Bulk and Single-Cell Next-Generation Sequencing: Individualizing Treatment for Colorectal Cancer

 ,
, 
Abstract
:1. Introduction
2. Advances and Limitations in the Management of Colorectal Cancer
3. Next-Generation Sequencing: Progress from Static to Saptiotemporal Genomic and Transcriptomic Analyses
3.1. Single-Biopsy Genomics and Transcriptomics
3.2. Bulk Inter- and Intra-Tumor Heterogeneity
3.3. Liquid Biopsies: Early Diagnosis, Drug Response Prediction and Patient Monitoring
3.4. Spatiotemporal Intra-Patient Heterogeneity
3.5. Translational Implications of Cell-by-Cell Cancer Variability
3.6. Functional Non-Coding Mutations and Regulatory Network Exploration
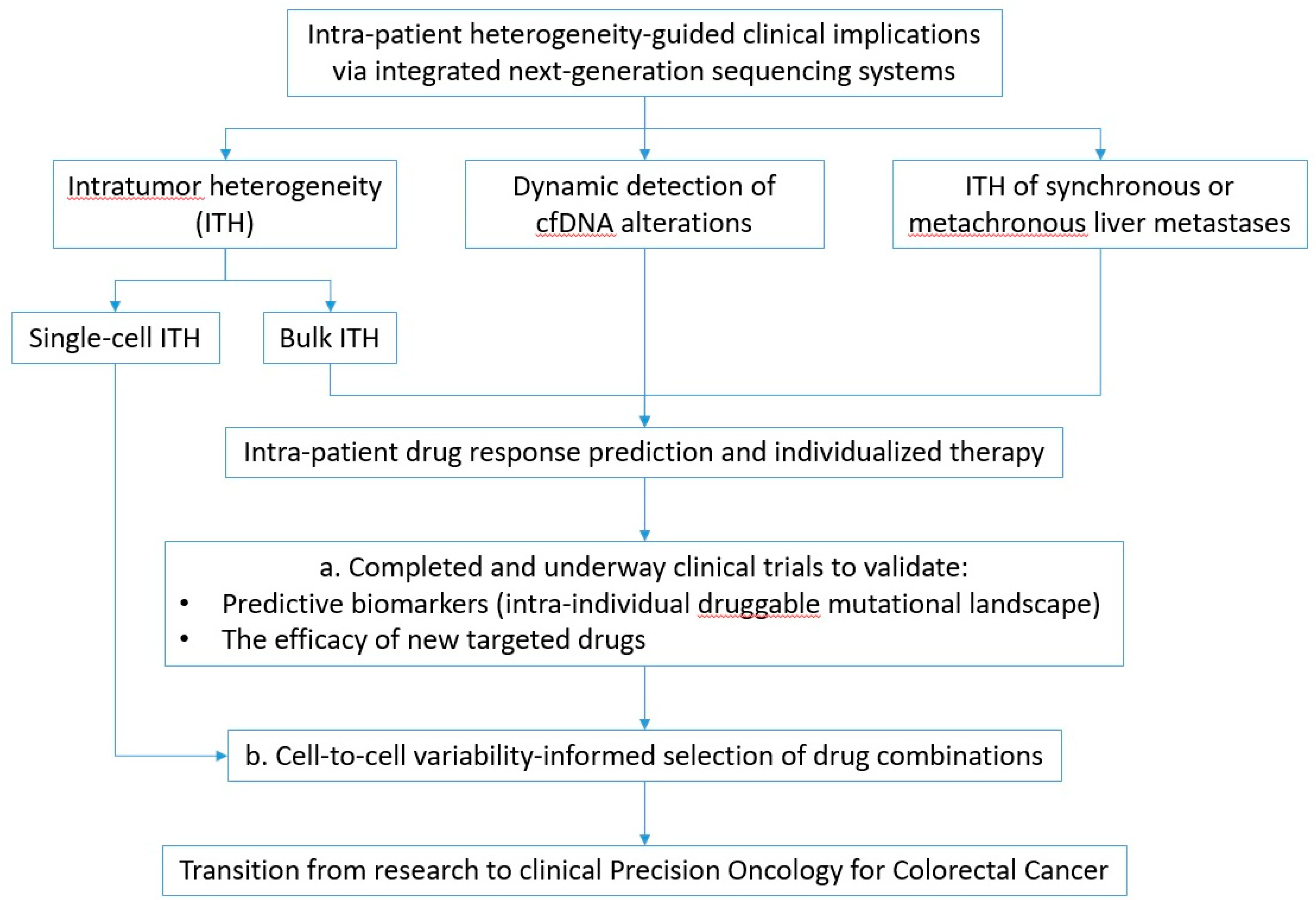
4. Future Perspectives
4.1. Emerging Clinical Trials in Precision Oncology
4.2. Pharmacogenomic Predictions at the Single-Cell Level: A New Horizon for Cancer Precision Medicine
4.3. Transcriptional Networks and Pharmaceutical Controllability
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA sequencing at 40: Past, present and future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Spielmann, M.; Qiu, X.; Huang, X.; Ibrahim, D.M.; Hill, A.J.; Zhang, F.; Mundlos, S.; Christiansen, L.; Steemers, F.J.; et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019, 566, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Elkon, R.; Agami, R. Characterization of noncoding regulatory DNA in the human genome. Nat. Biotechnol. 2017, 35, 732–746. [Google Scholar] [CrossRef] [PubMed]
- Haendel, M.A.; Chute, C.G.; Robinson, P.N. Classification, Ontology, and Precision Medicine. N. Engl. J. Med. 2018, 379, 1452–1462. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Hansen, A.R.; Ratain, M.J.; Siu, L.L. Tumour heterogeneity in the clinic. Nature 2013, 501, 355–364. [Google Scholar] [CrossRef]
- Swanton, C.; Soria, J.C.; Bardelli, A.; Biankin, A.; Caldas, C.; Chandarlapaty, S.; de Koning, L.; Dive, C.; Feunteun, J.; Leung, S.Y.; et al. Consensus on precision medicine for metastatic cancers: A report from the MAP conference. Ann. Oncol. 2016, 27, 1443–1448. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef]
- Roerink, S.F.; Sasaki, N.; Lee-Six, H.; Young, M.D.; Alexandrov, L.B.; Behjati, S.; Mitchell, T.J.; Grossmann, S.; Lightfoot, H.; Egan, D.A.; et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 2018, 556, 457–462. [Google Scholar] [CrossRef]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef]
- Kyrochristos, I.D.; Ziogas, D.E.; Roukos, D.H. Drug resistance: Origins, evolution and characterization of genomic clones and the tumor ecosystem to optimize precise individualized therapy. Drug Discov. Today 2019, 24, 1281–1294. [Google Scholar] [CrossRef]
- Klein, C.A. Selection and adaptation during metastatic cancer progression. Nature 2013, 501, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Orlando, G.; Law, P.J.; Cornish, A.J.; Dobbins, S.E.; Chubb, D.; Broderick, P.; Litchfield, K.; Hariri, F.; Pastinen, T.; Osborne, C.S.; et al. Promoter capture Hi-C-based identification of recurrent noncoding mutations in colorectal cancer. Nat. Genet. 2018, 50, 1375–1380. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network. Available online: https://www.nccn.org/ (accessed on 22 October 2019).
