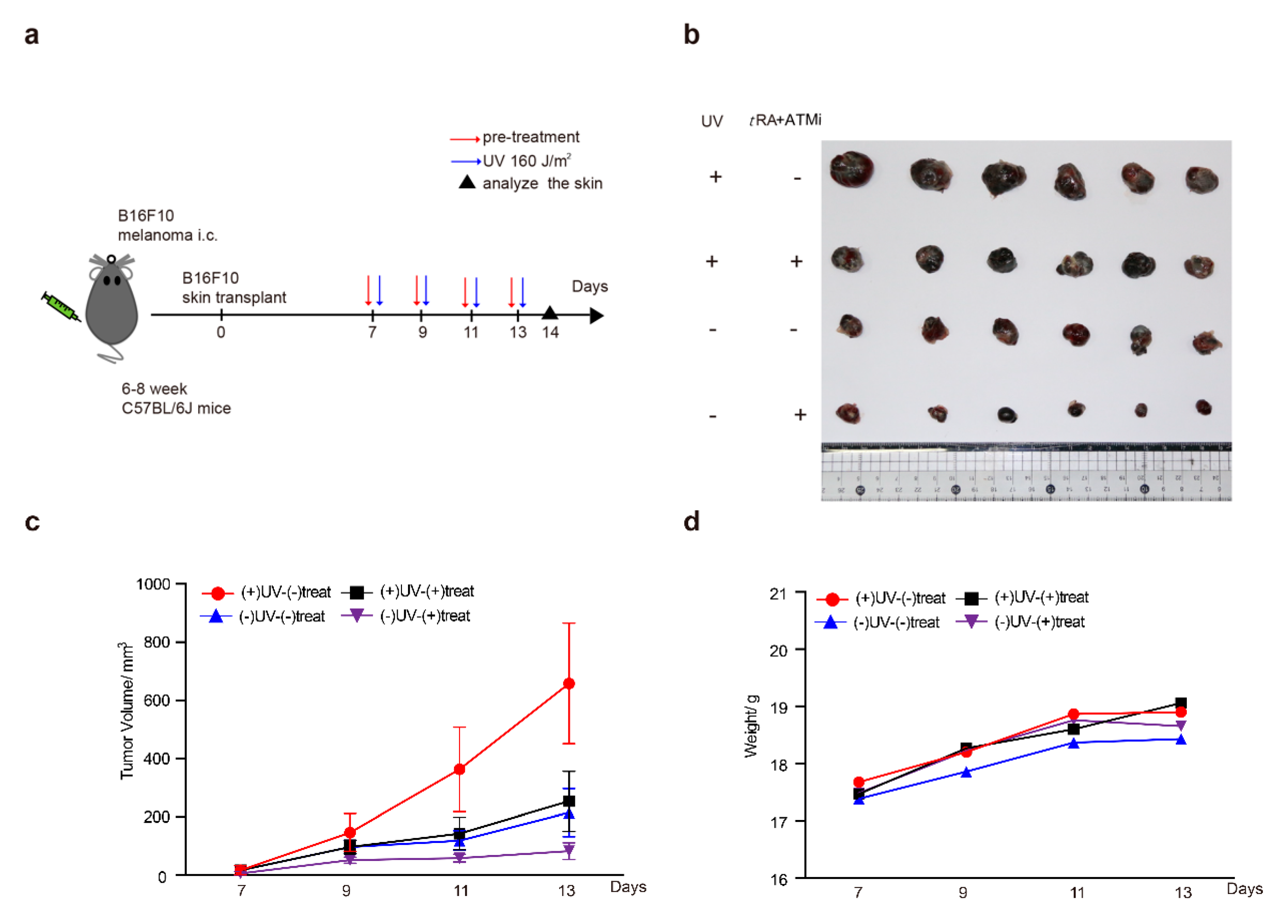
2.7. The ATM Inhibitor Greatly Improved the Effect of tRA in Inhibiting the Growth of a Melanoma Xenograft in Mice
3. Discussion
4. Materials and Methods
4.1. Regents and Antibodies
4.2. Cell Culture and UV Exposure
4.3. Animals and UVB Exposure
4.4. RNA Extraction and RT-PCR
4.5. Co-Immunoprecipitation Assay and Western Blotting Analysis
4.6. Measurement of VEGFA Secretion
4.7. H&E, Immunohistochemistry, and Immunofluorescent Staining
4.8. Dual-Luciferase Reporter Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Score | Visual Manifestation |
|---|---|
| 0 | Normal skin with a pink, smooth, and plump appearance |
| 0.5 | Mild erythema appearing at the exposed site |
| 1 | Moderate erythema, mild edema, and few scales |
| 1.5 | Obvious erythema and scales, with severe edema and mild roughness |
| 2 | Lots of scales, with hypertrophy and wrinkles |
References
- Bernard, J.J.; Gallo, R.L.; Krutmann, J. Photoimmunology: how ultraviolet radiation affects the immune system. Nat. Rev. Immunol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Zhang, T.; Wang, Q.; Reddy, K.; Chen, H.; Yao, K.; Wang, K.; Roh, E.; Zykova, T.; Ma, W.; et al. ADA-07 Suppresses Solar Ultraviolet-Induced Skin Carcinogenesis by Directly Inhibiting TOPK. Mol. Cancer Ther. 2017, 16, 1843–1854. [Google Scholar] [CrossRef] [PubMed]
- Viros, A.; Sanchez-Laorden, B.; Pedersen, M.; Furney, S.J.; Rae, J.; Hogan, K.; Ejiama, S.; Girotti, M.R.; Cook, M.; Dhomen, N.; et al. Ultraviolet radiation accelerates BRAF-driven melanomagenesis by targeting TP53. Nature 2014, 511, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Moyer, V.A. Behavioral counseling to prevent skin cancer: U.S. Preventive Services Task Force recommendation statement. Ann. Intern. Med. 2012, 157, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Riahi, R.R.; Bush, A.E.; Cohen, P.R. Topical Retinoids: Therapeutic Mechanisms in the Treatment of Photodamaged Skin. Am. J. Clin. Dermatol. 2016, 17, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Antille, C.; Tran, C.; Sorg, O.; Carraux, P.; Didierjean, L.; Saurat, J.H. Vitamin A exerts a photoprotective action in skin by absorbing ultraviolet B radiation. J. Investig. Dermatol. 2003, 121, 1163–1167. [Google Scholar] [CrossRef]
- Khalil, S.; Bardawil, T.; Stephan, C.; Darwiche, N.; Abbas, O.; Kibbi, A.G.; Nemer, G.; Kurban, M. Retinoids: a journey from the molecular structures and mechanisms of action to clinical uses in dermatology and adverse effects. J. Dermatol. Treat. 2017, 28, 684–696. [Google Scholar] [CrossRef]
