Structural Implications of STAT3 and STAT5 SH2 Domain Mutations


 , , , ,
, , , , 
Abstract
1. Introduction
2. Structure of STAT SH2 Domains
3. Disease-Associated Mutations in STAT3 and STAT5B SH2 Domains
3.1. Mutations in the pY Pocket
3.1.1. Mutations in Sheinerman Residues
3.1.2. Mutations Outside Sheinerman Residues
3.2. Mutations in the pY+3 Pocket
3.2.1. Mutations in the Hydrophobic System and βD Strand
3.2.2. Mutations in the Dimerization Interface
3.3. Mutations in the Additional Regions of the SH2 Domain
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Wingelhofer, B.; Neubauer, H.A.; Valent, P.; Han, X.; Constantinescu, S.N.; Gunning, P.T.; Müller, M.; Moriggl, R. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leuk. 2018, 32, 1713–1726. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; E Darnell, J. Stat3 as an Oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Tsutsui, M.; Yasuda, H.; Suto, H.; Imai, H.; Isobe, Y.; Sasaki, M.; Kojima, Y.; Oshimi, K.; Sugimoto, K. Frequent STAT3 activation is associated with Mcl-1 expression in nasal NK-cell lymphoma. Int. J. Lab. Hematol. 2010, 32, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Passerini, L.; Allan, S.E.; Battaglia, M.; Di Nunzio, S.; Alstad, A.N.; Levings, M.K.; Roncarolo, M.G.; Bacchetta, R. STAT5-signaling cytokines regulate the expression of FOXP3 in CD4+CD25+ regulatory T cells and CD4+ CD25- effector T cells. Int. Immunol. 2008, 20, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Mirmohammadsadegh, A.; Hassan, M.; Bardenheuer, W.; Marini, A.; Gustrau, A.; Nambiar, S.; Tannapfel, A.; Bojar, H.; Ruzicka, T.; Hengge, U.R. STAT5 Phosphorylation in Malignant Melanoma Is Important for Survival and Is Mediated Through SRC and JAK1 Kinases. J. Investig. Dermatol. 2006, 126, 2272–2280. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Briggs, J.; Park, S.; Niu, G.; Kortylewski, M.; Zhang, S.; Gritsko, T.; Turkson, J.; Kay, H.; Semenza, G.L.; et al. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 2005, 24, 5552–5560. [Google Scholar] [CrossRef]
- Matsumura, I.; Kitamura, T.; Wakao, H.; Tanaka, H.; Hashimoto, K.; Albanese, C.; Downward, J.; Pestell, R.G.; Kanakura, Y. Transcriptional regulation of the cyclin D1 promoter by STAT5: Its involvement in cytokine-dependent growth of hematopoietic cells. EMBO J. 1999, 18, 1367–1377. [Google Scholar] [CrossRef]
- Baik, M.; Yu, J.H.; Hennighausen, L. Growth hormone-STAT5 regulation of growth, hepatocellular carcinoma, and liver metabolism. Ann. New York Acad. Sci. 2011, 1229, 29–37. [Google Scholar] [CrossRef]
- Nichane, M.; Ren, X.; Bellefroid, E.J. Self-regulation of Stat3 activity coordinates cell-cycle progression and neural crest specification. EMBO J. 2010, 29, 55–67. [Google Scholar] [CrossRef]
- Ali, A.M.; Gómez-Biagi, R.F.; Rosa, D.A.; Lai, P.-S.; Heaton, W.L.; Park, J.S.; Eiring, A.M.; Vellore, N.A.; De Araujo, E.D.; Ball, D.P.; et al. Disarming an Electrophilic Warhead: Retaining Potency in Tyrosine Kinase Inhibitor (TKI)-Resistant CML Lines While Circumventing Pharmacokinetic Liabilities. ChemMedChem 2016, 11, 850–861. [Google Scholar] [CrossRef] [PubMed]
- De Araujo, E.D.; Manaswiyoungkul, P.; Erdogan, F.; Qadree, A.K.; Sina, D.; Tin, G.; Toutah, K.; Yuen, K.; Gunning, P.T.; Erodgan, F. A functional in vitro assay for screening inhibitors of STAT5B phosphorylation. J. Pharm. Biomed. Anal. 2019, 162, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Wingelhofer, B.; Maurer, B.; Heyes, E.C.; Cumaraswamy, A.A.; Berger-Becvar, A.; De Araujo, E.D.; Orlova, A.; Freund, P.; Ruge, F.; Park, J.; et al. Pharmacologic inhibition of STAT5 in acute myeloid leukemia. Leuk. 2018, 32, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Elumalai, N.; Berg, A.; Rubner, S.; Blechschmidt, L.; Song, C.; Natarajan, K.; Matysik, J.; Berg, T. Rational development of Stafib-2: A selective, nanomolar inhibitor of the transcription factor STAT5b. Sci. Rep. 2017, 7, 819. [Google Scholar] [CrossRef]
- Haftchenary, S.; Luchman, H.A.; Jouk, A.O.; Veloso, A.J.; Page, B.D.G.; Cheng, X.R.; Dawson, S.S.; Grinshtein, N.; Shahani, V.M.; Kerman, K.; et al. Potent Targeting of the STAT3 Protein in Brain Cancer Stem Cells: A Promising Route for Treating Glioblastoma. ACS Med. Chem. Lett. 2013, 4, 1102–1107. [Google Scholar] [CrossRef]
- Liu, B.A.; Engelmann, B.W.; Nash, P.D. The language of SH2 domain interactions defines phosphotyrosine-mediated signal transduction. FEBS Lett. 2012, 586, 2597–2605. [Google Scholar] [CrossRef]
- Gao, Q.; Hua, J.; Kimura, R.; Headd, J.J.; Fu, X.-Y.; Chin, Y.E. Identification of the Linker-SH2 Domain of STAT as the Origin of the SH2 Domain Using Two-dimensional Structural Alignment. Mol. Cell. Proteom. 2004, 3, 704–714. [Google Scholar] [CrossRef]
- Zhou, S. SH2 domains recognize specific phosphopeptide sequences. Cell 1993, 72, 767–778. [Google Scholar] [CrossRef]
- Songyang, Z.; Cantley, L.C. Recognition and specificity in protein tyrosine kinase-mediated signalling. Trends Biochem. Sci. 1995, 20, 470–475. [Google Scholar] [CrossRef]