- The national cancer act of 1971. J. Natl. Cancer Inst. 1972, 48, 577–584.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.H.; Cunningham, D.; Werner, B.; Vlachogiannis, G.; Spiteri, I.; Heide, T.; Mateos, J.F.; Vatsiou, A.; Lampis, A.; Damavandi, M.D.; et al. Longitudinal Liquid Biopsy and Mathematical Modeling of Clonal Evolution Forecast Time to Treatment Failure in the PROSPECT-C Phase II Colorectal Cancer Clinical Trial. Cancer Discov. 2018, 8, 1270–1285. [Google Scholar] [CrossRef] [PubMed]
- Mamlouk, S.; Childs, L.H.; Aust, D.; Heim, D.; Melching, F.; Oliveira, C.; Wolf, T.; Durek, P.; Schumacher, D.; Blaker, H.; et al. DNA copy number changes define spatial patterns of heterogeneity in colorectal cancer. Nat. Commun. 2017, 8, 14093. [Google Scholar] [CrossRef] [PubMed]
- Strickler, J.H.; Loree, J.M.; Ahronian, L.G.; Parikh, A.R.; Niedzwiecki, D.; Pereira, A.A.L.; McKinney, M.; Korn, W.M.; Atreya, C.E.; Banks, K.C.; et al. Genomic Landscape of Cell-Free DNA in Patients with Colorectal Cancer. Cancer Discov. 2018, 8, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular profiling of cancer patients enables personalized combination therapy: The I-PREDICT study. Nat. Med. 2019, 25, 744. [Google Scholar] [CrossRef]
- Rodon, J.; Soria, J.C.; Berger, R.; Miller, W.H.; Rubin, E.; Kugel, A.; Tsimberidou, A.; Saintigny, P.; Ackerstein, A.; Brana, I.; et al. Genomic and transcriptomic profiling expands precision cancer medicine: The WINTHER trial. Nat. Med. 2019, 25, 751. [Google Scholar] [CrossRef]
- Rothwell, D.G.; Ayub, M.; Cook, N.; Thistlethwaite, F.; Carter, L.; Dean, E.; Smith, N.; Villa, S.; Dransfield, J.; Clipson, A.; et al. Utility of ctDNA to support patient selection for early phase clinical trials: The TARGET study. Nat. Med. 2019, 25, 738. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Crick, F. Central dogma of molecular biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, M.; Almen, M.S.; Schioth, H.B. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discov. 2011, 10, 579–590. [Google Scholar] [CrossRef]
- Amin, M.B.; American Joint Committee on Cancer. AJCC Cancer Staging Manual, 8th ed.; Springer: New, York, NY, USA, 2017; p. xvii. 1024 p. [Google Scholar]
- Bosset, J.F.; Calais, G.; Mineur, L.; Maingon, P.; Stojanovic-Rundic, S.; Bensadoun, R.J.; Bardet, E.; Beny, A.; Ollier, J.C.; Bolla, M.; et al. Fluorouracil-based adjuvant chemotherapy after preoperative chemoradiotherapy in rectal cancer: Long-term results of the EORTC 22921 randomised study. Lancet Oncol. 2014, 15, 184–190. [Google Scholar] [CrossRef]
- Alberts, S.R.; Sargent, D.J.; Nair, S.; Mahoney, M.R.; Mooney, M.; Thibodeau, S.N.; Smyrk, T.C.; Sinicrope, F.A.; Chan, E.; Gill, S.; et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: A randomized trial. JAMA 2012, 307, 1383–1393. [Google Scholar] [CrossRef]
- Kerr, R.S.; Love, S.; Segelov, E.; Johnstone, E.; Falcon, B.; Hewett, P.; Weaver, A.; Church, D.; Scudder, C.; Pearson, S.; et al. Adjuvant capecitabine plus bevacizumab versus capecitabine alone in patients with colorectal cancer (QUASAR 2): An open-label, randomised phase 3 trial. Lancet Oncol. 2016, 17, 1543–1557. [Google Scholar] [CrossRef]
- Primrose, J.; Falk, S.; Finch-Jones, M.; Valle, J.; O’Reilly, D.; Siriwardena, A.; Hornbuckle, J.; Peterson, M.; Rees, M.; Iveson, T.; et al. Systemic chemotherapy with or without cetuximab in patients with resectable colorectal liver metastasis: The New EPOC randomised controlled trial. Lancet Oncol. 2014, 15, 601–611. [Google Scholar] [CrossRef]
- European Society for Medical Oncology. Available online: https://www.esmo.org/ (accessed on 22 October 2019).
