Treatment with a Combination of Metformin and 2-Deoxyglucose Upregulates Thrombospondin-1 in Microvascular Endothelial Cells: Implications in Anti-Angiogenic Cancer Therapy

 ,
,  ,
,
Abstract
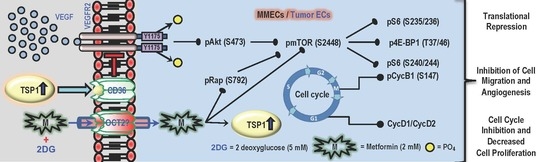
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
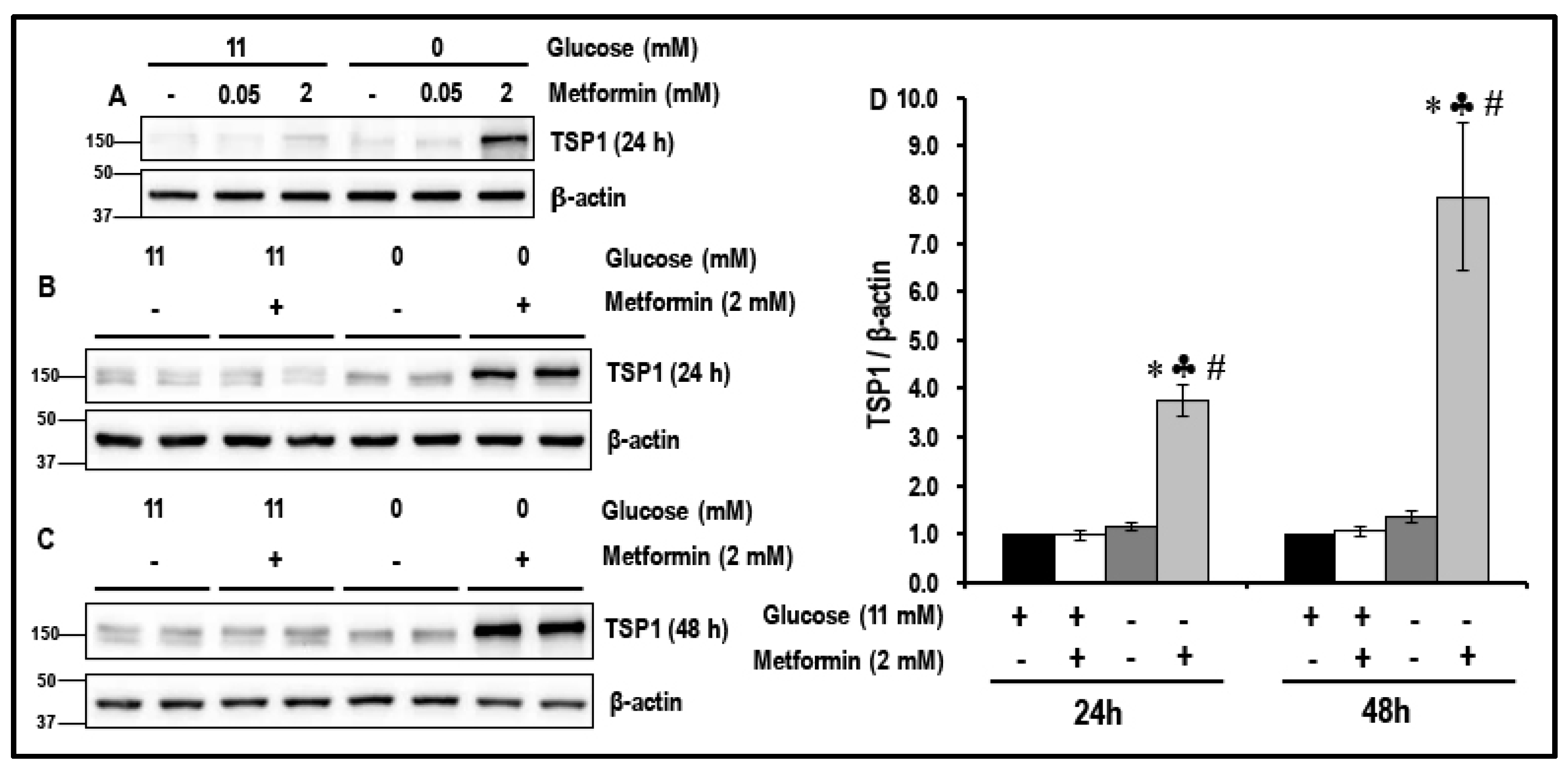
2.1. Metformin (2 mM) Increases the Levels of TSP1 in Glucose-Starved MMECs
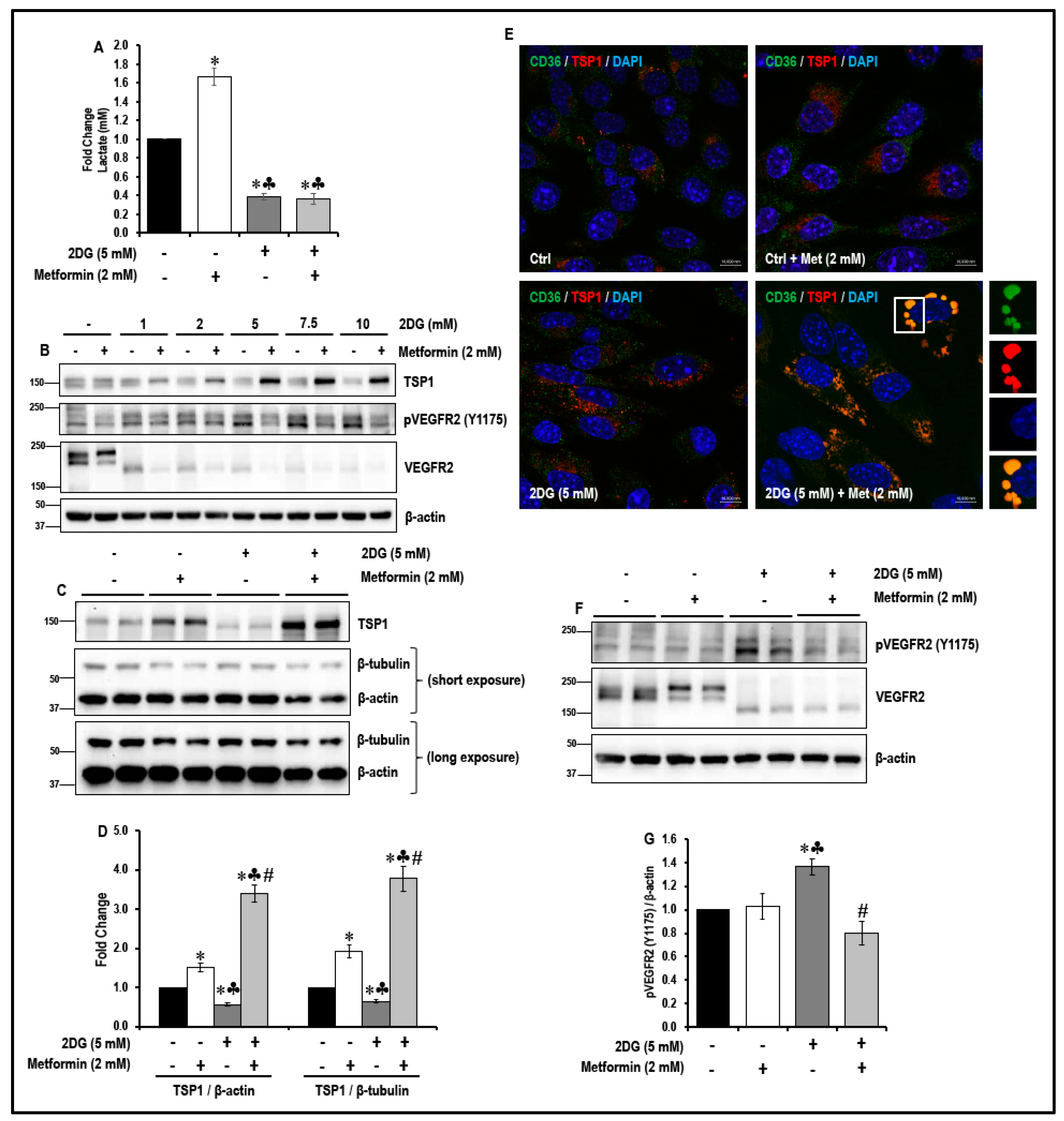
2.2. Treatment with a Combination of 2DG and Metformin Up-Regulates Expression of Anti-Angiogenic TSP-1 in MMECs
2.3. Treatment with a Combination of 2DG and Metformin Increases TSP1-Platelet Integral Membrane Glycoprotein IV/Scavenger Receptor Class B Member-3 (CD36) Co-Localization and Decreases VEGFR2 Phosphorylation in MMECs
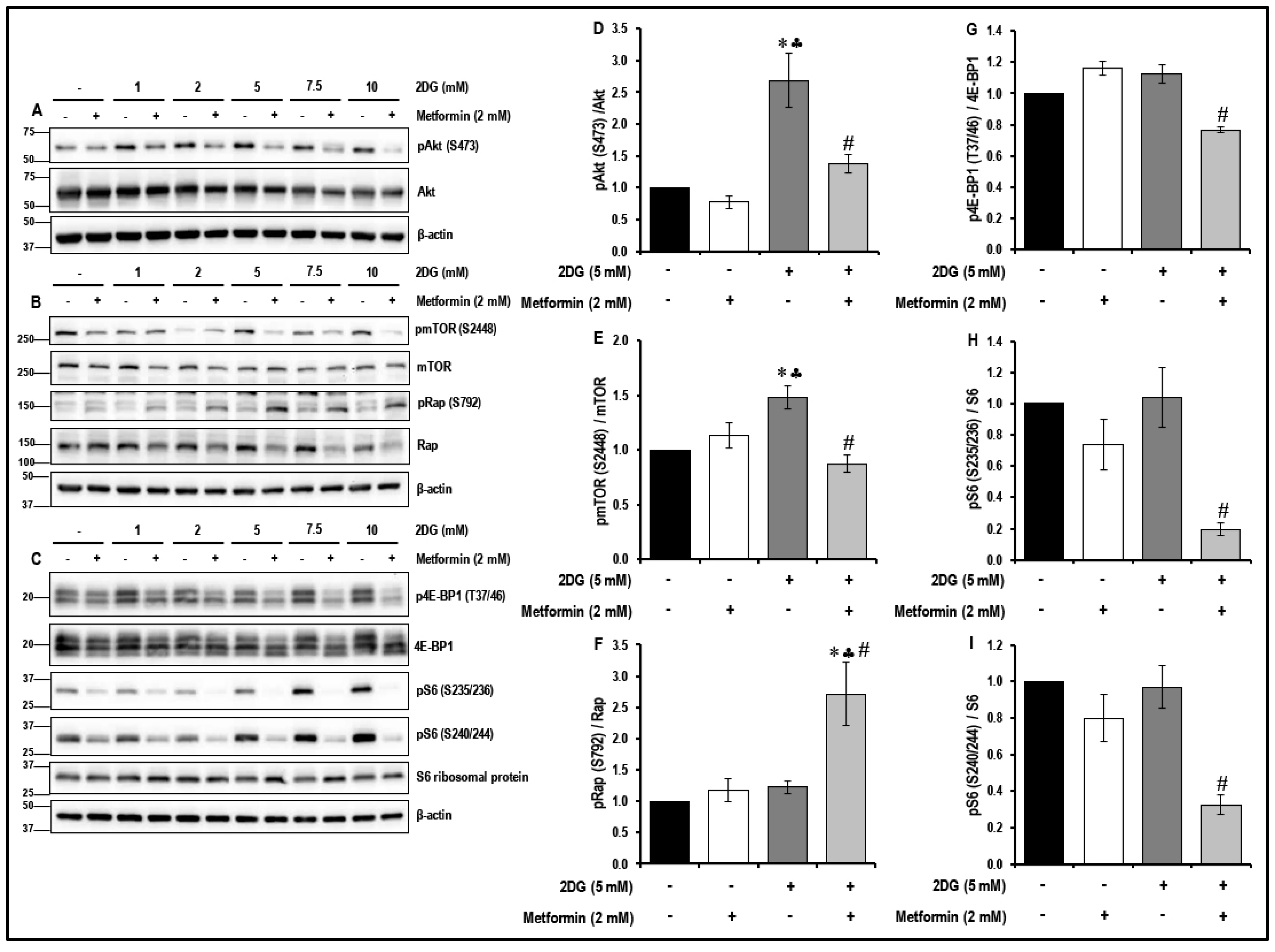
2.4. Treatment with a Combination of 2DG and Metformin Inhibits the Akt/mTOR Pathway in MMECs
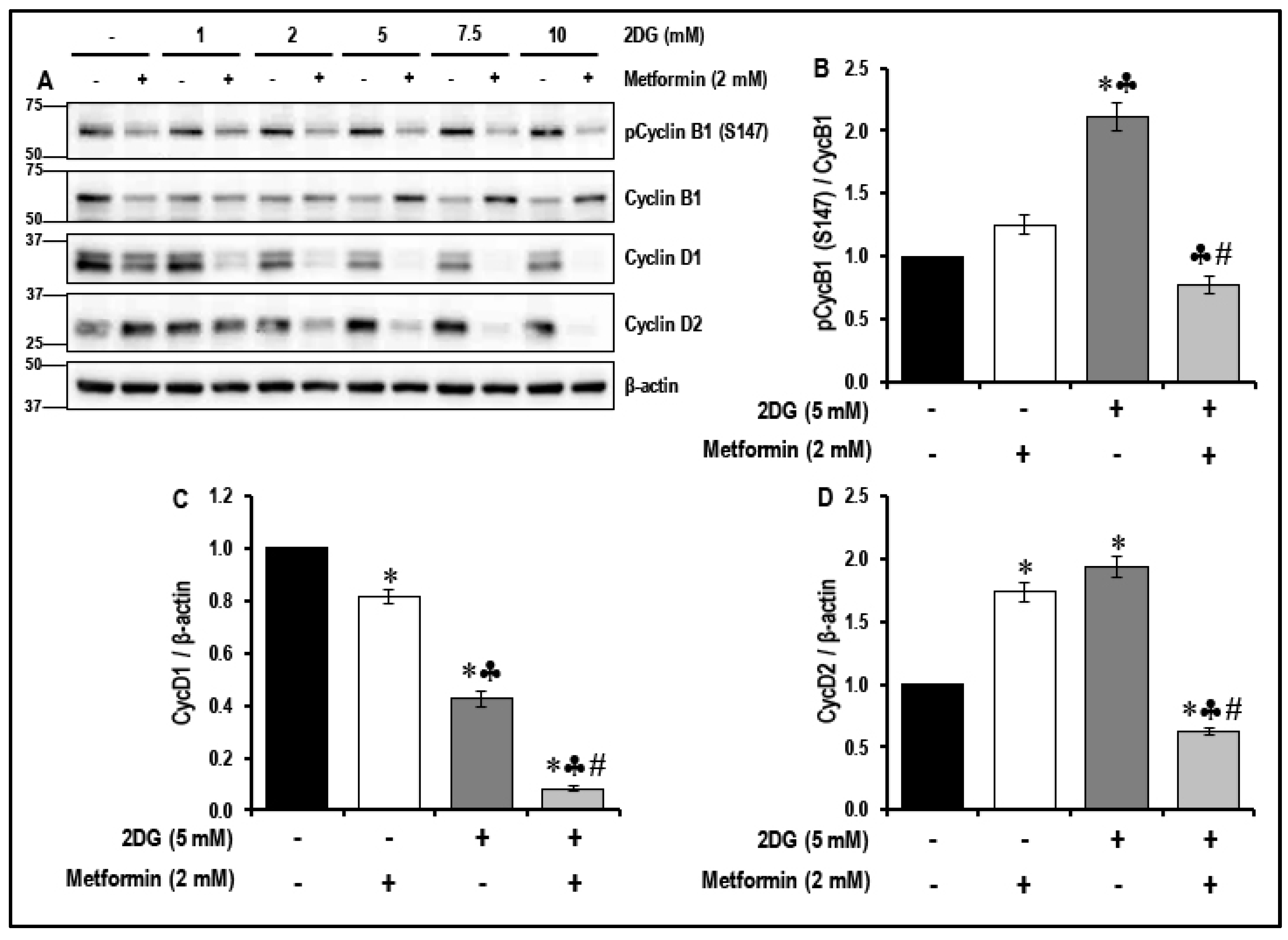
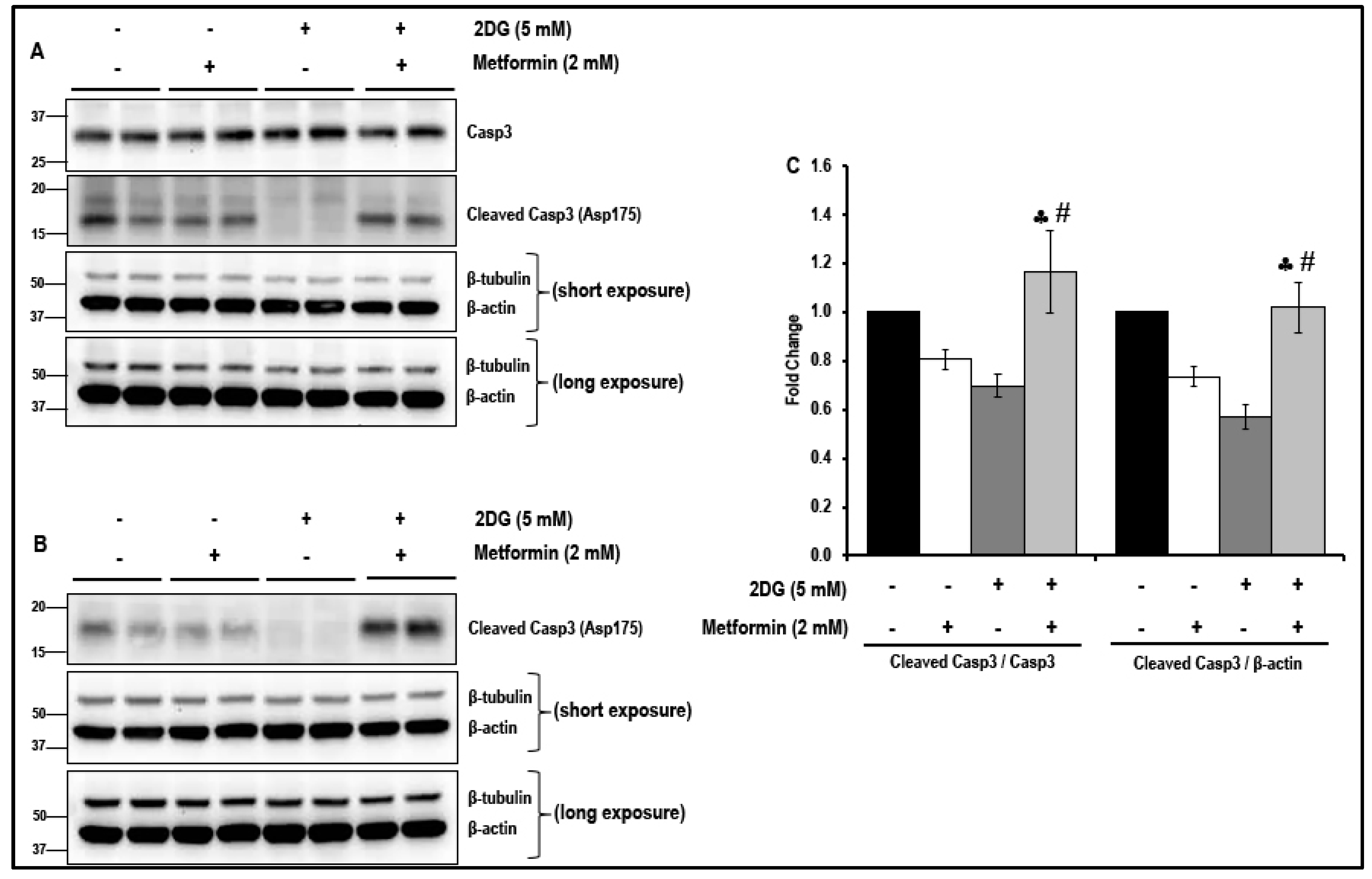
2.5. Treatment with a Combination of 2DG and Metformin Inhibits the Cell-Cycle-Related Proteins in MMECs
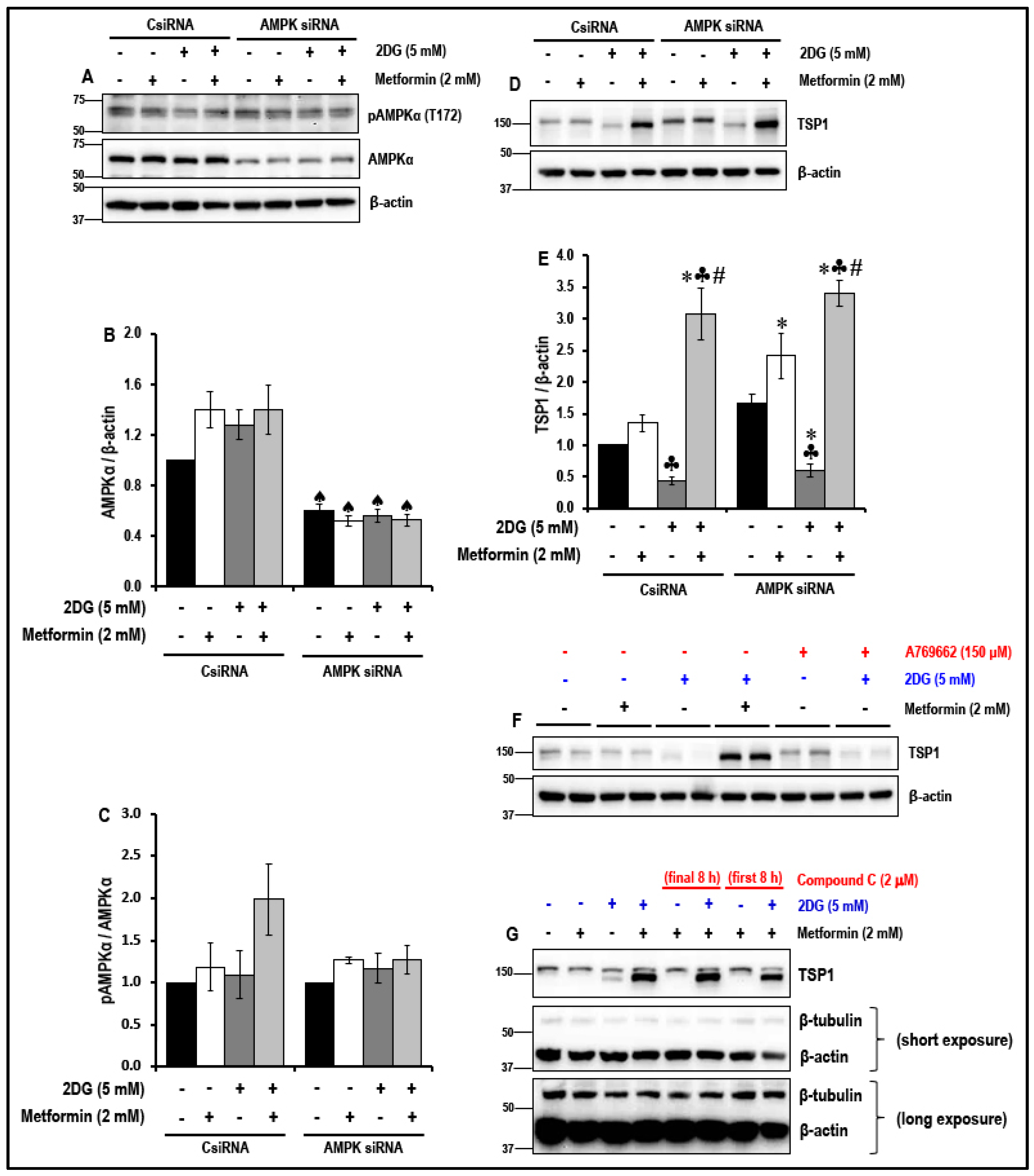
2.6. The Effect of the Treatment with a Combination of 2DG and Metformin on the Levels of Anti-Angiogenic TSP1 in MMECs is Independent of 5′ Adenosine Monophosphate-Activated Protein Kinase (AMPK) Activation
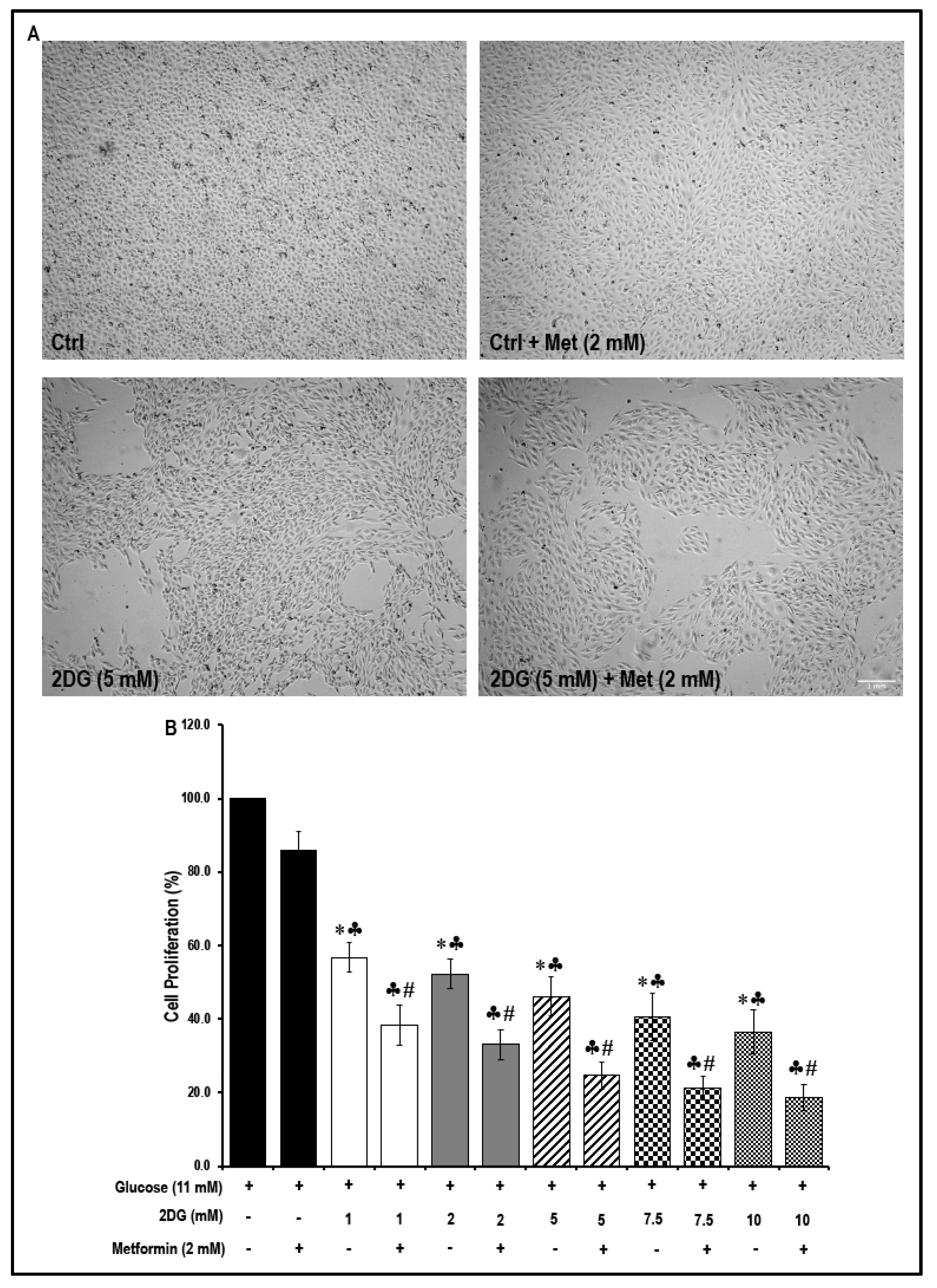
2.7. Treatment with a Combination of 2DG and Metformin Decreases Cell Proliferation
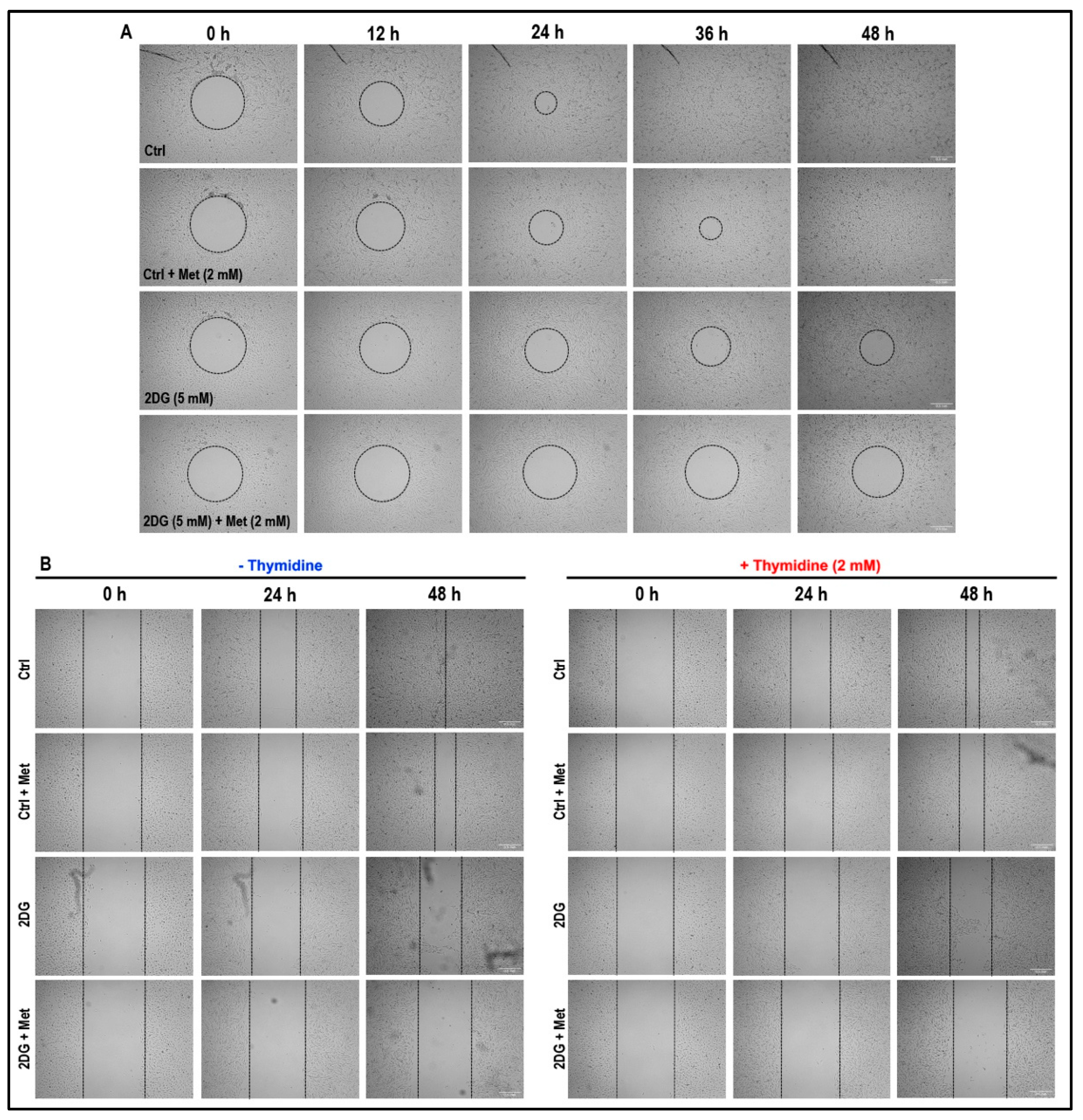
2.8. Treatment with a Combination of 2DG and Metformin Inhibits Cell Migration and Tubulogenesis in MMECs
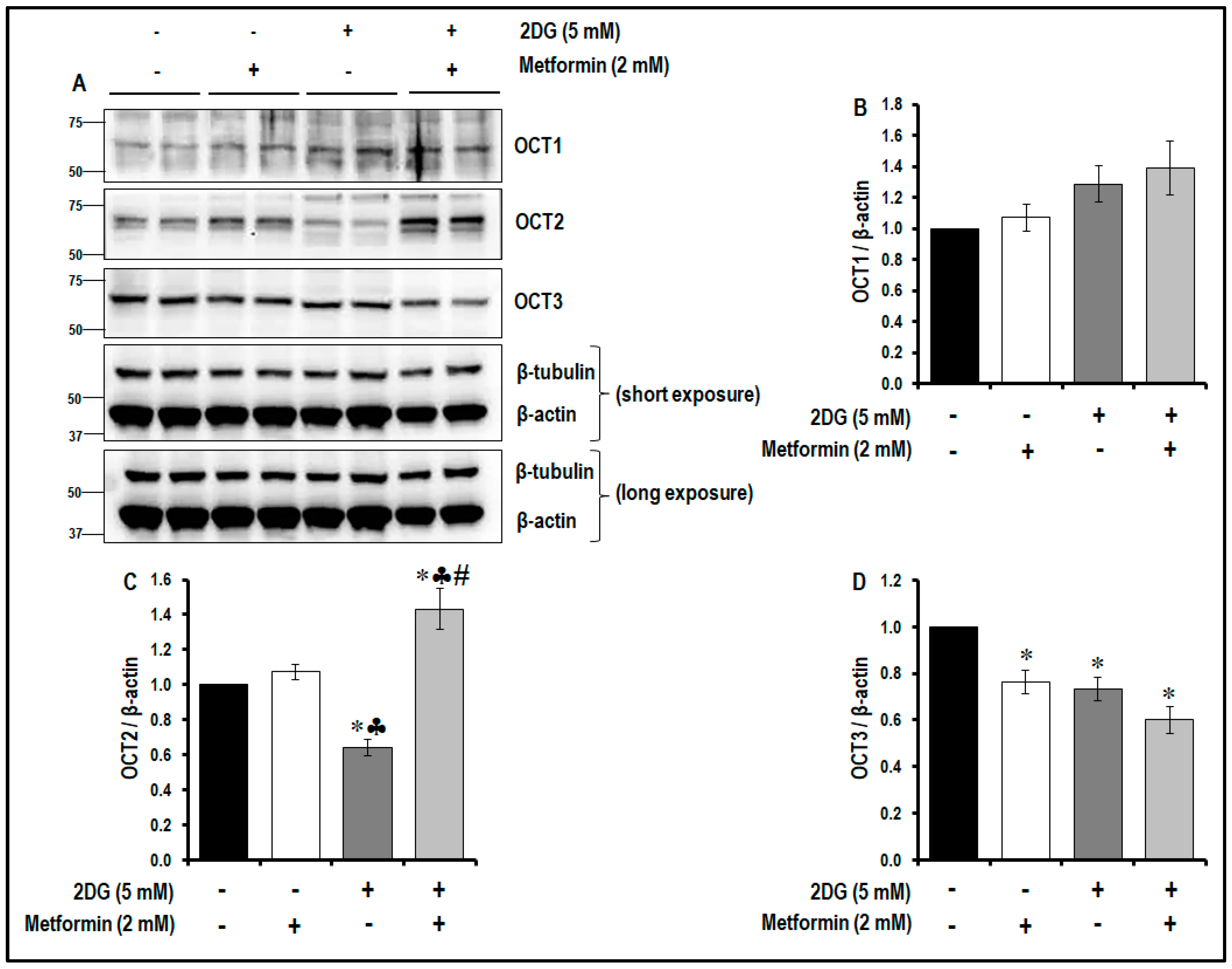
2.9. Treatment with a Combination of 2DG and Metformin Differentially Modulates the Levels of OCTs
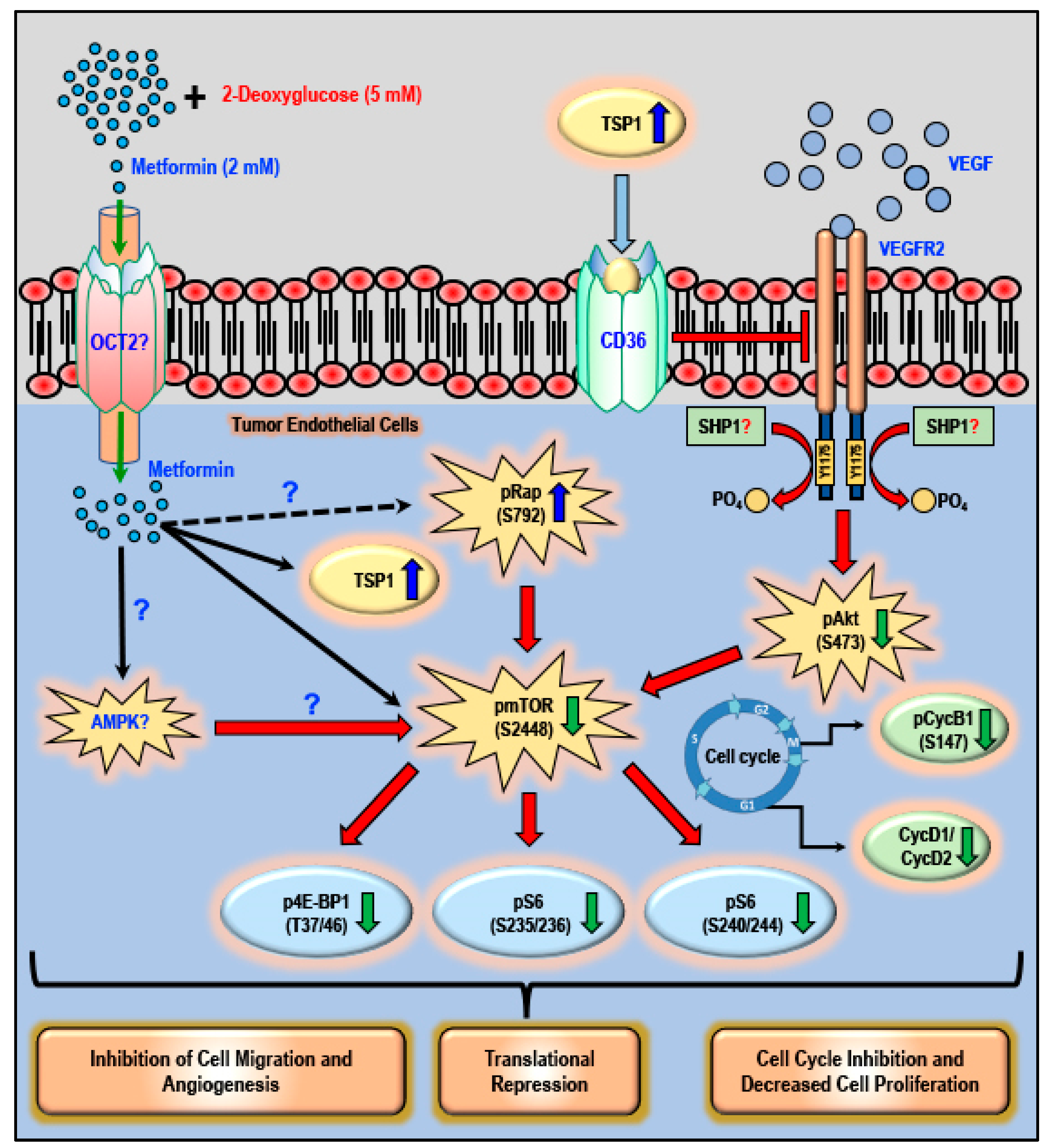
3. Discussion
4. Materials and Methods
4.1. Antibodies, Biochemicals, Chemicals and Reagents
4.2. Endothelial Cell Culture
4.3. Cell Treatments
4.4. Cell Proliferation Assay (MTS Assay)
4.5. Glycolysis Assay
4.6. Cell Migration Assay
4.7. Tubulogenesis Assay
4.8. AMPK Gene Silencing
4.9. Protein Isolation and Total Protein Estimation
4.10. Immunoblotting
4.11. Immunofluorescence Staining for CD36-TSP1 Co-Localization Studies
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| μΜ | Micromolar |
| μm | Micrometre |
| 2DG | 2-deoxyglucose |
| 4E-BP1 | Eukaryotic translation initiation factor 4E-binding protein 1 |
| Akt | Protein kinase B |
| AMPK | AMP activated protein kinase |
| CD36 | Platelet glycoprotein IV/scavenger receptor class B member 3 |
| CycB1 | Cyclin B1 |
| CycD1 | Cyclin D1 |
| CycD2 | Cyclin D2 |
| ECs | Endothelial cells |
| h | Hours |
| HER2 | Human epidermal growth factor receptor 2 |
| HIF1α | Hypoxia inducible factor-1 alpha |
| HUVECs | Human umbilical vein endothelial cells |
| MATE1/2 | Multidrug and toxin extrusion protein 1 and 2 |
| min | Minutes |
| mM | Millimolar |
| MMECs | Mouse microvascular endothelial cells |