- Gianti, E.; Zauhar, R.J. An SH2 domain model of STAT5 in complex with phospho-peptides define “STAT5 Binding Signatures. ” J. Comput. Mol. Des. 2015, 29, 451–470. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera?A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Nkansah, E.; Shah, R.; Collie, G.W.; Parkinson, G.N.; Palmer, J.; Rahman, K.M.; Bui, T.T.; Drake, A.F.; Husby, J.; Neidle, S.; et al. Observation of unphosphorylated STAT3 core protein binding to targetdsDNA by PEMSA and X-ray crystallography. FEBS Lett. 2013, 587, 833–839. [Google Scholar] [CrossRef] [PubMed]
- De Araujo, E.D.; Erdogan, F.; Neubauer, H.A.; Meneksedag-Erol, D.; Manaswiyoungkul, P.; Eram, M.S.; Seo, H.-S.; Qadree, A.K.; Israelian, J.; Orlova, A.; et al. Structural and functional consequences of the STAT5BN642H driver mutation. Nat. Commun. 2019, 10, 2517. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Ren, Z.; Parker, G.N.; Sondermann, H.; Pastorello, M.A.; Wang, W.; McMurray, J.S.; Demeler, B.; Darnell, J.E.; Chen, X. Structural Bases of Unphosphorylated STAT1 Association and Receptor Binding. Mol. Cell 2005, 17, 761–771. [Google Scholar] [CrossRef]
- Langenfeld, F.; Guarracino, Y.; Arock, M.; Trouve, A.; Tchertanov, L. How Intrinsic Molecular Dynamics Control Intramolecular Communication in Signal Transducers and Activators of Transcription Factor STAT5. PLoS ONE 2015, 10, e0145142. [Google Scholar] [CrossRef]
- Takeda, K.; Noguchi, K.; Shi, W.; Tanaka, T.; Matsumoto, M.; Yoshida, N.; Kishimoto, T.; Akira, S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. USA 1997, 94, 3801–3804. [Google Scholar] [CrossRef]
- Purvis, H.A.; Anderson, A.E.; Young, D.A.; Isaacs, J.D.; Hilkens, C.M.U. A Negative Feedback Loop Mediated by STAT3 Limits Human Th17 Responses. J. Immunol. 2014, 193, 1142–1150. [Google Scholar] [CrossRef]
- Haddad, E. STAT3: Too much may be worse than not enough! Blood 2015, 125, 583–584. [Google Scholar] [CrossRef]
- Laurence, A.D.J.; Uhlig, H.H. When half a glass of STAT3 is just not enough. Blood 2016, 128, 3020–3021. [Google Scholar] [CrossRef]
- Woellner, C.; Gertz, E.M.; Schäffer, A.A.; Lagos, M.; Perro, M.; Glocker, E.-O.; Pietrogrande, M.C.; Cossu, F.; Franco, J.L.; Matamoros, N.; et al. Mutations in STAT3 and diagnostic guidelines for hyper-IgE syndrome. J. Allergy Clin. Immunol. 2010, 125, 424–432. [Google Scholar] [CrossRef]
- Robinson, W.S.; Arnold, S.R.; Michael, C.F.; Vickery, J.D.; Schoumacher, R.A.; Pivnick, E.K.; Ward, J.C.; Nagabhushanam, V.; Lew, D.B. Case report of a young child with disseminated histoplasmosis and review of hyper immunoglobulin e syndrome (HIES). Clin. Mol. Allergy 2011, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.S.; Avery, D.T.; Chan, A.; Batten, M.; Bustamante, J.; Boisson-Dupuis, S.; Arkwright, P.D.; Kreins, A.Y.; Averbuch, D.; Engelhard, D.; et al. Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood 2012, 119, 3997–4008. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-Y.; Tian, W.; Shu, L.; Jiang, L.-P.; Zhan, Y.-Z.; Zhao, X.-D.; Cui, Y.-X.; Tang, X.-M.; Wu, D.-Q.; Yang, X.-Q.; et al. Clinical Features, STAT3 Gene Mutations and Th17 Cell Analysis in Nine Children with Hyper-IgE Syndrome in Mainland China. Scand. J. Immunol. 2013, 78, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Chandesris, M.O.; Melki, I.; Natividad, A.; Puel, A.; Fieschi, C.; Yun, L.; Thumerelle, C.; Oksenhendler, E.; Boutboul, D.; Thomas, C.; et al. Autosomal dominant STAT3 deficiency and hyper-IgE syndrome: Molecular, cellular, and clinical features from a French national survey. Medicine (United States) 2012, 91. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.F.; Renner, E.D.; Henderson, C.; Langenbeck, A.; Olivier, K.N.; Hsu, A.P.; Hagl, B.; Boos, A.; Davis, J.; Marciano, B.E.; et al. Lung Parenchyma Surgery in Autosomal Dominant Hyper-IgE Syndrome. J. Clin. Immunol. 2013, 33, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.M.; DeLeo, F.R.; Elloumi, H.Z.; Hsu, A.P.; Uzel, G.; Brodsky, N.; Freeman, A.F.; Demidowich, A.; Davis, J.; Turner, M.L.; et al. STAT3Mutations in the Hyper-IgE Syndrome. New Engl. J. Med. 2007, 357, 1608–1619. [Google Scholar] [CrossRef] [PubMed]
- Heimall, J.; Davis, J.; Shaw, P.A.; Hsu, A.P.; Gu, W.; Welch, P.; Holland, S.M.; Freeman, A.F. Paucity of genotype-phenotype correlations in STAT3 mutation positive Hyper IgE Syndrome (HIES). Clin. Immunol. 2011, 139, 75–84. [Google Scholar] [CrossRef]
- Moreira Varanese, I.; Seminario, A.; Diaz Balve, D.; Uriarte, I.; Belardinelli, G.; Gomez Racio, A.; di Giovanni, D.; Bezrodnik, L. New STAT3 mutation in a patient with Hyper IgE Syndrome (HIGES) without criteria in HIES STAT3 score. Transl. Biomed. 2010, 1. [Google Scholar] [CrossRef]
- Qiu, Z.-Y.; Fan, L.; Wang, L.; Qiao, C.; Wu, Y.-J.; Zhou, J.-F.; Xu, W.; Li, J.-Y. STAT3 mutations are frequent in T-cell large granular lymphocytic leukemia with pure red cell aplasia. J. Hematol. Oncol. 2013, 6, 82. [Google Scholar] [CrossRef]
- Yan, Y.; Olson, T.L.; Nyland, S.B.; Feith, D.J.; Loughran, T.P. Emergence of a STAT3 mutated NK clone in LGL leukemia. Leuk. Res. Rep. 2015, 4, 4–7. [Google Scholar] [CrossRef][Green Version]
- Kűcűk, C.; Jiang, B.; Hu, X.; Zhang, W.; Chan, J.K.C.; Xiao, W.; Lack, N.; Alkan, C.; Williams, J.C.; Avery, K.N.; et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from γδ-T or NK cells. Nat. Commun. 2015, 6, 6025. [Google Scholar] [CrossRef] [PubMed]
- Blombery, P.; Thompson, E.R.; Jones, K.; Arnau, G.M.; Lade, S.; Markham, J.F.; Li, J.; Deva, A.; Johnstone, R.W.; Khot, A.; et al. Whole exome sequencing reveals activating JAK1 and STAT3 mutations in breast implant-associated anaplastic large cell lymphoma anaplastic large cell lymphoma. Haematol. 2016, 101, e387–e390. [Google Scholar] [CrossRef] [PubMed]
