Deconvolution of the Genomic and Epigenomic Interaction Landscape of Triple-Negative Breast Cancer



Abstract
1. Introduction
2. Materials and Methods
2.1. Germline Mutations and Associated Genes
2.2. Gene Expression and DNA Methylation Data and Somatic Mutation Information
2.3. Bioinformatics Analysis of Gene Expression and DNA Methylation Data
2.4. Network and Pathway Analysis
3. Results
3.1. Discovery of a Signature of Aberrantly Methylated Genes Associated with TNBC
3.2. Discovery of a Signature of Differentially Expressed Genes Associated with TNBC
3.3. Discovery of a Signature of Aberrantly Methylated Genes Transcriptionally Associated with TNBC
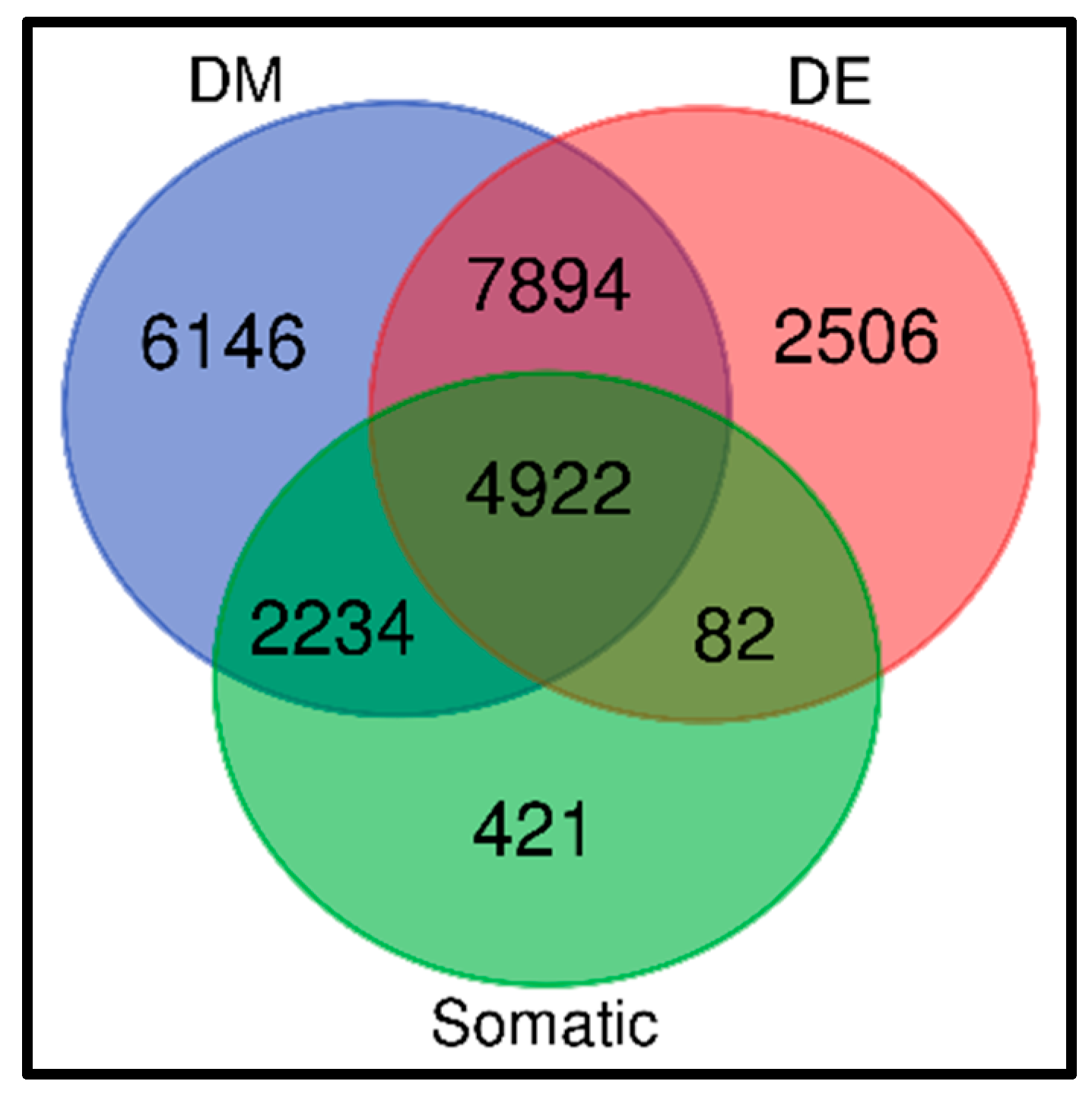
3.4. Discovery of a Signature of Genes Containing Both Somatic and Epigenetic Variation
3.5. Discovery of a Signature of Genes Containing Germline, Somatic and Epigenomic Variation
3.6. Molecular Networks and Signaling Pathways Enriched for Germline, Somatic and Epigenomic Variation
4. Discussion
4.1. Integrating Transcription with DNA Methylation Profiling
4.2. Integrating Somatic Variation with Epigenomic Variation
4.3. Oncogenic Interactions between Genes Containing Germline and Epigenetic Variation
4.4. Disease Networks and Pathways as Potential Therapeutic Targets
4.5. Limitations and Future Research Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Plasilova, M.L.; Hayse, B.; Killelea, B.K.; Horowitz, N.R.; Chagpar, A.B.; Lannin, D.R. Features of triple-negative breast cancer: Analysis of 38,813 cases from the national cancer database. Medicine 2016, 95, e4614. [Google Scholar] [CrossRef] [PubMed]
- Dietze, E.C.; Sistrunk, C.; Miranda-Carboni, G.; O’Regan, R.; Seewaldt, V.L. Triple-negative breast cancer in African-American women: Disparities versus biology. Nat. Rev. Cancer 2015, 15, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Dietze, E.C.; Chavez, T.A.; Seewaldt, V.L. Obesity and Triple-Negative Breast Cancer: Disparities, Controversies, and Biology. Am. J. Pathol. 2018, 188, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Sturtz, L.A.; Melley, J.; Mamula, K.; Shriver, C.D.; Ellsworth, R.E. Outcome disparities in African American women with triple negative breast cancer: A comparison of epidemiological and molecular factors between African American and Caucasian women with triple negative breast cancer. BMC Cancer. 2014, 4, 62. [Google Scholar] [CrossRef]