| mTOR | Mammalian target of rapamycin |
| nm | Nanometre |
| PMAT | Plasma membrane monoamine trasporter |
| Rap | Raptor |
| SHP1 | Src-homology 2-containing tyrosine phosphatase-1 |
| TECs | Tumor endothelial cells |
| TSP1 | Thrombospondin 1 |
| VEGF | Vascular endothelial growth factor |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
References
- Rojas, L.B.; Gomes, M.B. Metformin: An old but still the best treatment for type 2 diabetes. Diabetol. Metab. Syndr. 2013, 5, e6. [Google Scholar] [CrossRef] [PubMed]
- Coyle, C.; Cafferty, F.H.; Vale, C.; Langley, R.E. Metformin as an adjuvant treatment for cancer: A systematic review and meta-analysis. Ann. Oncol. 2016, 27, 2184–2195. [Google Scholar] [CrossRef] [PubMed]
- Emami Riedmaier, A.; Fisel, P.; Nies, A.T.; Schaeffeler, E.; Schwab, M. Metformin and cancer: From the old medicine cabinet to pharmacological pitfalls and prospects. Trends Pharmacol. Sci. 2013, 34, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef]
- Triggle, C.R.; Ding, H. Metformin is not just an antihyperglycaemic drug but also has protective effects on the vascular endothelium. Acta Physiol. (Oxf.) 2017, 219, 138–151. [Google Scholar] [CrossRef]
- Franciosi, M.; Lucisano, G.; Lapice, E.; Strippoli, G.F.; Pellegrini, F.; Nicolucci, A. Metformin therapy and risk of cancer in patients with type 2 diabetes: Systematic review. PLoS ONE 2013, 8, e71583. [Google Scholar] [CrossRef]
- Li, D. Diabetes and pancreatic cancer. Mol. Carcinog. 2012, 51, 64–74. [Google Scholar] [CrossRef]
- Christensen, M.M.; Hojlund, K.; Hother-Nielsen, O.; Stage, T.B.; Damkier, P.; Beck-Nielsen, H.; Brosen, K. Steady-state pharmacokinetics of metformin is independent of the OCT1 genotype in healthy volunteers. Eur. J. Clin. Pharmacol. 2015, 71, 691–697. [Google Scholar] [CrossRef]
- Samuel, S.M.; Varghese, E.; Varghese, S.; Busselberg, D. Challenges and perspectives in the treatment of diabetes associated breast cancer. Cancer Treat. Rev. 2018, 70, 98–111. [Google Scholar] [CrossRef]
- Kinaan, M.; Ding, H.; Triggle, C.R. Metformin: An Old Drug for the Treatment of Diabetes but a New Drug for the Protection of the Endothelium. Med. Princ. Pract. 2015, 24, 401–415. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, J.; Xi, M.; Jia, Y.; Wong, Y.; Zhao, J.; Ding, L.; Zhang, J.; Wen, A. Pharmacogenetic variation and metformin response. Curr. Drug Metab. 2013, 14, 1070–1082. [Google Scholar] [CrossRef] [PubMed]
- Checkley, L.A.; Rudolph, M.C.; Wellberg, E.A.; Giles, E.D.; Wahdan-Alaswad, R.S.; Houck, J.A.; Edgerton, S.M.; Thor, A.D.; Schedin, P.; Anderson, S.M.; et al. Metformin Accumulation Correlates with Organic Cation Transporter 2 Protein Expression and Predicts Mammary Tumor Regression in Vivo. Cancer Prev. Res. (Phila.) 2017, 10, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Zhang, Y.; Han, T.K.; Everett, R.S.; Thakker, D.R. Cation-selective transporters are critical to the AMPK-mediated antiproliferative effects of metformin in human breast cancer cells. Int. J. Cancer 2016, 138, 2281–2292. [Google Scholar] [CrossRef] [PubMed]