- McKinney, M.; Moffitt, A.B.; Gaulard, P.; Travert, M.; De Leval, L.; Nicolae, A.; Raffeld, M.; Jaffe, E.S.; Pittaluga, S.; Xi, L.; et al. The Genetic Basis of Hepatosplenic T-cell Lymphoma. Cancer Discov. 2017, 7, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, R.S.; Ma, L.; Monabati, A.; Zehnder, J.L.; Arber, D.A. STAT3 mutations are present in aggressive B-cell lymphomas including a subset of diffuse large B-cell lymphomas with CD30 expression. Haematol. 2014, 99, e105–e107. [Google Scholar] [CrossRef] [PubMed]
- Song, T.L.; Nairismägi, M.L.; Laurensia, Y.; Lim, J.Q.; Tan, J.; Li, Z.M.; Kizhakeyil, A.; Wijaya, G.C.; Huang, D.C.; Nagarajan, S.; et al. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood 2018, 132, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Schimke, L.F.; Sawalle-Belohradsky, J.; Roesler, J.; Wollenberg, A.; Rack, A.; Borte, M.; Rieber, N.; Cremer, R.; Maaß, E.; Dopfer, R.; et al. Diagnostic approach to the hyper-IgE syndromes: Immunologic and clinical key findings to differentiate hyper-IgE syndromes from atopic dermatitis. J. Allergy Clin. Immunol. 2010, 126, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Couronné, L.; Scourzic, L.; Pilati, C.; Della Valle, V.; Duffourd, Y.; Solary, E.; Vainchenker, W.; Merlio, J.-P.; Beylot-Barry, M.; Damm, F.; et al. STAT3 mutations identified in human hematologic neoplasms induce myeloid malignancies in a mouse bone marrow transplantation model. Haematol. 2013, 98, 1748–1752. [Google Scholar] [CrossRef]
- Kristensen, T.; Larsen, M.; Rewes, A.; Frederiksen, H.; Thomassen, M.; Møller, M.B. Clinical Relevance of Sensitive and Quantitative STAT3 Mutation Analysis Using Next-Generation Sequencing in T-Cell Large Granular Lymphocytic Leukemia. J. Mol. Diagn. 2014, 16, 382–392. [Google Scholar] [CrossRef]
- Renner, E.D.; Rylaarsdam, S.; Añover-Sombke, S.; Rack, A.L.; Reichenbach, J.; Carey, J.C.; Zhu, Q.; Jansson, A.F.; Barboza, J.; Schimke, L.F.; et al. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. J. Allergy Clin. Immunol. 2008, 122, 181–187. [Google Scholar] [CrossRef]
- Odio, C.D.; Milligan, K.L.; Mc Gowan, K.; Spergel, A.K.R.; Bishop, R.; Boris, L.; Urban, A.; Welch, P.; Heller, T.; Kleiner, D.; et al. Endemic mycoses in patients with STAT3-mutated hyper-IgE (Job) syndrome. J. Allergy Clin. Immunol. 2015, 136, 1411–1413. [Google Scholar] [CrossRef]
- Kumánovics, A.; Wittwer, C.T.; Pryor, R.J.; Augustine, N.H.; Leppert, M.F.; Carey, J.C.; Ochs, H.D.; Wedgwood, R.J.; Faville, R.J.; Quie, P.G.; et al. Rapid Molecular Analysis of the STAT3 Gene in Job Syndrome of Hyper-IgE and Recurrent Infectious Diseases. J. Mol. Diagn. 2010, 12, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Ives, M.L.; Ma, C.S.; Palendira, U.; Chan, A.; Bustamante, J.; Boisson-Dupuis, S.; Arkwright, P.D.; Engelhard, D.; Averbuch, D.; Magdorf, K.; et al. Signal transducer and activator of transcription 3 (STAT3) mutations underlying autosomal dominant hyper-IgE syndrome impair human CD8 + T-cell memory formation and function. J. Allergy Clin. Immunol. 2013, 132, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H.; Jakobsen, M.A.; Larsen, C.S. Identification of a novel STAT3 mutation in a patient with hyper-IgE syndrome. Scand. J. Infect. Dis. 2013, 45, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Bellon, M.; Lu, L.; Nicot, C. Constitutive activation of Pim1 kinase is a therapeutic target for adult T-cell leukemia. Blood 2016, 127, 2439–2450. [Google Scholar] [CrossRef]
- Giacomelli, M.; Tamassia, N.; Moratto, D.; Bertolini, P.; Bertulli, C.; Plebani, A.; Cassatella, M.; Ricci, G.; Bazzoni, F.; Badolato, R. SH2-domain mutations in STAT3 in hyper-IgE syndrome patients result in impairment of IL-10 function. Eur. J. Immunol. 2011, 41, 3075–3084. [Google Scholar] [CrossRef]
- Jiao, H.; Tóth, B.; Erdős, M.; Fransson, I.; Rakoczi, E.; Balogh, I.; Magyarics, Z.; Dérfalvi, B.; Csorba, G.; Szaflarska, A.; et al. Novel and recurrent STAT3 mutations in hyper-IgE syndrome patients from different ethnic groups. Mol. Immunol. 2008, 46, 202–206. [Google Scholar] [CrossRef]
- Shi, M.; He, R.; Feldman, A.L.; Viswanatha, D.S.; Jevremovic, D.; Chen, D.; Morice, W.G. STAT3 mutation and its clinical and histopathologic correlation in T-cell large granular lymphocytic leukemia. Hum. Pathol. 2018, 73, 74–81. [Google Scholar] [CrossRef]
- Goncalves, M.R.; Junior, P.R. Association between STAT3 mutations and phenotypic features in Hyper-IgE Syndrome. J. Allergy Clin. Immunol. 2019, 143, AB111. [Google Scholar] [CrossRef]
- Calderaro, J.; Nault, J.C.; Balabaud, C.; Couchy, G.; Saint-Paul, M.C.; Azoulay, D.; Mehdaoui, D.; Luciani, A.; Zafrani, E.S.; Bioulac-Sage, P.; et al. Inflammatory hepatocellular adenomas developed in the setting of chronic liver disease and cirrhosis. Mod. Pathol. 2016, 29, 43–50. [Google Scholar] [CrossRef]
- Dufva, O.; Kankainen, M.; Kelkka, T.; Sekiguchi, N.; Awad, S.A.; Eldfors, S.; Yadav, B.; Kuusanmäki, H.; Malani, D.; Andersson, E.I.; et al. Aggressive natural killer-cell leukemia mutational landscape and drug profiling highlight JAK-STAT signaling as therapeutic target. Nat. Commun. 2018, 9, 1567. [Google Scholar] [CrossRef]