- Darlenski, R.; Surber, C.; Fluhr, J.W. Topical retinoids in the management of photodamaged skin: from theory to evidence-based practical approach. Br. J. Dermatol. 2010, 163, 1157–1165. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, Y.K.; Eun, H.C.; Cho, K.H.; Chung, J.H. All-trans retinoic acid antagonizes UV-induced VEGF production and angiogenesis via the inhibition of ERK activation in human skin keratinocytes. J. Investig. Dermatol. 2006, 126, 2697–2706. [Google Scholar] [CrossRef]
- Hirakawa, S.; Fujii, S.; Kajiya, K.; Yano, K.; Detmar, M. Vascular endothelial growth factor promotes sensitivity to ultraviolet B-induced cutaneous photodamage. Blood 2005, 105, 2392–2399. [Google Scholar] [CrossRef]
- Ming, M.; Han, W.; Maddox, J.; Soltani, K.; Shea, C.R.; Freeman, D.M.; He, Y.Y. UVB-induced ERK/AKT-dependent PTEN suppression promotes survival of epidermal keratinocytes. Oncogene 2010, 29, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Rajput, S.; Tripathi, A.K.; Mandal, M. Targeted therapy against EGFR and VEGFR using ZD6474 enhances the therapeutic potential of UV-B phototherapy in breast cancer cells. Mol. Cancer. 2013, 12, 122. [Google Scholar] [CrossRef] [PubMed]
- Kini, A.R.; Peterson, L.A.; Tallman, M.S.; Lingen, M.W. Angiogenesis in acute promyelocytic leukemia: induction by vascular endothelial growth factor and inhibition by all-trans retinoic acid. Blood 2001, 97, 3919–3924. [Google Scholar] [CrossRef] [PubMed]
- Gee, M.F.; Tsuchida, R.; Eichler-Jonsson, C.; Das, B.; Baruchel, S.; Malkin, D. Vascular endothelial growth factor acts in an autocrine manner in rhabdomyosarcoma cell lines and can be inhibited with all-trans-retinoic acid. Oncogene 2005, 24, 8025–8037. [Google Scholar] [CrossRef]
- Batchelor, T.T.; Sorensen, A.G.; di Tomaso, E.; Zhang, W.T.; Duda, D.G.; Cohen, K.S.; Kozak, K.R.; Cahill, D.P.; Chen, P.J.; Zhu, M.; et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 2007, 11, 83–95. [Google Scholar] [CrossRef]
- Batchelor, T.T.; Duda, D.G.; di Tomaso, E.; Ancukiewicz, M.; Plotkin, S.R.; Gerstner, E.; Eichler, A.F.; Drappatz, J.; Hochberg, F.H.; Benner, T.; et al. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J. Clin. Oncol. 2010, 28, 2817–2823. [Google Scholar] [CrossRef]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef]
- Viallard, C.; Larrivee, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: Current status and future directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- Ferrara, N. VEGF and the quest for tumour angiogenesis factors. Nat. Rev. Cancer 2002, 2, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Xu, X.; Zhang, Q.; Fu, G.; Mo, Z.; Wang, G.S.; Kishi, S.; Yang, X.L. tRNA synthetase counteracts c-Myc to develop functional vasculature. eLife 2014, 3, e02349. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Shi, Y.; Zhang, H.M.; Swindell, E.C.; Marshall, A.G.; Guo, M.; Kishi, S.; Yang, X.L. Unique domain appended to vertebrate tRNA synthetase is essential for vascular development. Nat. Commun. 2012, 3, 681. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, T.J.; Duester, G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 2015, 16, 110–123. [Google Scholar] [CrossRef]
- Lo, J.A.; Fisher, D.E. The melanoma revolution: from UV carcinogenesis to a new era in therapeutics. Science 2014, 346, 945–949. [Google Scholar] [CrossRef]
- Ossio, R.; Roldan-Marin, R.; Martinez-Said, H.; Adams, D.J.; Robles-Espinoza, C.D. Melanoma: A global perspective. Nat. Rev. Cancer 2017, 17, 393–394. [Google Scholar] [CrossRef]
- Moon, H.; Donahue, L.R.; Choi, E.; Scumpia, P.O.; Lowry, W.E.; Grenier, J.K.; Zhu, J.; White, A.C. Melanocyte Stem Cell Activation and Translocation Initiate Cutaneous Melanoma in Response to UV Exposure. Cell Stem Cell 2017, 21, 665–678.e666. [Google Scholar] [CrossRef]
- Yoon, J.H.; McArthur, M.J.; Park, J.; Basu, D.; Wakamiya, M.; Prakash, L.; Prakash, S. Error-Prone Replication through UV Lesions by DNA Polymerase theta Protects against Skin Cancers. Cell 2019, 176, 1295–1309.e1215. [Google Scholar] [CrossRef]
- Linos, E.; Pagoto, S. USPSTF Recommendations for Behavioral Counseling for Skin Cancer Prevention: Throwing Shade on UV Radiation. JAMA Intern. Med. 2018, 178, 609–611. [Google Scholar] [CrossRef]
- Simon, T.; Gagliano, T.; Giamas, G. Direct Effects of Anti-Angiogenic Therapies on Tumor Cells: VEGF Signaling. Trends Mol. Med. 2017, 23, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Presta, L.G.; Chen, H.; O’Connor, S.J.; Chisholm, V.; Meng, Y.G.; Krummen, L.; Winkler, M.; Ferrara, N. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997, 57, 4593–4599. [Google Scholar] [PubMed]
- van Beijnum, J.R.; Nowak-Sliwinska, P.; Huijbers, E.J.; Thijssen, V.L.; Griffioen, A.W. The great escape; the hallmarks of resistance to antiangiogenic therapy. Pharmacol. Rev. 2015, 67, 441–461. [Google Scholar] [CrossRef]
- Ilhan-Mutlu, A.; Osswald, M.; Liao, Y.; Gommel, M.; Reck, M.; Miles, D.; Mariani, P.; Gianni, L.; Lutiger, B.; Nendel, V.; et al. Bevacizumab Prevents Brain Metastases Formation in Lung Adenocarcinoma. Mol. Cancer Ther. 2016, 15, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.K.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Haworth, L.; Sherry, R.M.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Steinberg, S.M.; Chen, H.X.; Rosenberg, S.A. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N. Engl. J. Med. 2003, 349, 427–434. [Google Scholar] [CrossRef]