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Yue, F.; Cheng, Y.; Breschi, A.; Vierstra, J.; Wu, W.; Ryba, T.; Sandstrom, R.; Ma, Z.; Davis, C.; Pope, B.D.; et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature 2014, 515, 355–364. [Google Scholar] [CrossRef]
- Mod, E.C.; Roy, S.; Ernst, J.; Kharchenko, P.V.; Kheradpour, P.; Negre, N.; Eaton, M.L.; Landolin, J.M.; Bristow, C.A.; Ma, L.; et al. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 2010, 330, 1787–1797. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Program—National Cancer Institute. Available online: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga (accessed on 13 October 2019).
- International Cancer Genome Consortium. Available online: https://icgc.org/ (accessed on 16 October 2019).
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Park, P.J. ChIP-seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hause, R.J.; Pritchard, C.C.; Shendure, J.; Salipante, S.J. Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 2016, 22, 1342–1350. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Huyghe, J.R.; Bien, S.A.; Harrison, T.A.; Kang, H.M.; Chen, S.; Schmit, S.L.; Conti, D.V.; Qu, C.; Jeon, J.; Edlund, C.K.; et al. Discovery of common and rare genetic risk variants for colorectal cancer. Nat. Genet. 2019, 51, 76–87. [Google Scholar] [CrossRef]
- Chubb, D.; Broderick, P.; Dobbins, S.E.; Frampton, M.; Kinnersley, B.; Penegar, S.; Price, A.; Ma, Y.P.; Sherborne, A.L.; Palles, C.; et al. Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nat. Commun. 2016, 7, 11883. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136. [Google Scholar] [CrossRef]
- Zhang, W.; Bojorquez-Gomez, A.; Velez, D.O.; Xu, G.; Sanchez, K.S.; Shen, J.P.; Chen, K.; Licon, K.; Melton, C.; Olson, K.M.; et al. A global transcriptional network connecting noncoding mutations to changes in tumor gene expression. Nat. Genet. 2018, 50, 613–620. [Google Scholar] [CrossRef]
- Liu, Y.; Sethi, N.S.; Hinoue, T.; Schneider, B.G.; Cherniack, A.D.; Sanchez-Vega, F.; Seoane, J.A.; Farshidfar, F.; Bowlby, R.; Islam, M.; et al. Comparative Molecular Analysis of Gastrointestinal Adenocarcinomas. Cancer Cell 2018, 33, 721–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingo, E.; Camps, C.; Kaisaki, P.J.; Parsons, M.J.; Mouradov, D.; Pentony, M.M.; Makino, S.; Palmieri, M.; Ward, R.L.; Hawkins, N.J.; et al. Mutation burden and other molecular markers of prognosis in colorectal cancer treated with curative intent: Results from the QUASAR 2 clinical trial and an Australian community-based series. Lancet Gastroenterol. Hepatol. 2018, 3, 635–643. [Google Scholar] [CrossRef] [Green Version]
- Schell, M.J.; Yang, M.; Teer, J.K.; Lo, F.Y.; Madan, A.; Coppola, D.; Monteiro, A.N.; Nebozhyn, M.V.; Yue, B.; Loboda, A.; et al. A multigene mutation classification of 468 colorectal cancers reveals a prognostic role for APC. Nat. Commun. 2016, 7, 11743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Li, Q.; Wu, X. Construction and analysis for differentially expressed long non-coding RNAs and MicroRNAs mediated competing endogenous RNA network in colon cancer. PLoS ONE 2018, 13, e0192494. [Google Scholar] [CrossRef] [PubMed]
- Smeets, D.; Miller, I.S.; O’Connor, D.P.; Das, S.; Moran, B.; Boeckx, B.; Gaiser, T.; Betge, J.; Barat, A.; Klinger, R.; et al. Copy number load predicts outcome of metastatic colorectal cancer patients receiving bevacizumab combination therapy. Nat. Commun. 2018, 9, 4112. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385. [Google Scholar] [CrossRef] [Green Version]
- Katainen, R.; Dave, K.; Pitkanen, E.; Palin, K.; Kivioja, T.; Valimaki, N.; Gylfe, A.E.; Ristolainen, H.; Hanninen, U.A.; Cajuso, T.; et al. CTCF/cohesin-binding sites are frequently mutated in cancer. Nat. Genet. 2015, 47, 818–821. [Google Scholar] [CrossRef]
- Schutte, M.; Risch, T.; Abdavi-Azar, N.; Boehnke, K.; Schumacher, D.; Keil, M.; Yildiriman, R.; Jandrasits, C.; Borodina, T.; Amstislavskiy, V.; et al. Molecular dissection of colorectal cancer in pre-clinical models identifies biomarkers predicting sensitivity to EGFR inhibitors. Nat. Commun. 2017, 8, 14262. [Google Scholar] [CrossRef]
- Guda, K.; Veigl, M.L.; Varadan, V.; Nosrati, A.; Ravi, L.; Lutterbaugh, J.; Beard, L.; Willson, J.K.; Sedwick, W.D.; Wang, Z.J.; et al. Novel recurrently mutated genes in African American colon cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 1149–1154. [Google Scholar] [CrossRef] [Green Version]
- Joung, J.G.; Oh, B.Y.; Hong, H.K.; Al-Khalidi, H.; Al-Alem, F.; Lee, H.O.; Bae, J.S.; Kim, J.; Cha, H.U.; Alotaibi, M.; et al. Tumor Heterogeneity Predicts Metastatic Potential in Colorectal Cancer. Clin. Cancer Res. 2017, 23, 7209–7216. [Google Scholar] [CrossRef] [PubMed]