- Kataoka, K.; Nagata, Y.; Kitanaka, A.; Shiraishi, Y.; Shimamura, T.; Yasunaga, J.-I.; Totoki, Y.; Chiba, K.; Sato-Otsubo, A.; Nagae, G.; et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat. Genet. 2015, 47, 1304–1315. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; Sahasrabuddhe, A.A.; Rolland, D.C.M.; Velusamy, T.; Chung, F.; Schaller, M.; Bailey, N.G.; Betz, B.L.; Miranda, R.N.; Porcu, P.; et al. Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK–STAT pathway in Sézary syndrome. Nat. Commun. 2015, 6, 8470. [Google Scholar] [CrossRef] [PubMed]
- Ishida, F.; Matsuda, K.; Sekiguchi, N.; Makishima, H.; Taira, C.; Momose, K.; Nishina, S.; Senoo, N.; Sakai, H.; Ito, T.; et al. STAT3 gene mutations and their association with pure red cell aplasia in large granular lymphocyte leukemia. Cancer Sci. 2014, 105, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, A.K.; Fataccioli, V.; Castellano, G.; Martin-Garcia, N.; Pelletier, L.; Ammerpohl, O.; Bergmann, J.; Bhat, J.; Pau, E.C.; Martín-Subero, J.I.; et al. DNA methylation profiling of hepatosplenic t-cell lymphoma. Haematologica 2019, 104, e104–e107. [Google Scholar] [CrossRef] [PubMed]
- Koskela, H.L.M.; Eldfors, S.; Ellonen, P.; van Adrichem, A.J.; Kuusanmäki, H.; Andersson, E.I.; Lagström, S.; Clemente, M.J.; Olson, T.; Jalkanen, S.E.; et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N. Engl. J. Med. 2012, 366, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Pilati, C.; Amessou, M.; Bihl, M.P.; Balabaud, C.; Van Nhieu, J.T.; Paradis, V.; Nault, J.C.; Izard, T.; Bioulac-Sage, P.; Couchy, G.; et al. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J. Exp. Med. 2011, 208, 1359–1366. [Google Scholar] [CrossRef]
- Jerez, A.; Clemente, M.J.; Makishima, H.; Koskela, H.; Leblanc, F.; Ng, K.P.; Olson, T.; Przychodzen, B.; Afable, M.; Gomez-Segui, I.; et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood 2012, 120, 3048–3057. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Ma, L.; Merker, J.D.; Martinez, B.; Zehnder, J.L.; Arber, D.A. STAT3 mutations are frequent in CD30+ T-cell lymphomas and T-cell large granular lymphocytic leukemia. Leukemia 2013, 27, 2244–2247. [Google Scholar] [CrossRef]
- Nicolae, A.; Xi, L.; Pittaluga, S.; Abdullaev, Z.; Pack, S.D.; Chen, J.; Waldmann, T.A.; Jaffe, E.S.; Raffeld, M. Frequent STAT5B mutations in γδ hepatosplenic T-cell lymphomas. Leuk. 2014, 28, 2244–2248. [Google Scholar] [CrossRef]
- Ma, C.S.; Chew, G.Y.; Simpson, N.; Priyadarshi, A.; Wong, M.; Grimbacher, B.; Fulcher, D.A.; Tangye, S.G.; Cook, M.C. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J. Exp. Med. 2008, 205, 1551–1557. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Haapaniemi, E.; Russell, M.A.; Caswell, R.; Allen, H.L.; De Franco, E.; McDonald, T.J.; Rajala, H.; Ramelius, A.; Barton, J.; et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat. Genet. 2014, 46, 812–814. [Google Scholar] [CrossRef] [PubMed]
- Fasan, A.; Kern, W.; Grossmann, V.; Haferlach, C.; Haferlach, T.; Schnittger, S. STAT3 mutations are highly specific for large granular lymphocytic leukemia. Leukemia 2013, 27, 1598–1600. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Lee, S.H.; Yoo, N.J.; Lee, S.H. STAT3 exon 21 mutation is rare in common human cancers. Acta Oncol. 2013, 52, 1221–1222. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Y.; Li, Q.; Chen, T.T.; Guo, X.; Ge, J.; Yuan, L.X. Destructive pulmonary staphylococcal infection in a boy with hyper-IgE syndrome: A novel mutation in the signal transducer and activator of transcription 3 (STAT3) gene (p.Y657S). Eur. J. Pediatr. 2011, 170, 661–666. [Google Scholar] [CrossRef]
- Milner, J.D.; Vogel, T.P.; Forbes, L.; Ma, C.A.; Stray-Pedersen, A.; Niemela, J.E.; Lyons, J.J.; Engelhardt, K.R.; Zhang, Y.; Topcagic, N.; et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood 2015, 125, 591–599. [Google Scholar] [CrossRef]
- Saikia, B.; Goel, S.; Suri, D.; Minz, R.W.; Rawat, A.; Singh, S. Novel Mutation in SH2 Domain of STAT3 (p.M660T) in Hyper-IgE Syndrome with Sterno-Clavicular and Paravertebral Abscesses. Indian J. Pediatr. 2017, 203, 244–495. [Google Scholar] [CrossRef]
- Teramo, A.; Barila, G.; Calabretto, G.; Ercolin, C.; Lamy, T.; Moignet, A.; Roussel, M.; Pastoret, C.; Leoncin, M.; Gattazzo, C.; et al. STAT3 mutation impacts biological and clinical features of T-LGL leukemia. Oncotarget 2017, 8, 61876–61889. [Google Scholar] [CrossRef]
- Crescenzo, R.; Abate, F.; Lasorsa, E.; Tabbo’, F.; Gaudiano, M.; Chiesa, N.; Di Giacomo, F.; Spaccarotella, E.; Barbarossa, L.; Ercole, E.; et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 2015, 27, 516–532. [Google Scholar] [CrossRef]
- Iwanaga, E.; Nakamura, M.; Nanri, T.; Kawakita, T.; Horikawa, K.; Mitsuya, H.; Asou, N. Acute promyelocytic leukemia harboring a STAT5B-RARA fusion gene and a G596V missense mutation in the STAT5B SH2 domain of the STAT5B-RARA. Eur. J. Haematol. 2009, 83, 499–501. [Google Scholar] [CrossRef]
- Kiel, M.J.; Velusamy, T.; Rolland, D.; Sahasrabuddhe, A.A.; Chung, F.; Bailey, N.G.; Schrader, A.; Li, B.; Li, J.Z.; Ozel, A.B.; et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood 2014, 124, 1460–1472. [Google Scholar] [CrossRef]