- Martin-Castillo, B.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J.A. Metformin and cancer: Doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle 2010, 9, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Wahdan-Alaswad, R.; Fan, Z.; Edgerton, S.M.; Liu, B.; Deng, X.S.; Arnadottir, S.S.; Richer, J.K.; Anderson, S.M.; Thor, A.D. Glucose promotes breast cancer aggression and reduces metformin efficacy. Cell Cycle 2013, 12, 3759–3769. [Google Scholar] [CrossRef]
- Samuel, S.M.; Ghosh, S.; Majeed, Y.; Arunachalam, G.; Emara, M.M.; Ding, H.; Triggle, C.R. Metformin represses glucose starvation induced autophagic response in microvascular endothelial cells and promotes cell death. Biochem. Pharmacol. 2017, 132, 118–132. [Google Scholar] [CrossRef]
- Menendez, J.A.; Oliveras-Ferraros, C.; Cufi, S.; Corominas-Faja, B.; Joven, J.; Martin-Castillo, B.; Vazquez-Martin, A. Metformin is synthetically lethal with glucose withdrawal in cancer cells. Cell Cycle 2012, 11, 2782–2792. [Google Scholar] [CrossRef]
- Dalby, K.N.; Tekedereli, I.; Lopez-Berestein, G.; Ozpolat, B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy 2010, 6, 322–329. [Google Scholar] [CrossRef]
- Das, C.K.; Mandal, M.; Kögel, D. Pro-survival autophagy and cancer cell resistance to therapy. Cancer Metastasis Rev. 2018, 37, 749–766. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Laurent, K.; Giuliano, S.; Larbret, F.; Ponzio, G.; Gounon, P.; Le Marchand-Brustel, Y.; Giorgetti-Peraldi, S.; Cormont, M.; Bertolotto, C.; et al. Targeting cancer cell metabolism: The combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010, 70, 2465–2475. [Google Scholar] [CrossRef]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Goveia, J.; Stapor, P.; Carmeliet, P. Principles of targeting endothelial cell metabolism to treat angiogenesis and endothelial cell dysfunction in disease. EMBO Mol. Med. 2014, 6, 1105–1120. [Google Scholar] [CrossRef]
- McDonald, D.M.; Choyke, P.L. Imaging of angiogenesis: From microscope to clinic. Nat. Med. 2003, 9, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef]
- Niu, G.; Chen, X. Vascular endothelial growth factor as an anti-angiogenic target for cancer therapy. Curr. Drug Targets 2010, 11, 1000–1017. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Tan, B.K.; Adya, R.; Chen, J.; Farhatullah, S.; Heutling, D.; Mitchell, D.; Lehnert, H.; Randeva, H.S. Metformin decreases angiogenesis via NF-kappaB and Erk1/2/Erk5 pathways by increasing the antiangiogenic thrombospondin-1. Cardiovasc. Res. 2009, 83, 566–574. [Google Scholar] [CrossRef]
- Arbiser, J.L.; Larsson, H.; Claesson-Welsh, L.; Bai, X.; LaMontagne, K.; Weiss, S.W.; Soker, S.; Flynn, E.; Brown, L.F. Overexpression of VEGF 121 in immortalized endothelial cells causes conversion to slowly growing angiosarcoma and high level expression of the VEGF receptors VEGFR-1 and VEGFR-2 in vivo. Am. J. Pathol. 2000, 156, 1469–1476. [Google Scholar] [CrossRef]