- Brannon, A.R.; Vakiani, E.; Sylvester, B.E.; Scott, S.N.; McDermott, G.; Shah, R.H.; Kania, K.; Viale, A.; Oschwald, D.M.; Vacic, V.; et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014, 15, 454. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ding, J.; Ma, Z.; Sun, R.; Seoane, J.A.; Scott Shaffer, J.; Suarez, C.J.; Berghoff, A.S.; Cremolini, C.; Falcone, A.; et al. Quantitative evidence for early metastatic seeding in colorectal cancer. Nat. Genet. 2019, 51, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Tan, I.B.; Malik, S.; Ramnarayanan, K.; McPherson, J.R.; Ho, D.L.; Suzuki, Y.; Ng, S.B.; Yan, S.; Lim, K.H.; Koh, D.; et al. High-depth sequencing of over 750 genes supports linear progression of primary tumors and metastases in most patients with liver-limited metastatic colorectal cancer. Genome Biol. 2015, 16, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, R.; Schell, M.J.; Teer, J.K.; Greenawalt, D.M.; Yang, M.; Yeatman, T.J. Co-evolution of somatic variation in primary and metastatic colorectal cancer may expand biopsy indications in the molecular era. PLoS ONE 2015, 10, e0126670. [Google Scholar] [CrossRef] [PubMed]
- Naxerova, K.; Reiter, J.G.; Brachtel, E.; Lennerz, J.K.; van de Wetering, M.; Rowan, A.; Cai, T.; Clevers, H.; Swanton, C.; Nowak, M.A.; et al. Origins of lymphatic and distant metastases in human colorectal cancer. Science 2017, 357, 55–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovaleva, V.; Geissler, A.L.; Lutz, L.; Fritsch, R.; Makowiec, F.; Wiesemann, S.; Hopt, U.T.; Passlick, B.; Werner, M.; Lassmann, S. Spatio-temporal mutation profiles of case-matched colorectal carcinomas and their metastases reveal unique de novo mutations in metachronous lung metastases by targeted next generation sequencing. Mol. Cancer 2016, 15, 63. [Google Scholar] [CrossRef] [Green Version]
- Ishaque, N.; Abba, M.L.; Hauser, C.; Patil, N.; Paramasivam, N.; Huebschmann, D.; Leupold, J.H.; Balasubramanian, G.P.; Kleinheinz, K.; Toprak, U.H.; et al. Whole genome sequencing puts forward hypotheses on metastasis evolution and therapy in colorectal cancer. Nat. Commun. 2018, 9, 4782. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Niida, A.; Uchi, R.; Hirata, H.; Komatsu, H.; Sakimura, S.; Hayashi, S.; Nambara, S.; Kuroda, Y.; Ito, S.; et al. A temporal shift of the evolutionary principle shaping intratumor heterogeneity in colorectal cancer. Nat. Commun. 2018, 9, 2884. [Google Scholar] [CrossRef]
- Uchi, R.; Takahashi, Y.; Niida, A.; Shimamura, T.; Hirata, H.; Sugimachi, K.; Sawada, G.; Iwaya, T.; Kurashige, J.; Shinden, Y.; et al. Integrated Multiregional Analysis Proposing a New Model of Colorectal Cancer Evolution. PLoS Genet. 2016, 12, e1005778. [Google Scholar] [CrossRef]
- Arnadottir, S.S.; Jeppesen, M.; Lamy, P.; Bramsen, J.B.; Nordentoft, I.; Knudsen, M.; Vang, S.; Madsen, M.R.; Thastrup, O.; Thastrup, J.; et al. Characterization of genetic intratumor heterogeneity in colorectal cancer and matching patient-derived spheroid cultures. Mol. Oncol. 2018, 12, 132–147. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Jung, S.H.; An, C.H.; Lee, S.H.; Baek, I.P.; Kim, M.S.; Park, S.W.; Rhee, J.K.; Lee, S.H.; Chung, Y.J. Subclonal Genomic Architectures of Primary and Metastatic Colorectal Cancer Based on Intratumoral Genetic Heterogeneity. Clin. Cancer Res. 2015, 21, 4461–4472. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Ye, Z.; Zhong, X.; Li, L.; Wang, C.; Myers, R.E.; Palazzo, J.P.; Fortuna, D.; Yan, A.; Waldman, S.A.; et al. Multiregion whole-exome sequencing of matched primary and metastatic tumors revealed genomic heterogeneity and suggested polyclonal seeding in colorectal cancer metastasis. Ann. Oncol. 2017, 28, 2135–2141. [Google Scholar] [CrossRef] [PubMed]
- Angelova, M.; Mlecnik, B.; Vasaturo, A.; Bindea, G.; Fredriksen, T.; Lafontaine, L.; Buttard, B.; Morgand, E.; Bruni, D.; Jouret-Mourin, A.; et al. Evolution of Metastases in Space and Time under Immune Selection. Cell 2018, 175, 751–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zill, O.A.; Banks, K.C.; Fairclough, S.R.; Mortimer, S.A.; Vowles, J.V.; Mokhtari, R.; Gandara, D.R.; Mack, P.C.; Odegaard, J.I.; Nagy, R.J.; et al. The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin. Cancer Res. 2018, 24, 3528–3538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GRAIL. Available online: https://grail.com/press-releases/grail-announces-positive-new-data-with-multi-cancer-early-detection-blood-test-from-ccga-study/ (accessed on 27 September 2019).