- Roberti, A.; Dobay, M.P.; Bisig, B.; Vallois, D.; Boéchat, C.; Lanitis, E.; Bouchindhomme, B.; Parrens, M.-C.; Bossard, C.; Quintanilla-Martinez, L.; et al. Type II enteropathy-associated T-cell lymphoma features a unique genomic profile with highly recurrent SETD2 alterations. Nat. Commun. 2016, 7, 12602–12613. [Google Scholar] [CrossRef] [PubMed]
- Chia, D.J.; Subbian, E.; Buck, T.M.; Hwa, V.; Rosenfeld, R.G.; Skach, W.R.; Shinde, U.; Rotwein, P. Aberrant folding of a mutant Stat5b causes growth hormone insensitivity and proteasomal dysfunction. J. Biol. Chem. 2006, 281, 6552–6558. [Google Scholar] [CrossRef] [PubMed]
- Schrader, A.; Crispatzu, G.; Oberbeck, S.; Mayer, P.; Pützer, S.; Von Jan, J.; Vasyutina, E.; Warner, K.; Weit, N.; Pflug, N.; et al. Actionable perturbations of damage responses by TCL1/ATM and epigenetic lesions form the basis of T-PLL. Nat. Commun. 2018, 9, 697. [Google Scholar] [CrossRef] [PubMed]
- Rajala, H.L.M.; Eldfors, S.; Kuusanmäki, H.; Van Adrichem, A.J.; Olson, T.; Lagström, S.; Andersson, E.I.; Jerez, A.; Clemente, M.J.; Yan, Y.; et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood 2013, 121, 4541–4550. [Google Scholar] [CrossRef] [PubMed]
- Babushok, D.V.; Perdigones, N.; Perin, J.C.; Olson, T.S.; Ye, W.; Roth, J.J.; Lind, C.; Cattier, C.; Li, Y.; Hartung, H.; et al. Emergence of clonal hematopoiesis in the majority of patients with acquired aplastic anemia. Cancer Genet. 2015, 208, 115–128. [Google Scholar] [CrossRef]
- Lopez, C.; Bergmann, A.K.; Paul, U.; Penas, E.M.M.; Nagel, I.; Betts, M.; Johansson, P.; Ritgen, M.; Baumann, T.; Aymerich, M.; et al. Genes encoding members of the JAK-STAT pathway or epigenetic regulators are recurrently mutated in T-cell prolymphocytic leukaemia. Br. J. Haematol. 2016, 173, 265–273. [Google Scholar] [CrossRef]
- Andersson, E.I.; Tanahashi, T.; Sekiguchi, N.; Gasparini, V.R.; Bortoluzzi, S.; Kawakami, T.; Matsuda, K.; Mitsui, T.; Eldfors, S.; Bortoluzzi, S.; et al. High incidence of activating STAT5B mutations in CD4-positive T-cell large granular lymphocyte leukemia. Blood 2016, 128, 2465–2468. [Google Scholar] [CrossRef]
- Andersson, E.I.; Pützer, S.; Yadav, B.; Dufva, O.; Khan, S.; He, L.; Sellner, L.; Schrader, A.; Crispatzu, G.; Oleś, M.; et al. Discovery of novel drug sensitivities in T-PLL by high-throughput ex vivo drug testing and mutation profiling. Leukemia 2018, 32, 774–787. [Google Scholar] [CrossRef]
- Bandapalli, O.R.; Schuessele, S.; Kunz, J.B.; Rausch, T.; Stütz, A.M.; Tal, N.; Geron, I.; Gershman, N.; Izraeli, S.; Eilers, J.; et al. The activating STAT5B N642H mutation is a common abnormality in pediatric T-cell acute lymphoblastic leukemia and confers a higher risk of relapse. Haematol. 2014, 99, e188–e192. [Google Scholar] [CrossRef]
- Atak, Z.K.; Gianfelici, V.; Hulselmans, G.; De Keersmaecker, K.; Devasia, A.G.; Geerdens, E.; Mentens, N.; Chiaretti, S.; Durinck, K.; Uyttebroeck, A.; et al. Comprehensive Analysis of Transcriptome Variation Uncovers Known and Novel Driver Events in T-Cell Acute Lymphoblastic Leukemia. PLoS Genet. 2013, 9, e1003997. [Google Scholar]
- Nairismägi, M.-L.; Tan, J.; Lim, J.Q.; Nagarajan, S.; Ng, C.C.Y.; Rajasegaran, V.; Huang, D.; Lim, W.K.; Laurensia, Y.; Wijaya, G.C.; et al. JAK-STAT and G-protein-coupled receptor signaling pathways are frequently altered in epitheliotropic intestinal T-cell lymphoma. Leuk. 2016, 30, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Lavallée, V.-P.; Krošl, J.; Lemieux, S.; Boucher, G.; Gendron, P.; Pabst, C.; Boivin, I.; Marinier, A.; Guidos, C.J.; Meloche, S.; et al. Chemo-genomic interrogation of CEBPA mutated AML reveals recurrent CSF3R mutations and subgroup sensitivity to JAK inhibitors. Blood 2016, 127, 3054–3061. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Shen, J.; Yang, Y.; Tang, H.; Shi, M.; Liu, J.; Liu, Z.; Shi, X.; Yi, Y. CSF3R T618I, ASXL1 G942 fs and STAT5B N642H trimutation co-contribute to a rare chronic neutrophilic leukaemia manifested by rapidly progressive leucocytosis, severe infections, persistent fever and deep venous thrombosis. Br. J. Haematol. 2018, 180, 892–894. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Rabionet, R.; Espinet, B.; Zapata, L.; Puiggros, A.; Melero, C.; Puig, A.; Sarria-Trujillo, Y.; Ossowski, S.; García-Muret, M.P.; et al. Identification of Gene Mutations and Fusion Genes in Patients with Sézary Syndrome. J. Investig. Dermatol. 2016, 136, 1490–1499. [Google Scholar] [CrossRef]
- Ma, X.; Wen, L.; Wu, L.; Wang, Q.; Yao, H.; Wang, Q.; Ma, L.; Chen, S. Rare occurrence of a STAT5B N642H mutation in adult T-cell acute lymphoblastic leukemia. Cancer Genet. 2015, 208, 52–53. [Google Scholar] [CrossRef]
- Cross, N.C.P.; Hoade, Y.; Tapper, W.J.; Carreno-Tarragona, G.; Fanelli, T.; Jawhar, M.; Naumann, N.; Pieniak, I.; Lübke, J.; Ali, S.; et al. Recurrent activating STAT5B N642H mutation in myeloid neoplasms with eosinophilia. Leukemia 2019, 33, 415–425. [Google Scholar] [CrossRef]
- Ma, C.A.; Xi, L.; Cauff, B.; DeZure, A.; Freeman, A.F.; Hambleton, S.; Kleiner, G.; Leahy, T.R.; O’Sullivan, M.; Makiya, M.; et al. Somatic STAT5b gain-of-function mutations in early onset nonclonal eosinophilia, urticaria, dermatitis, and diarrhea. Blood 2017, 129, 650–653. [Google Scholar] [CrossRef]
- Baer, C.; Muehlbacher, V.; Kern, W.; Haferlach, C.; Haferlach, T. Molecular genetic characterization of myeloid/lymphoid neoplasms associated with eosinophilia and rearrangement of PDGFRA, PDGFRB, FGFR1 or PCM1-JAK2. Haematol. 2018, 103, e348–e350. [Google Scholar] [CrossRef]