- Kabbinavar, F.; Hurwitz, H.I.; Fehrenbacher, L.; Meropol, N.J.; Novotny, W.F.; Lieberman, G.; Griffing, S.; Bergsland, E. Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J. Clin. Oncol. 2003, 21, 60–65. [Google Scholar] [CrossRef]
- Ebos, J.M.; Kerbel, R.S. Antiangiogenic therapy: Impact on invasion, disease progression, and metastasis. Nat. Rev. Clin. Oncol. 2011, 8, 210–221. [Google Scholar] [CrossRef]
- Amsterdam, A.; Nissen, R.M.; Sun, Z.; Swindell, E.C.; Farrington, S.; Hopkins, N. Identification of 315 genes essential for early zebrafish development. Proc. Natl. Acad. Sci. USA 2004, 101, 12792–12797. [Google Scholar] [CrossRef]
- Fukui, H.; Hanaoka, R.; Kawahara, A. Noncanonical activity of seryl-tRNA synthetase is involved in vascular development. Circ. Res. 2009, 104, 1253–1259. [Google Scholar] [CrossRef]
- Herzog, W.; Muller, K.; Huisken, J.; Stainier, D.Y. Genetic evidence for a noncanonical function of seryl-tRNA synthetase in vascular development. Circ. Res. 2009, 104, 1260–1266. [Google Scholar] [CrossRef] [PubMed]
- Stiff, T.; Walker, S.A.; Cerosaletti, K.; Goodarzi, A.A.; Petermann, E.; Concannon, P.; O′Driscoll, M.; Jeggo, P.A. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. EMBO J. 2006, 25, 5775–5782. [Google Scholar] [CrossRef] [PubMed]
- Gibson, S.L.; Bindra, R.S.; Glazer, P.M. Hypoxia-induced phosphorylation of Chk2 in an ataxia telangiectasia mutated-dependent manner. Cancer Res. 2005, 65, 10734–10741. [Google Scholar] [CrossRef] [PubMed]
- Elvers, I.; Hagenkort, A.; Johansson, F.; Djureinovic, T.; Lagerqvist, A.; Schultz, N.; Stoimenov, I.; Erixon, K.; Helleday, T. CHK1 activity is required for continuous replication fork elongation but not stabilization of post-replicative gaps after UV irradiation. Nucleic Acids Res. 2012, 40, 8440–8448. [Google Scholar] [CrossRef] [PubMed]
- Mongiardi, M.P.; Radice, G.; Piras, M.; Stagni, V.; Pacioni, S.; Re, A.; Putti, S.; Ferre, F.; Farsetti, A.; Pallini, R.; et al. Axitinib exposure triggers endothelial cells senescence through ROS accumulation and ATM activation. Oncogene 2019, 38, 5413–5424. [Google Scholar] [CrossRef] [PubMed]
- Okuno, Y.; Nakamura-Ishizu, A.; Otsu, K.; Suda, T.; Kubota, Y. Pathological neoangiogenesis depends on oxidative stress regulation by ATM. Nat. Med. 2012, 18, 1208–1216. [Google Scholar] [CrossRef]
- Bolis, M.; Garattini, E.; Paroni, G.; Zanetti, A.; Kurosaki, M.; Castrignano, T.; Garattini, S.K.; Biancardi, F.; Barzago, M.M.; Gianni, M.; et al. Network-guided modeling allows tumor-type independent prediction of sensitivity to all-trans-retinoic acid. Ann. Oncol. 2017, 28, 611–621. [Google Scholar] [CrossRef]