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Dittmar, R.L.; Xia, S.; Zhang, H.; Du, M.; Huang, C.C.; Druliner, B.R.; Boardman, L.; Wang, L. Cell-free DNA copy number variations in plasma from colorectal cancer patients. Mol. Oncol. 2017, 11, 1099–1111. [Google Scholar] [CrossRef]
- Peeters, M.; Price, T.; Boedigheimer, M.; Kim, T.W.; Ruff, P.; Gibbs, P.; Thomas, A.; Demonty, G.; Hool, K.; Ang, A. Evaluation of Emergent Mutations in Circulating Cell-Free DNA and Clinical Outcomes in Patients with Metastatic Colorectal Cancer Treated with Panitumumab in the ASPECCT Study. Clin. Cancer Res. 2019, 25, 1216–1225. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.W.; Peeters, M.; Thomas, A.; Gibbs, P.; Hool, K.; Zhang, J.; Ang, A.L.; Bach, B.A.; Price, T. Impact of Emergent Circulating Tumor DNA RAS Mutation in Panitumumab-Treated Chemoresistant Metastatic Colorectal Cancer. Clin. Cancer Res. 2018, 24, 5602–5609. [Google Scholar] [CrossRef] [Green Version]
- Tie, J.; Kinde, I.; Wang, Y.; Wong, H.L.; Roebert, J.; Christie, M.; Tacey, M.; Wong, R.; Singh, M.; Karapetis, C.S.; et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann. Oncol. 2015, 26, 1715–1722. [Google Scholar] [CrossRef]
- Frenel, J.S.; Carreira, S.; Goodall, J.; Roda, D.; Perez-Lopez, R.; Tunariu, N.; Riisnaes, R.; Miranda, S.; Figueiredo, I.; Nava-Rodrigues, D.; et al. Serial Next-Generation Sequencing of Circulating Cell-Free DNA Evaluating Tumor Clone Response to Molecularly Targeted Drug Administration. Clin. Cancer Res. 2015, 21, 4586–4596. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siravegna, G.; Lazzari, L.; Crisafulli, G.; Sartore-Bianchi, A.; Mussolin, B.; Cassingena, A.; Martino, C.; Lanman, R.B.; Nagy, R.J.; Fairclough, S.; et al. Radiologic and Genomic Evolution of Individual Metastases during HER2 Blockade in Colorectal Cancer. Cancer Cell 2018, 34, 148–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrantonio, F.; Vernieri, C.; Siravegna, G.; Mennitto, A.; Berenato, R.; Perrone, F.; Gloghini, A.; Tamborini, E.; Lonardi, S.; Morano, F.; et al. Heterogeneity of Acquired Resistance to Anti-EGFR Monoclonal Antibodies in Patients with Metastatic Colorectal Cancer. Clin. Cancer Res. 2017, 23, 2414–2422. [Google Scholar] [CrossRef] [PubMed]
- Beije, N.; Helmijr, J.C.; Weerts, M.J.; Beaufort, C.M.; Wiggin, M.; Marziali, A.; Verhoef, C.; Sleijfer, S.; Jansen, M.P.; Martens, J.W. Somatic mutation detection using various targeted detection assays in paired samples of circulating tumor DNA, primary tumor and metastases from patients undergoing resection of colorectal liver metastases. Mol. Oncol. 2016, 10, 1575–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suva, M.L.; Tirosh, I. Single-Cell RNA Sequencing in Cancer: Lessons Learned and Emerging Challenges. Mol. Cell 2019, 75, 7–12. [Google Scholar] [CrossRef]
- Bian, S.; Hou, Y.; Zhou, X.; Li, X.; Yong, J.; Wang, Y.; Wang, W.; Yan, J.; Hu, B.; Guo, H.; et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science 2018, 362, 1060–1063. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017, 49, 708–718. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, X.Y.; Hu, Z.; Hou, Q.; Zhang, H.; Li, Y.; Li, S.; Yue, J.; Jiang, Z.; Weissman, S.M.; et al. Evolution and heterogeneity of non-hereditary colorectal cancer revealed by single-cell exome sequencing. Oncogene 2017, 36, 2857–2867. [Google Scholar] [CrossRef]
- Leung, M.L.; Davis, A.; Gao, R.; Casasent, A.; Wang, Y.; Sei, E.; Vilar, E.; Maru, D.; Kopetz, S.; Navin, N.E. Single-cell DNA sequencing reveals a late-dissemination model in metastatic colorectal cancer. Genome Res. 2017, 27, 1287–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Yu, J.; Yao, X.; Wu, W.K.; Lu, Y.; Tang, S.; Li, X.; Bao, L.; Li, X.; Hou, Y.; et al. Discovery of biclonal origin and a novel oncogene SLC12A5 in colon cancer by single-cell sequencing. Cell Res. 2014, 24, 701–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewey, F.E.; Murray, M.F.; Overton, J.D.; Habegger, L.; Leader, J.B.; Fetterolf, S.N.; O’Dushlaine, C.; Van Hout, C.V.; Staples, J.; Gonzaga-Jauregui, C.; et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 2016, 354, aaf6814. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA Cancer J. Clin. 2009, 59, 225–249. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 7 October 2019).