- Hasenstein, J.R.; Kasmerchak, K.; Buehler, D.; Hafez, G.R.; Cleary, K.; Moody, J.S.; Kozak, K.R. Efficacy of Tie2 receptor antagonism in angiosarcoma. Neoplasia 2012, 14, 131–140. [Google Scholar] [CrossRef]
- Taylor, K.L.; Oates, R.K.; Grane, R.; Leaman, D.W.; Borden, E.C.; Lindner, D.J. IFN-alpha1,8 inhibits tumor-induced angiogenesis in murine angiosarcomas. J. Interferon Cytokine Res. 2006, 26, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.Y.; Ramakrishnan, D.P.; Silverstein, R.L. Thrombospondin-1 modulates VEGF signaling via CD36 by recruiting SHP-1 to VEGFR2 complex in microvascular endothelial cells. Blood 2013, 122, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Polet, F.; Feron, O. Endothelial cell metabolism and tumour angiogenesis: Glucose and glutamine as essential fuels and lactate as the driving force. J. Intern. Med. 2013, 273, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Pan, J.; Yu, L.; Liu, H.; Shu, X.; Sun, L.; Lou, J.; Yang, Z.; Ran, Y. Tumor endothelial cells promote metastasis and cancer stem cell-like phenotype through elevated Epiregulin in esophageal cancer. Am. J. Cancer Res. 2016, 6, 2277–2288. [Google Scholar] [PubMed]
- Hida, K.; Maishi, N.; Torii, C.; Hida, Y. Tumor angiogenesis–characteristics of tumor endothelial cells. Int. J. Clin. Oncol. 2016, 21, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Maishi, N.; Ohba, Y.; Akiyama, K.; Ohga, N.; Hamada, J.; Nagao-Kitamoto, H.; Alam, M.T.; Yamamoto, K.; Kawamoto, T.; Inoue, N.; et al. Tumour endothelial cells in high metastatic tumours promote metastasis via epigenetic dysregulation of biglycan. Sci. Rep. 2016, 6, e28039. [Google Scholar] [CrossRef]
- Verdegem, D.; Moens, S.; Stapor, P.; Carmeliet, P. Endothelial cell metabolism: Parallels and divergences with cancer cell metabolism. Cancer Metab. 2014, 2, e19. [Google Scholar] [CrossRef]
- Eelen, G.; de Zeeuw, P.; Simons, M.; Carmeliet, P. Endothelial cell metabolism in normal and diseased vasculature. Circ. Res. 2015, 116, 1231–1244. [Google Scholar] [CrossRef]
- Dudley, A.C. Tumor endothelial cells. Cold Spring Harb Perspect. Med. 2012, 2, a006536. [Google Scholar] [CrossRef]
- Cantelmo, A.R.; Conradi, L.C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef] [PubMed]
- Schoors, S.; De Bock, K.; Cantelmo, A.R.; Georgiadou, M.; Ghesquiere, B.; Cauwenberghs, S.; Kuchnio, A.; Wong, B.W.; Quaegebeur, A.; Goveia, J.; et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014, 19, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Colen, C.B.; Seraji-Bozorgzad, N.; Marples, B.; Galloway, M.P.; Sloan, A.E.; Mathupala, S.P. Metabolic remodeling of malignant gliomas for enhanced sensitization during radiotherapy: An in vitro study. Neurosurgery 2006, 59, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Colen, C.B.; Shen, Y.; Ghoddoussi, F.; Yu, P.; Francis, T.B.; Koch, B.J.; Monterey, M.D.; Galloway, M.P.; Sloan, A.E.; Mathupala, S.P. Metabolic targeting of lactate efflux by malignant glioma inhibits invasiveness and induces necrosis: An in vivo study. Neoplasia 2011, 13, 620–632. [Google Scholar] [CrossRef]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef]