- Hahnen, E.; Hauke, J.; Engel, C.; Neidhardt, G.; Rhiem, K.; Schmutzler, R.K. Germline Mutations in Triple-Negative Breast Cancer. Breast Care 2017, 12, 15–19. [Google Scholar] [CrossRef]
- Ellsworth, D.L.; Turner, C.E.; Ellsworth, R.E. A Review of the Hereditary Component of Triple Negative Breast Cancer: High- and Moderate-Penetrance Breast Cancer Genes, Low-Penetrance Loci, and the Role of Nontraditional Genetic Elements. J. Oncol. 2019, 2019, 4382606. [Google Scholar] [CrossRef]
- Pidsley, R.; Lawrence, M.G.; Zotenko, E.; Niranjan, B.; Statham, A.; Song, J.; Chabanon, R.M.; Qu, W.; Wang, H.; Richards, M.; et al. Enduring epigenetic landmarks define the cancer microenvironment. Genome Res. 2018, 28, 625–638. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Coyle, K.; Dean, C.; Thomas, M.; Vidovic, D.; Giacomantonio, C.; Helyer, L.; Marcato, P. DNA Methylation Predicts the Response of Triple-Negative Breast Cancers to All-Trans Retinoic Acid. Cancers 2018, 10, 397. [Google Scholar] [CrossRef]
- Yu, J.; Qin, B.; Moyer, A.M.; Nowsheen, S.; Liu, T.; Qin, S.; Zhuang, Y.; Liu, D.; Lu, S.W.; Kalari, K.R.; et al. DNA methyltransferase expression in triple-negative breast cancer predicts sensitivity to decitabine. J. Clin. Investig. 2018, 128, 2376–2388. [Google Scholar] [CrossRef]
- Perou, C.M. Molecular Stratification of Triple-Negative Breast Cancers. Oncologist 2010, 15 (Suppl. 5), 39–48. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Pietenpol, J.A. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J. Pathol. 2014, 232, 142–150. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Prat, A.; Parker, J.S. Standardized versus research-based PAM50 intrinsic subtyping of breast cancer. Clin. Transl. Oncol. 2019. [Google Scholar] [CrossRef]
- Ohnstad, H.O.; Borgen, E.; Falk, R.S.; Lien, T.G.; Aaserud, M.; Sveli, M.A.T.; Kyte, J.A.; Kristensen, V.N.; Geitvik, G.A.; Schlichting, E.; et al. Prognostic value of PAM50 and risk of recurrence score in patients with early-stage breast cancer with long-term follow-up. Breast Cancer Res. 2017, 19, 120. [Google Scholar] [CrossRef]
- Xin, L.; Liu, Y.H.; Martin, T.A.; Jiang, W.G. The Era of Multigene Panels Comes? The Clinical Utility of Oncotype DX and MammaPrint. World J. Oncol. 2017, 8, 34–40. [Google Scholar] [CrossRef]
- Wu, J.; Mamidi, T.K.K.; Zhang, L.; Hicks, C. Integrating Germline and Somatic Mutation Information for the Discovery of Biomarkers in Triple-Negative Breast Cancer. Int. J. Environ. Res. Public Health. 2019, 16, 1055. [Google Scholar] [CrossRef]