- Eirew, P.; Steif, A.; Khattra, J.; Ha, G.; Yap, D.; Farahani, H.; Gelmon, K.; Chia, S.; Mar, C.; Wan, A.; et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 2015, 518, 422–426. [Google Scholar] [CrossRef]
- Roukos, D.H. Spatiotemporal diversification of intrapatient genomic clones and early drug development concepts realize the roadmap of precision cancer medicine. Drug Discov. Today 2017, 22, 1148–1164. [Google Scholar] [CrossRef]
- Zhang, Y.; Chang, L.; Yang, Y.; Fang, W.; Guan, Y.; Wu, A.; Hong, S.; Zhou, H.; Chen, G.; Chen, X.; et al. Intratumor heterogeneity comparison among different subtypes of non-small-cell lung cancer through multi-region tissue and matched ctDNA sequencing. Mol. Cancer 2019, 18, 7. [Google Scholar] [CrossRef]
- Kyrochristos, I.D.; Roukos, D.H. Comprehensive intra-individual genomic and transcriptional heterogeneity: Evidence-based Colorectal Cancer Precision Medicine. Cancer Treat. Rev. 2019, 80, 101894. [Google Scholar] [CrossRef]
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.T.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, M.N.; Kulkarni, A.S.; Liu, A.; et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019, 178, 160–175. [Google Scholar] [CrossRef]
- Gasperini, M.; Hill, A.J.; McFaline-Figueroa, J.L.; Martin, B.; Kim, S.; Zhang, M.D.; Jackson, D.; Leith, A.; Schreiber, J.; Noble, W.S.; et al. A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell 2019, 176, 377–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaneau, O.; Zazhytska, M.; Borel, C.; Giannuzzi, G.; Rey, G.; Howald, C.; Kumar, S.; Ongen, H.; Popadin, K.; Marbach, D.; et al. Chromatin three-dimensional interactions mediate genetic effects on gene expression. Science 2019, 364, eaat8266. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Jeng, E.E.; Hess, G.T.; Morgens, D.W.; Li, A.; Bassik, M.C. Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat. Biotechnol. 2017, 35, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Raj, B.; Wagner, D.E.; McKenna, A.; Pandey, S.; Klein, A.M.; Shendure, J.; Gagnon, J.A.; Schier, A.F. Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat. Biotechnol. 2018, 36, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Roukos, D.H. Crossroad between linear and nonlinear transcription concepts in the discovery of next-generation sequencing systems-based anticancer therapies. Drug Discov. Today 2016, 21, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Dugger, S.A.; Platt, A.; Goldstein, D.B. Drug development in the era of precision medicine. Nat. Rev. Drug Discov. 2018, 17, 183–196. [Google Scholar] [CrossRef]
- Bashor, C.J.; Patel, N.; Choubey, S.; Beyzavi, A.; Kondev, J.; Collins, J.J.; Khalil, A.S. Complex signal processing in synthetic gene circuits using cooperative regulatory assemblies. Science 2019, 364, 593–597. [Google Scholar] [CrossRef]

{kind=link}
| Patients/Samples | Technology | Findings and Potential Clinical Implications | Ref. |
|---|---|---|---|
| 5930 (18 cancer types) | WES | MSI-positive tumors were found in 14/18 cancer types and MSI had prognostic significance | [40] |
| 4151 | RNAseq, Affymetrix and Agilent gene expression platforms | Four consensus molecular subtypes were identified potentially informing patient classification | [41] |
| 1439 | WGS | 40 new independent association signals were discovered prompting further research for rare variants | [42] |
| 1006 (familial) | WES | 16% of familial CRCs had highly penetrant rare mutations including 3 novel candidate cancer driver genes (POT1, POLE2, MRE11) | [43] |
| 999 (601 PTs, 533 MTs) | tNGS | Right- and left-sided CRCs harbored distinct oncogenic mutations, potentially explaining differences in survival | [44] |
| 930 from 22 cancer types | WGS, RNAseq | A network of 193 non-coding loci was identified, affecting gene expression and warranting further research on functional mutation significance | [45] |
| 921 (multiple GI cancer types) | WES | 5 major GI adenocarcinoma subtypes were identified, with potential prognostic relevance | [46] |
| 511 from QUASAR 2 trial | tNGS | TP53, KRAS, BRAF and GNAS mutations were independent adverse prognostic factors and total mutation burden correlated with favorable survival, while MSI was not associated with survival | [47] |
| 468 | tNGS (1,321 gene panel) | 17 genes correlated to prognosis and absence of APC mutations was associated with worse prognosis | [48] |
| 341 | RNAseq | 20 dysregulated lncRNAs were identified, potentially related to tumorigenesis and/or progression, 9 of which correlated to OS, and a CRC-specific RNA network was constructed | [49] |
| 274 pts and mouse xenografts | WES, WGS | CNA analysis revealed 3 clusters overlapping with consensus molecular subtypes and high chromosomal instability predicted better response to BVZ combination therapy | [50] |