- Huang, C.C.; Wang, S.Y.; Lin, L.L.; Wang, P.W.; Chen, T.Y.; Hsu, W.M.; Lin, T.K.; Liou, C.W.; Chuang, J.H. Glycolytic inhibitor 2-deoxyglucose simultaneously targets cancer and endothelial cells to suppress neuroblastoma growth in mice. Dis. Model. Mech. 2015, 8, 1247–1254. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- Arunachalam, G.; Samuel, S.M.; Marei, I.; Ding, H.; Triggle, C.R. Metformin modulates hyperglycaemia-induced endothelial senescence and apoptosis through SIRT1. Br. J. Pharmacol. 2014, 171, 523–535. [Google Scholar] [CrossRef]
- Ghosh, S.; Lakshmanan, A.P.; Hwang, M.J.; Kubba, H.; Mushannen, A.; Triggle, C.R.; Ding, H. Metformin improves endothelial function in aortic tissue and microvascular endothelial cells subjected to diabetic hyperglycaemic conditions. Biochem. Pharmacol. 2015, 98, 412–421. [Google Scholar] [CrossRef]
- Dykens, J.A.; Jamieson, J.; Marroquin, L.; Nadanaciva, S.; Billis, P.A.; Will, Y. Biguanide-induced mitochondrial dysfunction yields increased lactate production and cytotoxicity of aerobically-poised HepG2 cells and human hepatocytes in vitro. Toxicol. Appl. Pharmacol. 2008, 233, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, L.M.; Mukherjee, A.; Eckert, M.A.; Johnson, A.; Mills, K.A.; Pan, S.; Shridhar, V.; Lengyel, E.; Romero, I.L. Hyperglycemia-induced metabolic compensation inhibits metformin sensitivity in ovarian cancer. Oncotarget 2015, 6, 23548–23560. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, G.; Wang, Y.; Tang, S.; Sun, X.; Feng, X.; Li, Y.; Bao, G.; Li, P.; Mao, X.; et al. Suppression of tumor angiogenesis by metformin treatment via a mechanism linked to targeting of HER2/HIF-1alpha/VEGF secretion axis. Oncotarget 2015, 6, 44579–44592. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Laurent, K.; Loubat, A.; Giorgetti-Peraldi, S.; Colosetti, P.; Auberger, P.; Tanti, J.F.; Le Marchand-Brustel, Y.; Bost, F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008, 27, 3576–3586. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Le Marchand-Brustel, Y.; Tanti, J.F.; Bost, F. Metformin in cancer therapy: A new perspective for an old antidiabetic drug? Mol. Cancer Ther. 2010, 9, 1092–1099. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Tanti, J.F.; Bost, F. The combination of metformin and 2-deoxyglucose inhibits autophagy and induces AMPK-dependent apoptosis in prostate cancer cells. Autophagy 2010, 6, 670–671. [Google Scholar] [CrossRef]
- Zhu, J.; Zheng, Y.; Zhang, H.; Sun, H. Targeting cancer cell metabolism: The combination of metformin and 2-Deoxyglucose regulates apoptosis in ovarian cancer cells via p38 MAPK/JNK signaling pathway. Am. J. Transl. Res. 2016, 8, 4812–4821. [Google Scholar]