- Cicconi, L.; Lo-Coco, F. Current management of newly diagnosed acute promyelocytic leukemia. Ann. Oncol. 2016, 27, 1474–1481. [Google Scholar] [CrossRef]
- Abu, J.; Batuwangala, M.; Herbert, K.; Symonds, P. Retinoic acid and retinoid receptors: potential chemopreventive and therapeutic role in cervical cancer. Lancet Oncol. 2005, 6, 712–720. [Google Scholar] [CrossRef]
- Okuno, M.; Kojima, S.; Matsushima-Nishiwaki, R.; Tsurumi, H.; Muto, Y.; Friedman, S.L.; Moriwaki, H. Retinoids in cancer chemoprevention. Curr. Cancer Drug Targets 2004, 4, 285–298. [Google Scholar] [CrossRef]
- Bogos, K.; Renyi-Vamos, F.; Kovacs, G.; Tovari, J.; Dome, B. Role of retinoic receptors in lung carcinogenesis. J. Exp. Clin. Cancer Res. 2008, 27, 18. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bauer, R.; Udonta, F.; Wroblewski, M.; Ben-Batalla, I.; Santos, I.M.; Taverna, F.; Kuhlencord, M.; Gensch, V.; Pasler, S.; Vinckier, S.; et al. Blockade of Myeloid-Derived Suppressor Cell Expansion with All-Trans Retinoic Acid Increases the Efficacy of Antiangiogenic Therapy. Cancer Res. 2018, 78, 3220–3232. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.A.; Kerbel, R.S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 2018, 15, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef]
- Kelly, P.N. The Cancer Immunotherapy Revolution. Science 2018, 359, 1344–1345. [Google Scholar] [CrossRef]
- Rotte, A.; Jin, J.Y.; Lemaire, V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann. Oncol. 2018, 29, 71–83. [Google Scholar] [CrossRef]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef]












© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.; Lu, H.; Wang, Q.; Xiang, R. Targeting Angiogenesis by Blocking the ATM–SerRS–VEGFA Pathway for UV-Induced Skin Photodamage and Melanoma Growth. Cancers 2019, 11, 1847. https://doi.org/10.3390/cancers11121847
Song Y, Lu H, Wang Q, Xiang R. Targeting Angiogenesis by Blocking the ATM–SerRS–VEGFA Pathway for UV-Induced Skin Photodamage and Melanoma Growth. Cancers. 2019; 11(12):1847. https://doi.org/10.3390/cancers11121847
Chicago/Turabian StyleSong, Yadong, Hongyan Lu, Qiong Wang, and Rong Xiang. 2019. "Targeting Angiogenesis by Blocking the ATM–SerRS–VEGFA Pathway for UV-Induced Skin Photodamage and Melanoma Growth" Cancers 11, no. 12: 1847. https://doi.org/10.3390/cancers11121847
APA StyleSong, Y., Lu, H., Wang, Q., & Xiang, R. (2019). Targeting Angiogenesis by Blocking the ATM–SerRS–VEGFA Pathway for UV-Induced Skin Photodamage and Melanoma Growth. Cancers, 11(12), 1847. https://doi.org/10.3390/cancers11121847