- Morales, J.; Welter, D.; Bowler, E.H.; Cerezo, M.; Harris, L.W.; McMahon, A.C.; Hall, P.; Junkins, H.A.; Milano, A.; Hastings, E.; et al. A standardized framework for representation of ancestry data in genomics studies, with application to the NHGRI-EBI GWAS Catalog. Genome Biol. 2018, 19, 21. [Google Scholar] [CrossRef]
- The NHGRI-EBI Catalog of Published Genome-Wide Association Studies. Available online: https://www.ebi.ac.uk/gwas/ (accessed on 7 September 2019).
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. Cancer Genome Atlas Research Network. Nat Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- International Cancer Genome Consortium; Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabé, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; et al. The International Cancer Genome Consortium International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef]
- Hicks, C.; Kumar, R.; Pannuti, A.; Backus, K.; Brown, A.; Monico, J.; Miele, L. An integrative genomics approach for associating GWAS information with triple-negative breast cancer. Cancer Inf. 2013, 12, CIN–S10413. [Google Scholar] [CrossRef]
- Ioannidis, J.P.; Boffetta, P.; Little, J.; O’Brien, T.R.; Uitterlinden, A.G.; Vineis, P.; Balding, D.J.; Chokkalingam, A.; Dolan, S.M.; Flanders, W.D.; et al. Assessment of cumulative evidence on genetic associations: Interim guidelines. Int. J. Epidemiol. 2007, 37, 120–132. [Google Scholar] [CrossRef]
- Khoury, M.J.; Bertram, L.; Boffetta, P.; Butterworth, A.S.; Chanock, S.J.; Dolan, S.M.; Fortier, I.; Garcia-Closas, M.; Gwinn, M.; Higgins, J.P.; et al. Genome-wide association studies, field synopses, and the development of the knowledge base on genetic variation and human diseases. Am. J. Epidemiol. 2009, 170, 269–279. [Google Scholar] [CrossRef]
- Sagoo, G.S.; Little, J.; Higgins, J.P. Systematic reviews of genetic association studies. PLoS Med. 2009, 6, e1000028. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: Explanation and elaboration. PLoS Med. 2009, 6, e1000100. [Google Scholar] [CrossRef]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2018, 47, D1005–D1012. [Google Scholar] [CrossRef]
- National Cancer Institute. Available online: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga (accessed on 7 September 2019).