- Silverstein, R.L.; Baird, M.; Lo, S.K.; Yesner, L.M. Sense and antisense cDNA transfection of CD36 (glycoprotein IV) in melanoma cells. Role of CD36 as a thrombospondin receptor. J. Biol. Chem. 1992, 267, 16607–16612. [Google Scholar]
- Dawson, D.W.; Pearce, S.F.; Zhong, R.; Silverstein, R.L.; Frazier, W.A.; Bouck, N.P. CD36 mediates the In vitro inhibitory effects of thrombospondin-1 on endothelial cells. J. Cell Biol. 1997, 138, 707–717. [Google Scholar] [CrossRef]
- Jimenez, B.; Volpert, O.V.; Crawford, S.E.; Febbraio, M.; Silverstein, R.L.; Bouck, N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat. Med. 2000, 6, 41–48. [Google Scholar] [CrossRef]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, e51. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Nagy, J.A.; Manseau, E.J.; Phung, T.L.; Dvorak, H.F.; Benjamin, L.E. Rapamycin inhibition of the Akt/mTOR pathway blocks select stages of VEGF-A164-driven angiogenesis, in part by blocking S6Kinase. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1172–1178. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Trinh, X.B.; Tjalma, W.A.; Vermeulen, P.B.; Van den Eynden, G.; Van der Auwera, I.; Van Laere, S.J.; Helleman, J.; Berns, E.M.; Dirix, L.Y.; van Dam, P.A. The VEGF pathway and the AKT/mTOR/p70S6K1 signalling pathway in human epithelial ovarian cancer. Br. J. Cancer 2009, 100, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, H.B.; Fumagalli, S.; Dennis, P.B.; Reinhard, C.; Pearson, R.B.; Thomas, G. Rapamycin suppresses 5′TOP mRNA translation through inhibition of p70s6k. EMBO J. 1997, 16, 3693–3704. [Google Scholar] [CrossRef] [PubMed]
- Ikezoe, T.; Nishioka, C.; Bandobashi, K.; Yang, Y.; Kuwayama, Y.; Adachi, Y.; Takeuchi, T.; Koeffler, H.P.; Taguchi, H. Longitudinal inhibition of PI3K/Akt/mTOR signaling by LY294002 and rapamycin induces growth arrest of adult T-cell leukemia cells. Leuk. Res. 2007, 31, 673–682. [Google Scholar] [CrossRef] [PubMed]
- McGillicuddy, F.C.; O’Toole, D.; Hickey, J.A.; Gallagher, W.M.; Dawson, K.A.; Keenan, A.K. TGF-beta1-induced thrombospondin-1 expression through the p38 MAPK pathway is abolished by fluvastatin in human coronary artery smooth muscle cells. Vasc. Pharmacol. 2006, 44, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Nagata, D.; Mogi, M.; Walsh, K. AMP-activated protein kinase (AMPK) signaling in endothelial cells is essential for angiogenesis in response to hypoxic stress. J. Biol. Chem. 2003, 278, 31000–31006. [Google Scholar] [CrossRef]