- Simpson, H.M.; Khan, R.Z.; Song, C.; Sharma, D.; Sadashivaiah, K.; Furusawa, A.; Liu, X.; Nagaraj, S.; Sengamalay, N.; Sadzewicz, L.; et al. Concurrent Mutations in ATM and Genes Associated with Common γ Chain Signaling in Peripheral T Cell Lymphoma. PLoS ONE 2015, 10, e0141906. [Google Scholar] [CrossRef]
- Jiang, L.; Gu, Z.-H.; Yan, Z.-X.; Zhao, X.; Xie, Y.-Y.; Zhang, Z.-G.; Pan, C.-M.; Hu, Y.; Cai, C.-P.; Dong, Y.; et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat. Genet. 2015, 47, 1061–1066. [Google Scholar] [CrossRef]
- Kontro, M.; Kuusanmäki, H.; Eldfors, S.; Burmeister, T.; Andersson, E.I.; Bruserud, O.; Brümmendorf, T.H.; Edgren, H.; Gjertsen, B.T.; Itälä-Remes, M.; et al. Novel activating STAT5B mutations as putative drivers of T-cell acute lymphoblastic leukemia. Leukemia 2014, 28, 1738–1742. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.P.; Nicolae, A.; Meeker, H.; Raffeld, M.; Xi, L.; Jegalian, A.G.; Miller, D.C.; Pittaluga, S.; Jaffe, E.S. Primary CNS T-cell Lymphomas: A Clinical, Morphologic, Immunophenotypic, and Molecular Analysis. Am. J. Surg. Pathol. 2015, 39, 1719–1729. [Google Scholar] [CrossRef] [PubMed]
- Varco-Merth, B.; Feigerlová, E.; Shinde, U.; Rosenfeld, R.G.; Hwa, V.; Rotwein, P. Severe growth deficiency is associated with STAT5b mutations that disrupt protein folding and activity. Mol. Endocrinol. 2013, 27, 150–161. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hwa, V. STAT5B deficiency: Impacts on human growth and immunity. Growth Horm. IGF Res. 2016, 28, 16–20. [Google Scholar] [CrossRef]
- Cui, Y.; Riedlinger, G.; Miyoshi, K.; Tang, W.; Li, C.; Deng, C.-X.; Robinson, G.W.; Hennighausen, L. Inactivation of Stat5 in Mouse Mammary Epithelium during Pregnancy Reveals Distinct Functions in Cell Proliferation, Survival, and Differentiation. Mol. Cell. Boil. 2004, 24, 8037–8047. [Google Scholar] [CrossRef]
- Hoelbl, A.; Kovacic, B.; Kerenyi, M.A.; Simma, O.; Warsch, W.; Cui, Y.; Beug, H.; Hennighausen, L.; Moriggl, R.; Sexl, V. Clarifying the role of Stat5 in lymphoid development and Abelson-induced transformation. Blood 2006, 107, 4898–4906. [Google Scholar] [CrossRef]
- Klammt, J.; Neumann, D.; Gevers, E.F.; Andrew, S.F.; Schwartz, I.D.; Rockstroh, D.; Colombo, R.; Sanchez, M.A.; Vokurkova, D.; Kowalczyk, J.; et al. Dominant-negative STAT5B mutations cause growth hormone insensitivity with short stature and mild immune dysregulation. Nat. Commun. 2018, 9, 2105. [Google Scholar] [CrossRef]
- Kanai, T.; Jenks, J.; Nadeau, K.C. The STAT5b Pathway Defect and Autoimmunity. Front. Immunol. 2012, 3, 3. [Google Scholar] [CrossRef]
- Sallmyr, A.; Fan, J.; Datta, K.; Kim, K.-T.; Grosu, D.; Shapiro, P.; Small, D.; Rassool, F. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: Implications for poor prognosis in AML. Blood 2008, 111, 3173–3182. [Google Scholar] [CrossRef]
- Warsch, W.; Kollmann, K.; Eckelhart, E.; Fajmann, S.; Cerny-Reiterer, S.; Hölbl, A.; Gleixner, K.V.; Dworzak, M.; Mayerhofer, M.; Hoermann, G.; et al. High STAT5 levels mediate imatinib resistance and indicate disease progression in chronic myeloid leukemia. Blood 2011, 117, 3409–3420. [Google Scholar] [CrossRef]
- Ribeiro, D.; Melão, A.; Van Boxtel, R.; Santos, C.I.; Silva, A.; Silva, M.C.; Cardoso, B.A.; Coffer, P.J.; Barata, J.T. STAT5 is essential for IL-7–mediated viability, growth, and proliferation of T-cell acute lymphoblastic leukemia cells. Blood Adv. 2018, 2, 2199–2213. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Bunting, K.D. STAT5 activation in B-cell acute lymphoblastic leukemia: Damned if you do, damned if you don’t. Cancer Cell Microenviron. 2016, 3, 1186. [Google Scholar]
- Chiorazzi, N. Cell proliferation and death: Forgotten features of chronic lymphocytic leukemia B cells. Best Pr. Res. Clin. Haematol. 2007, 20, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Rajala, H.L.M.; Mustjoki, S. STAT5b in LGL leukemia—A novel therapeutic target? Oncotarget 2013, 4, 808–809. [Google Scholar] [CrossRef][Green Version]
- Mitchell, T.J.; Whittaker, S.J.; John, S. Dysregulated expression of COOH-terminally truncated Stat5 and loss of IL2-inducible Stat5-dependent gene expression in Sezary Syndrome. Cancer Res. 2003, 63, 9048–9054. [Google Scholar]
- Zhang, Q.; Wang, H.Y.; Liu, X.; A Wasik, M. STAT5A is epigenetically silenced by the tyrosine kinase NPM1-ALK and acts as a tumor suppressor by reciprocally inhibiting NPM1-ALK expression. Nat. Med. 2007, 13, 1341–1348. [Google Scholar] [CrossRef]
- Kollmann, S.; Grundschober, E.; Maurer, B.; Warsch, W.; Grausenburger, R.; Edlinger, L.; Huuhtanen, J.; Lagger, S.; Hennighausen, L.; Valent, P.; et al. Twins with different personalities: STAT5B-but not STAT5A-has a key role in BCR/ABL-induced leukemia. Leuk. 2019, 33, 1583–1597. [Google Scholar] [CrossRef]
- He, J.; Shi, J.; Xu, X.; Zhang, W.; Wang, Y.; Chen, X.; Du, Y.; Zhu, N.; Zhang, J.; Wang, Q.; et al. STAT3 mutations correlated with hyper-IgE syndrome lead to blockage of IL-6/STAT3 signalling pathway. J. Biosci. 2012, 37, 243–257. [Google Scholar] [CrossRef]
- Pham, H.T.T.; Maurer, B.; Prchal-Murphy, M.; Grausenburger, R.; Grundschober, E.; Javaheri, T.; Nivarthi, H.; Boersma, A.; Kolbe, T.; Elabd, M.; et al. STAT5B N642H is a driver mutation for T cell neoplasia. J. Clin. Investig. 2018, 128, 387–401. [Google Scholar] [CrossRef]