| 276 pts | 224 WES, 97 WGS, 215 RNAseq |
| [51] |
| 233 (4,742 from 21 cancer types) | WES | 4 novel genes with clear connections to cancer were identified | [7] |
| 230 (9423 from 33 cancer types) | WES | Up to 75% of CRCs harbored drug targets, while 59 novel cancer drivers were identified in the total cohort | [52] |
| 213 pts and cell lines | WGS, ChIP-seq | Functional non-coding point mutations at cohesin binding sites (CBSs) were frequent, similarly to other cancers, putatively driving tumorigenesis | [53] |
| 106 pts plus organoids and xenografts | tNGS, WES, WGS, RNAseq | Models retain genetic and transcriptomic tumor characteristics enabling research for improving therapeutic response prediction | [54] |
| 103 pts | tNGS, WES | 20 new recurrently mutated genes were identified | [55] |
| Patients (Samples) | Technology | Findings and Potential Clinical Implications | Ref. |
|---|---|---|---|
| 88 pts (46 matched PT and MTs and 42 non-metastatic PTs) | WES | Computationally calculated tumor heterogeneity was highly variable, with 70% sub-clone consistency between PT and LM, while high heterogeneity correlated to worse outcomes | [56] |
| 69 pts (Matched PT and MT samples) | tNGS (WGS on 4) | KRAS, NRAS, and BRAF mutations were 100% consistent and recurrent alterations were highly similar, suggesting that NGS of either PT or MT could suffice | [57] |
| 27 pts (97 samples from PT and MTs and 68 samples from a single PT) | tNGS (100 gene panel) | Inter- and intra-tumor variability was due to CNAs, which were highly discordant between PT and MT | [18] |
| 23 (118 MR tissue samples from matched PT and MTs) | WES | Although extensive inter- and intratumor heterogeneity was identified, matched PT and MTs were highly concordant for driver mutations, suggesting the early acquisition of aggressive alterations responsible for metastasis, while the modeof tumor evolution and sub-clonality correlated with disease stage | [58] |
| 18 (Matched PT and LM samples) | tNGS | 79.3% of SNVs in the PT were detected in the LM, while 81.7% of LM mutations were found in the PT, suggesting linear progression | [59] |
| 18 (Matched PT and MT samples) | tNGS | While concordance was 93.5%, most tumors showed at least one discordance due to co-evolution, suggesting that sampling over therapy could be useful | [60] |
| 17 (213 matched PT, LN and MT) | Polyguanine-repeat analysis | In 65% and 35% of cases, LN and distant metastases originated from distinct and single PT subclones respectively | [61] |
| 14 pts (70 MR samples from PT and matched liver and/or lung MTs) | tNGS | RAS status was preserved in MTs, while emerging mutations in other genes were also identified | [62] |
| 12 (Matched PT and MT) | WGS |
| [63] |
| 10 early CRC (53 MR samples) | MR-WES | This study supports a shift from Darwinian to neutral evolution during CRC progression | [64] |
| 9 (75 MR PT and 2 LM samples) | MR-WES | All cancers exhibited high ITH due to neutral evolution and drug resistance was attributed to pre-existing minor subclones | [65] |
| 6 (3-5 biopsies per patient) | MR-WES, RNAseq | Although ITH was universal, transcriptomics-guided classification could be independent of ITH | [66] |
| 5 pts (35 MR PT and LM samples) | MR-WES of the PT and MT | Branching evolution was identified, with prevalent CNA-based ITH as a putative source of metastasis | [67] |
| 4 (23 MR PT and MT samples) | MR-WES |
| [68] |
| 2 (36 spatiotemporal PT and MT samples) | WES |
| [69] |
| Patients (Samples) | Technology | Findings and Potential Clinical Implications | Ref. |
|---|---|---|---|
| Static cf/ctDNA next-generation sequencing analysis | |||
| 21,807 (>50 advanced cancer types) | tNGS | Driver gene cfDNA mutation profiles were similar to tumor NGS, while differences were attributed to clonal evolution over therapy leading to resistance | [70] |
| 1422 (sub-study, 21 tumor types) | tNGS, WGS, WGBS | Sensitivity for 12 cancers including CRC was 76% and 74% for stage I-III CRC | NCT02889978 [71] |
| 1397 (advanced CRC) | tNGS | Mutation frequencies in ctDNA were similar to tissue, and multiple distinct resistant mutations were identified in single patients | [19] |
| 1005 (8 cancer types) | CancerSEEK | Sensitivity was 65% and stage-dependent for CRC, suggesting the need for improvement before clinical applicability | [72] |
| 100 (TARGET study, diverse advanced cancers, 23 CRC) | tNGS | Druggable mutations were identified in 41/100 pts, 11/41 received matched therapy and all 11 achieved PR or stable disease | [22] |
| 80 pts | WGS | Recurrent CNVs were identified in multiple chromosomal regions and correlated with stage and prognosis | [73] |