- Stahmann, N.; Woods, A.; Spengler, K.; Heslegrave, A.; Bauer, R.; Krause, S.; Viollet, B.; Carling, D.; Heller, R. Activation of AMP-activated protein kinase by vascular endothelial growth factor mediates endothelial angiogenesis independently of nitric-oxide synthase. J. Biol. Chem. 2010, 285, 10638–10652. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brule, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Dallaglio, K.; Bruno, A.; Cantelmo, A.R.; Esposito, A.I.; Ruggiero, L.; Orecchioni, S.; Calleri, A.; Bertolini, F.; Pfeffer, U.; Noonan, D.M.; et al. Paradoxic effects of metformin on endothelial cells and angiogenesis. Carcinogenesis 2014, 35, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Li, Y.; Liu, X.; Zhou, T.; Sun, H.; Edwards, P.; Gao, H.; Yu, F.S.; Qiao, X. Metformin suppresses retinal angiogenesis and inflammation in vitro and in vivo. PLoS ONE 2018, 13, e0193031. [Google Scholar] [CrossRef]
- Ying, Y.; Ueta, T.; Jiang, S.; Lin, H.; Wang, Y.; Vavvas, D.; Wen, R.; Chen, Y.G.; Luo, Z. Metformin inhibits ALK1-mediated angiogenesis via activation of AMPK. Oncotarget 2017, 8, 32794–32806. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.W.; Deng, Y.P.; Han, X.; Ren, G.F.; Cai, J.; Jiang, G.J. Metformin improves the angiogenic functions of endothelial progenitor cells via activating AMPK/eNOS pathway in diabetic mice. Cardiovasc. Diabetol. 2016, 15, e88. [Google Scholar] [CrossRef]
- Goransson, O.; McBride, A.; Hawley, S.A.; Ross, F.A.; Shpiro, N.; Foretz, M.; Viollet, B.; Hardie, D.G.; Sakamoto, K. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J. Biol. Chem. 2007, 282, 32549–32560. [Google Scholar] [CrossRef]
- Dasgupta, B.; Seibel, W. Compound C/Dorsomorphin: Its Use and Misuse as an AMPK Inhibitor. In AMPK: Methods and Protocols; Neumann, D., Viollet, B., Eds.; Springer New York: New York, NY, USA, 2018; pp. 195–202. [Google Scholar] [CrossRef]
- Liu, X.; Chhipa, R.R.; Nakano, I.; Dasgupta, B. The AMPK inhibitor compound C is a potent AMPK-independent antiglioma agent. Mol. Cancer Ther. 2014, 13, 596–605. [Google Scholar] [CrossRef]
- Merchan, J.R.; Kovács, K.; Railsback, J.W.; Kurtoglu, M.; Jing, Y.; Piña, Y.; Gao, N.; Murray, T.G.; Lehrman, M.A.; Lampidis, T.J. Antiangiogenic Activity of 2-Deoxy-D-Glucose. PLoS ONE 2010, 5, e13699. [Google Scholar] [CrossRef]
- Dwarakanath, B.; Singh, D.; Banerji, A.; Sarin, R.; Venkataramana, N.; Jalali, R.; Vishwanath, P.; Mohanti, B.; Tripathi, R.; Kalia, V.; et al. Clinical studies for improving radiotherapy with 2-deoxy-D-glucose: Present status and future prospects. J. Cancer Res. Ther. 2009, 5, 21–26. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Conradi, L.C.; Brajic, A.; Cantelmo, A.R.; Bouche, A.; Kalucka, J.; Pircher, A.; Bruning, U.; Teuwen, L.A.; Vinckier, S.; Ghesquiere, B.; et al. Tumor vessel disintegration by maximum tolerable PFKFB3 blockade. Angiogenesis 2017, 20, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Draoui, N.; de Zeeuw, P.; Carmeliet, P. Angiogenesis revisited from a metabolic perspective: Role and therapeutic implications of endothelial cell metabolism. Open Biol. 2017, 7, e170219. [Google Scholar] [CrossRef] [PubMed]
- Pannirselvam, M.; Verma, S.; Anderson, T.J.; Triggle, C.R. Cellular basis of endothelial dysfunction in small mesenteric arteries from spontaneously diabetic (db/db -/-) mice: Role of decreased tetrahydrobiopterin bioavailability. Br. J. Pharmacol. 2002, 136, 255–263. [Google Scholar] [CrossRef]