- De Araujo, E.D.; Manaswiyoungkul, P.; Israelian, J.; Park, J.; Yuen, K.; Farhangi, S.; Berger-Becvar, A.; Abu-Jazar, L.; Gunning, P.T. High-throughput thermofluor-based assays for inhibitor screening of STAT SH2 domains. J. Pharm. Biomed. Anal. 2017, 143, 159–167. [Google Scholar] [CrossRef]
- Kofoed, E.M.; Hwa, V.; Woods, K.A.; Buckway, C.K.; Tsubaki, J.; Pratt, K.L.; Bezrodnik, L.; Tepper, A.; Little, B.; Jasper, H.; et al. Growth Hormone Insensitivity Associated with aSTAT5bMutation. New Engl. J. Med. 2003, 349, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Bocchini, C.E.; Nahmod, K.; Katsonis, P.; Kim, S.; Kasembeli, M.M.; Freeman, A.; Lichtarge, O.; Makedonas, G.; Tweardy, D.J. Protein stabilization improves STAT3 function in autosomal dominant hyper-IgE syndrome. Blood 2016, 128, 3061–3072. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, L.; Genini, D.; Laurini, E.; Merulla, J.; Perez, L.; Fermeglia, M.; Carbone, G.M.; Pricl, S.; Catapano, C.V. Hitting the right spot: Mechanism of action of OPB-31121, a novel and potent inhibitor of the Signal Transducer and Activator of Transcription 3 (STAT3). Mol. Oncol. 2015, 9, 1194–1206. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, P.; Zimmerman, O.; Paulson, M.; Sampaio, E.P.; Freeman, A.F.; Sowerwine, K.J.; Hurt, D.; Alcántara-Montiel, J.C.; Hsu, A.P.; Holland, S.M. Distinct mutations at the same positions of STAT3 cause either loss or gain of function. J. Allergy Clin. Immunol. 2016, 138, 1222–1224. [Google Scholar] [CrossRef] [PubMed]
- Avvisati, G.; Tallman, M.S. All-trans retinoic acid in acute promyelocytic leukaemia. Best Pr. Res. Clin. Haematol. 2003, 16, 419–432. [Google Scholar] [CrossRef]
- Yao, L.; Wen, L.; Wang, N.; Liu, T.; Xu, Y.; Ruan, C.; Wu, D.; Chen, S. Identification of novel recurrent STAT3-RARA fusions in acute promyelocytic leukemia lacking t(15;17)(q22;q12)/PML-RARA. Blood 2018, 131, 935–939. [Google Scholar] [CrossRef] [PubMed]


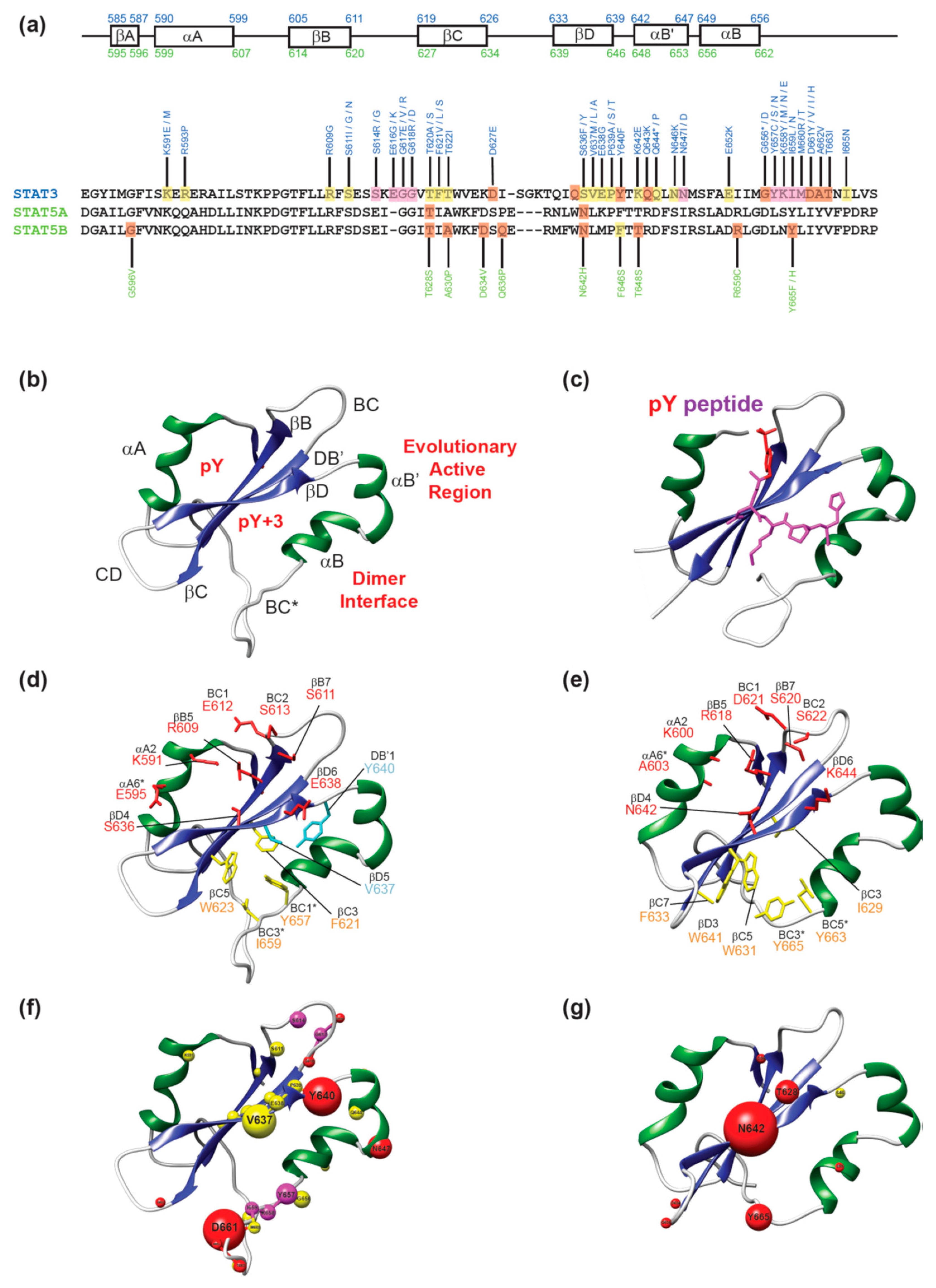{kind=link}
| Mutation | Position | Location | Residue Relevance | Nucleotide Substitution | Cases | Pathology | Type | Ref |
|---|---|---|---|---|---|---|---|---|
| K591E | αA2 | pY | Sheinerman | 1771A>G | 1 | AD-HIES | Germline | [30] |
| K591M | αA2 | pY | Sheinerman | 1772A>T | 1 | AD-HIES | Germline | [31] |
| R593P | αA4 | pY | - | 1778G>C | 1 | AD-HIES | Germline | [32] |
| R609G | βB5 | pY | Sheinerman& Signature | 1825A>G | 1 | AD-HIES | Germline | [33] |
| S611G | βB7 | pY | Sheinerman | 1831A>G | 2 | AD-HIES | Germline | [34,35] |
| S611N | βB7 | pY | & Signature Sheinerman | 1832G>A | 2 | AD-HIES | Germline | [36,37] |
| S611I | βB7 | pY | Sheinerman & Signature | 1832G>T | 1 | AD-HIES | Germline | [38] |
| S614G | BC3 | pY | Sheinerman | 18040A>G | 1 | AD-HIES | Germline | [34] |
| S614R | BC3 | pY | Sheinerman | 1842C>G | 1 | T-LGLL | Somatic | [39,40,41,42,43] |
| 2 | NK-LGLL | |||||||
| 1 | ALK-ALCL | |||||||
| 1 | HSTL | |||||||
| E616G | BC5 | pY | BC loop | 1847A>G | 1 | DLBCL, NOS | Somatic | [44] |
| E616K | BC5 | pY | BC loop | 1846G>A | 1 | NKTL | Somatic | [45] |
| G617E | BC6 | pY | BC loop | 1850G>A | 1 | AD-HIES | Germline | [46] |
| G617V | BC6 | pY | BC loop | 1850G>T | 1 | AD-HIES | Germline | [34] |
| G617R | BC6 | pY | BC loop | 1849G>A | 1 | DLBCL, NOS | Somatic | [44] |
| G618R | BC6 | pY | BC loop | 1852G>C | 1 | ALK-ALCL | Somatic | [47,48] |
| 1 | T-LGLL | |||||||
| G618D | BC6 | pY | BC loop | 1853G>A | 2 | AD-HIES | Germline | [35,37] |
| T620A | βC2 | pY | - | 1858A>G | 2 | AD-HIES | Germline | [34,49] |
| T620S | βC2 | pY | - | 1859C>G | 1 | AD-HIES | Germline | [33] |
| F621V | βC3 | pY+3 | Hydro. Sys. | 1861T>G | 3 | AD-HIES | Germline | [36,37,50] |
| F621L | βC3 | pY+3 | Hydro. Sys. | 1863C>G | 2 | AD-HIES | Germline | [51,52] |
| F621S | βC3 | pY+3 | Hydro. Sys. | 1862T>C | 1 | AD-HIES | Germline | [53] |
| T622I | βC4 | pY | - | 1865C>T | 4 | AD-HIES | Germline | [30,34,36] |
| D627E | CD1 | - | 1881C>A | 1 | ATLL | Somatic | [54] | |
| S636F | βD4 | pY | Sheinerman | 1907C>T, | 1 | AD-HIES | Germline | [49] |
| S636Y | βD4 | pY | Sheinerman | 1907C>A | 1 | AD-HIES | Germline | [30] |
| V637M | βD5 | pY+3 | Sel. Filter | 1909G>A | 43 | AD-HIES | Germline | [30,32,33,34,35,36,37,49,52,55,56] |
| V637L | βD5 | pY+3 | Sel. Filter | 1909G>T | 1 | AD-HIES | Germline | [36] |
| V637A | βD5 | pY+3 | Sel. Filter | 1910T>C | 1 | AD-HIES | Germline | [30] |
| E638G | βD6 | pY | Sheinerman | 1913A>G | 4 | AD-HIES | Germline | [49,51,57,58] |
| P639A | βD7 | pY+3 | - | 1915C>G | 1 | AD-HIES | Germline | [37] |
| P639S | βD7 | pY+3 | - | 1915C>T* | 2 | AD-HIES | Germline | [30,34] |
| P639T | βD7 | pY+3 | - | 1915C>A | 1 | AD-HIES | Germline | [35] |
| Y640F | DB’1 | pY+3 | - | 1919A>T | 56 | T-LGLL | Somatic | [43,44,47,48,57,59,60,61,62,63,64,65,66,67,68,69] |
| 2 | IHT | |||||||
| 3 | CLPD-NKs | |||||||
| 2 | NK-LGLL | |||||||
| 1 | DLBCL, NOS | |||||||
| 1 | ANKL | |||||||
| 1 | NKTL | |||||||
| 1 | Sezary | |||||||
| 3 | HSTL | |||||||
| K642E | αB’1 | - | Dimer Inter | 1924A>G | 1 | AD-HIES | Germline | [34] |
| Q643K | αB’2 | - | Dimer Inter | 1927C>A | 1 | T-LGLL | Somatic | [68] |