| Consecutive liquid biopsies before and after systemic therapy | |||
| 261 (ASPECCT study, plasma samples before and after panitumumab) | tNGS |
| [74] |
| 238 (ASPECCT study, plasma samples before and after panitumumab) | tNGS | 79% of baseline samples were WT and 21% mutant RAS (associated with worse outcomes), while 32% of baseline-WT tumors had emergent RAS mutations | [75] |
| 53 (159 serial samples over chemotherapy) | tNGS | Mutational concordance between tumor and cfDNA was 92.3%, while cfDNA levels were predictive of clinical response | [76] |
| 39 various metastatic cancers, 12 CRC (159 total serial samples over targeted therapy) | tNGS | Monitoring of plasma mutation allele identified potential clonal responses to targeted therapy associated with progression, suggesting potential prognostic and predictive utility | [77] |
| Patients (Samples) | Technology | Findings and Potential Clinical Implications | Ref. |
|---|---|---|---|
| 100 (Matched PT and plasma samples after anti-EGFR) | BEAMing, tNGS | Resistant circulating mutations were detected (KRAS, NRAS, MET, ERBB2, FLT3, EGFR, MAP2K1), while treatment cessation led to re-emergence of sensitivity | [78] |
| 83 diverse advanced cancers (14 CRC, Static PT and ctDNA) | tNGS, ctDNA-tNGS | 30% of pts achieved disease control and targeting of more drug targets correlated with significantly favorable clinical outcomes, supporting individualized drug combinations | NCT02534675 [20] |
| 47 (archived PT, double MT samples at baseline, PR and progression and serial plasma samples) | tNGS, cfDNA-tNGS |
| NCT02994888 [17] |
| 33 (Serial liquid biopsies over HER2 blockade and diverse PT and MT samples) | WES, ctDNA-tNGS | ERBB2, RAS and PIK3CA mutations correlated to HER2-targeted therapy resistance and liquid biopsies identified primary resistance with >85% sensitivity, suggesting utility for decision-making | [79] |
| 22 (archived and post-progression tissue after anti-EGFR and static ctDNA) | tNGS | RAS mutations and HER2/MET amplification were the most prominent mechanisms of resistance in both tissue and ctDNA, suggesting utility for decision-making | [80] |
| 12 (Matched PT, MT and plasma samples) | tNGS | Limited concordance between ctDNA and PT/MT was identified, suggesting the need for refinement | [81] |
| 7 (diverse tumor samples over anti-EGFR, matched ctDNA, mouse xenografts) | WES, WGS, CNA, BEAMing | MET amplifications within rare pre-existing subclones confer resistance in KRAS-WT tumors during anti-EGFR therapy | [82] |
| Patients/Samples | Technology | Findings and Potential Translational Implications | Ref. |
|---|---|---|---|
| 12 pts (1,900 single cells and bulk multi-regional PT and MT) | Multiomics including single-cell Trio-seq and bulk MR-WGS |
| [84] |
| 11 pts and 7 cell lines (590 patient-derived and 561 cell line-derived single cells) | Single-cell RNAseq and RCA algorithm | Single-cell transcriptomics enabled more detailed sub-classification of CRC subtypes than bulk RNAseq, correlating to prognosis | [85] |
| 3 pts (Single cell-derived clonal organoids) | tNGS, WGS, RNAseq | All three colorectal cancers contained cells resistant to common drugs, while drug sensitivity was variable even among closely related single cell-derived clones, suggesting late emergence of resistance | [8] |
| 2 pts (6 bulk samples and 336 single cells from CRC, normal epithelium and polyps) | WES, single-cell WES |
| [86] |
| 2 pts (360 single-cells and bulk PT and LM samples) | Single-cell tNGS, bulk WES | Monoclonal and polyclonal seeding was identified, while rare cell sub-populations were found to correlate with progression and metastasis, although a late-dissemination model was identified | [87] |
| 1 pt (63 single cells) | WES | Two distinct clones were identified, one major with early APC and TP53 mutations and one minor with CDC27 and PABPC1 mutations, highlighting the ability of single-cell NGS to identify rare mutations | [88] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kyrochristos, I.D.; Ziogas, D.E.; Goussia, A.; Glantzounis, G.K.; Roukos, D.H. Bulk and Single-Cell Next-Generation Sequencing: Individualizing Treatment for Colorectal Cancer. Cancers 2019, 11, 1809. https://doi.org/10.3390/cancers11111809
Kyrochristos ID, Ziogas DE, Goussia A, Glantzounis GK, Roukos DH. Bulk and Single-Cell Next-Generation Sequencing: Individualizing Treatment for Colorectal Cancer. Cancers. 2019; 11(11):1809. https://doi.org/10.3390/cancers11111809
Chicago/Turabian StyleKyrochristos, Ioannis D., Demosthenes E. Ziogas, Anna Goussia, Georgios K. Glantzounis, and Dimitrios H. Roukos. 2019. "Bulk and Single-Cell Next-Generation Sequencing: Individualizing Treatment for Colorectal Cancer" Cancers 11, no. 11: 1809. https://doi.org/10.3390/cancers11111809