- Arbiser, J.L.; Moses, M.A.; Fernandez, C.A.; Ghiso, N.; Cao, Y.; Klauber, N.; Frank, D.; Brownlee, M.; Flynn, E.; Parangi, S.; et al. Oncogenic H-ras stimulates tumor angiogenesis by two distinct pathways. Proc. Natl. Acad. Sci. USA 1997, 94, 861–866. [Google Scholar] [CrossRef]
- Hamada, Y.; Gonda, K.; Takeda, M.; Sato, A.; Watanabe, M.; Yambe, T.; Satomi, S.; Ohuchi, N. In vivo imaging of the molecular distribution of the VEGF receptor during angiogenesis in a mouse model of ischemia. Blood 2011, 118, 93–100. [Google Scholar] [CrossRef][Green Version]
- Tucker, G.T.; Casey, C.; Phillips, P.J.; Connor, H.; Ward, J.D.; Woods, H.F. Metformin kinetics in healthy subjects and in patients with diabetes mellitus. Br. J. Clin. Pharmacol. 1981, 12, 235–246. [Google Scholar] [CrossRef]
- Varghese, S.; Samuel, S.M.; Varghese, E.; Kubatka, P.; Busselberg, D. High Glucose Represses the Anti-Proliferative and Pro-Apoptotic Effect of Metformin in Triple Negative Breast Cancer Cells. Biomolecules 2019, 9, 16. [Google Scholar] [CrossRef]
- Arunachalam, G.; Lakshmanan, A.P.; Samuel, S.M.; Triggle, C.R.; Ding, H. Molecular Interplay between microRNA-34a and Sirtuin1 in Hyperglycemia-Mediated Impaired Angiogenesis in Endothelial Cells: Effects of Metformin. J. Pharmacol. Exp. Ther. 2016, 356, 314–323. [Google Scholar] [CrossRef]











© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathews Samuel, S.; Satheesh, N.J.; Ghosh, S.; Büsselberg, D.; Majeed, Y.; Ding, H.; Triggle, C.R. Treatment with a Combination of Metformin and 2-Deoxyglucose Upregulates Thrombospondin-1 in Microvascular Endothelial Cells: Implications in Anti-Angiogenic Cancer Therapy. Cancers 2019, 11, 1737. https://doi.org/10.3390/cancers11111737
Mathews Samuel S, Satheesh NJ, Ghosh S, Büsselberg D, Majeed Y, Ding H, Triggle CR. Treatment with a Combination of Metformin and 2-Deoxyglucose Upregulates Thrombospondin-1 in Microvascular Endothelial Cells: Implications in Anti-Angiogenic Cancer Therapy. Cancers. 2019; 11(11):1737. https://doi.org/10.3390/cancers11111737
Chicago/Turabian StyleMathews Samuel, Samson, Noothan Jyothi Satheesh, Suparna Ghosh, Dietrich Büsselberg, Yasser Majeed, Hong Ding, and Chris R. Triggle. 2019. "Treatment with a Combination of Metformin and 2-Deoxyglucose Upregulates Thrombospondin-1 in Microvascular Endothelial Cells: Implications in Anti-Angiogenic Cancer Therapy" Cancers 11, no. 11: 1737. https://doi.org/10.3390/cancers11111737
APA StyleMathews Samuel, S., Satheesh, N. J., Ghosh, S., Büsselberg, D., Majeed, Y., Ding, H., & Triggle, C. R. (2019). Treatment with a Combination of Metformin and 2-Deoxyglucose Upregulates Thrombospondin-1 in Microvascular Endothelial Cells: Implications in Anti-Angiogenic Cancer Therapy. Cancers, 11(11), 1737. https://doi.org/10.3390/cancers11111737