| Q644P | αB’3 | - | Dimer Inter | 1929A>C | 3 | AD-HIES | Germline | [52,70] |
| Q644del | αB’3 | - | Dimer Inter | 1930del CAG | 2 | AD-HIES | Germline | [35,36] |
| N646K | αB’5 | pY+3 | Dimer Inter | 1938C>G | 2 | EOAD | Germline | [71] |
| N647D | αB’6 | pY+3 | Dimer Inter | 1939A>G | 8 | AD-HIES | Germline | [36,72] |
| N647I | αB’6 | pY+3 | Dimer Inter | 1940A>T | 3 | CLPD-NK | Somatic | [43,57,65,67] |
| 6 | T-LGLL | |||||||
| 1 | HSTL | |||||||
| E652K | αB3 | pY+3 | Dimer Inter | 1954G>A | 1 | AD-HIES | Germline | [36] |
| G656D | αB7 | pY+3 | Dimer Inter | 1967G>A | 1 | T-LGLL | Somatic | [73] |
| G656_Y657insF | αB7 | pY+3 | Hydro. Sys. | 1968C>T; 1969_1970insTTT | 1 | IHCA | Somatic | [66] |
| Y657C | BC1* | pY+3 | Hydro. Sys. | 1970A>G | 5 | AD-HIES | Germline | [30,34,36,58] |
| Y657S | BC1* | pY+3 | Hydro. Sys. | 1970A>C | 1 | AD-HIES | Germline | [74] |
| Y657N | BC1* | pY+3 | Hydro. Sys. | 1969T>A | 1 | AD-HIES | Germline | [52] |
| Y657ins | BC1* | pY+3 | Hydro. Sys. | - | 1 | TCL | Somatic | [47] |
| Y657dup | BC1* | pY+3 | Hydro. Sys. | - | 3 | T-LGLL | Somatic | [48,65,72] |
| Y657_M660dup | BC1* | pY+3 | Hydro. Sys. | 1969T_1980G dup | 1 | IHCA | Somatic | [66] |
| K658M | BC2* | pY+3 | Dimer Inter | 1973A>T | 2 | T-LGLL | Somatic | [67,72] |
| K658N | BC2* | pY+3 | Dimer Inter | 1974G>T | 1 | T-LGLL EOAD | Somatic/Germline | [65,71] |
| K658Y | BC2* | pY+3 | Dimer Inter | 1972A>T; 1974G>T | 1 | IHCA | Somatic | [66] |
| K658E | BC2* | pY+3 | Dimer Inter | 1972A>G | 1 | AD-HIES | Germline | [75] |
| I659N | BC3* | pY+3 | Hydro. Sys. | 1976A>T | 1 | AD-HIES | Germline | [58] |
| I659L | BC3* | pY+3 | Hydro. Sys. | 1975A>C | 2 | T-LGLL | Somatic | [57,72] |
| M660R | BC4* | pY+3 | Dimer Inter | 1979T>G | 1 | AD-HIES | Germline | [55] |
| M660T | BC4* | pY+3 | Dimer Inter | 1978T>A | 1 | AD-HIES | Germline | [76] |
| D661I | BC5* | - | Dimer Inter | 1981G>A; 1982A>T | 1 | CLPD-NK | Somatic | [67] |
| D661Y | BC5* | - | Dimer Inter | 1981G>T | 1 | NK-LGL | Somatic | [41,47,48,60,63,65,67,68,72,77] |
| 56 | T-LGL | |||||||
| 1 | HSTL | |||||||
| 10 | NKTL | |||||||
| D661ins | BC5* | - | Dimer Inter | - | 1 | T-LGLL | Somatic | [47] |
| D661V | BC5* | - | Dimer Inter | 1981A>T | 10 | T-LGLL | Somatic | [65,67] |
| D661H | BC5* | - | Dimer Inter | 1981G>C | 1 | T-LGLL | Somatic | [65] |
| A662V | BC6* | - | Dimer Inter | 1985C>T | 1 | ALK-ALCL | Somatic | [78] |
| T663I | BC7* | - | Dimer Inter | 1988, 1989>TT | 2 | DLBCL/B1 | Germline | [44,75] |
| I665N | BC9* | - | Dimer Inter | 1998T>A | 2 | AD-HIES | Germline | [34] |
| V667L | BC11* | - | Dimer Inter | 1999C>G | 1 | NKTL | Somatic | [45] |
| S668F | BC12* | - | Dimer Inter | 2003C>T | 3 | AD-HIES | Germline | [30,34,37] |
| S668Y | BC12* | - | Dimer Inter | 2003C>A | 1 | AD-HIES | Germline | [34] |
| Mutation | Position | Location | Residue Relevance | Nucleotide Substitution | Cases | Pathology | Type | Ref. |
|---|---|---|---|---|---|---|---|---|
| G596V | βA2 | - | - | 1787G>T | 1 | APL | Somatic | [79] |
| T628S | βC2 | pY | - | 1883C>G | 11 | T-PLL | Somatic | [43,80,81] |
| 1 | MEITL | |||||||
| 3 | HSTL | |||||||
| 1 | Eosinophilia | |||||||
| A630P | βC4 | pY | - | 1888G>C | 1 | GHI | Germline * | [82] |
| D634V | βC8 | pY | - | 1901A>T | 1 | T-PLL | Somatic | [83] |
| Q636P | CD2 | pY+3 | - | 1907A>C | 1 | MEITL | Somatic | [81] |
| N642H | βD4 | pY | Sheinerman | 1924A>C | 39 | MEITL | Somatic | [41,62,69,77,80,81,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102] |
| 33 | T-PLL | |||||||
| 29 | Eosinophilia | |||||||
| 28 | T-ALL | |||||||
| 7 | HSTL | |||||||
| 11 | LGLL | |||||||
| 3 | PCTL | |||||||
| 3 | Sézary | |||||||
| 1 | AAA | |||||||
| 1 | CNL | |||||||
| 1 | PTCL, NOS | |||||||
| 1 | AML | |||||||
| F646S | DB’1 | pY+3 | - | 1937T>C | 1 | GHI | Germline * | [103] |
| T648S | DB’3 | pY+3 | - | 1942A>T | 1 | T-ALL | Somatic | [101] |
| R659C | αB’4 | Dimer Inter | - | 1975C>T | 1 | T-PLL | Somatic | [80] |
| Y665F | BC3* | Dimer Inter | Hydro. Sys. | 1994A>T | 6 | T-PLL | Somatic | [43,80,101] |
| 3 | HSTL | |||||||
| 2 | T-ALL | |||||||
| 2 | NKTL | |||||||
| 5 | LGLL | |||||||
| Y665H | BC3* | Dimer Inter | Hydro. Sys. | 1993T>C | 2 | T-PLL | Somatic | [80] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Araujo, E.D.; Orlova, A.; Neubauer, H.A.; Bajusz, D.; Seo, H.-S.; Dhe-Paganon, S.; Keserű, G.M.; Moriggl, R.; Gunning, P.T. Structural Implications of STAT3 and STAT5 SH2 Domain Mutations. Cancers 2019, 11, 1757. https://doi.org/10.3390/cancers11111757
de Araujo ED, Orlova A, Neubauer HA, Bajusz D, Seo H-S, Dhe-Paganon S, Keserű GM, Moriggl R, Gunning PT. Structural Implications of STAT3 and STAT5 SH2 Domain Mutations. Cancers. 2019; 11(11):1757. https://doi.org/10.3390/cancers11111757
Chicago/Turabian Stylede Araujo, Elvin D., Anna Orlova, Heidi A. Neubauer, Dávid Bajusz, Hyuk-Soo Seo, Sirano Dhe-Paganon, György M. Keserű, Richard Moriggl, and Patrick T. Gunning. 2019. "Structural Implications of STAT3 and STAT5 SH2 Domain Mutations" Cancers 11, no. 11: 1757. https://doi.org/10.3390/cancers11111757
APA Stylede Araujo, E. D., Orlova, A., Neubauer, H. A., Bajusz, D., Seo, H.-S., Dhe-Paganon, S., Keserű, G. M., Moriggl, R., & Gunning, P. T. (2019). Structural Implications of STAT3 and STAT5 SH2 Domain Mutations. Cancers, 11(11), 1757. https://doi.org/10.3390/cancers11111757