- National Cancer Institute, Genomic Data Commons. Available online: https://gdc.cancer.gov/ (accessed on 7 September 2019).
- Marabita, F.; Almgren, M.; Lindholm, M.E.; Ruhrmann, S.; Fagerström-Billai, F.; Jagodic, M.; Sundberg, C.J.; Ekström, T.J.; Teschendorff, A.E.; Tegnér, J.; et al. An evaluation of analysis pipelines for DNA methylation profiling using the Illumina HumanMethylation450 BeadChip platform. Epigenetics 2013, 8, 333–346. [Google Scholar] [CrossRef]
- Liu, J.; Siegmund, K.D. An evaluation of processing methods for HumanMethylation450 BeadChip data. BMC Genom. 2016, 17, 469. [Google Scholar] [CrossRef]
- Maksimovic, J.; Gordon, L.; Oshlack, A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 2012, 13, R44. [Google Scholar] [CrossRef]
- Wang, T.; Guan, W.; Lin, J.; Boutaoui, N.; Canino, G.; Luo, J.; Celedón, J.C.; Chen, W. A systematic study of normalization methods for Infinium 450K methylation data using whole-genome bisulfite sequencing data. Epigenetics 2015, 10, 662–669. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Benjamini, Y.; Yosef, H. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Ensemble Biomart Browser. Available online: https://useast.ensembl.org/info/data/biomart/index.html (accessed on 7 September 2019).
- TCGAvisualize-Starburst Tool. Available online: https://rdrr.io/bioc/TCGAbiolinks/man/TCGAvisualize_starburst.html (accessed on 7 September 2019).
- Ingenuity Pathways Analysis (IPA) System. Redwood, CA: Ingenuity Systems. Available online: http://www.ingenuity.com/ (accessed on 7 September 2019).
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Lips, E.H.; Michaut, M.; Hoogstraat, M.; Mulder, L.; Besselink, N.J.; Koudijs, M.J.; Cuppen, E.; Voest, E.E.; Bernards, R.; Nederlof, P.M.; et al. Next generation sequencing of triple negative breast cancer to find predictors for chemotherapy response. Breast Cancer Res. 2015, 17, 134. [Google Scholar] [CrossRef]
- Saravia, C.H.; Flores, C.; Schwarz, L.J.; Bravo, L.; Zavaleta, J.; Araujo, J.; Neciosup, S.; Pinto, J.A. Patterns of Mutation Enrichment in Metastatic Triple-Negative Breast Cancer. Clin. Med. Insights Oncol. 2019, 13, 1179554919868482. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Lakshmanan, I.; Ponnusamy, M.P.; Chakraborty, S.; Jain, M.; Pai, P.; Smith, L.M.; Lele, S.M.; Batra, S.K. MUC4 overexpression augments cell migration and metastasis through EGFR family proteins in triple negative breast cancer cells. PLoS ONE 2013, 8, e54455. [Google Scholar] [CrossRef]
- Kumar, M.A.; Naushad, S.M.; Narasimgu, N.; Naik, S.N.; Kadali, S.; Shanker, U.; Narasu, M.L. Whole exome sequencing of breast cancer (TNBC) cases from India: Association of MSH6 and BRIP1 variants with TNBC risk and oxidative DNA damage. Mol. Biol. Rep. 2018, 45, 1413–1419. [Google Scholar] [CrossRef]
- Elfgen, C.; Reeve, K.; Moskovszky, L.; Güth, U.; Bjelic-Radisic, V.; Fleisch, M.; Tausch, C.; Varga, Z. Prognostic impact of PIK3CA protein expression in triple negative breast cancer and its subtypes. J. Cancer Res. Clin. Oncol. 2019, 145, 2051–2059. [Google Scholar] [CrossRef]
- Komatsu, M.; Yoshimaru, T.; Matsuo, T.; Kiyotani, K.; Miyoshi, Y.; Tanahashi, T.; Rokutan, K.; Yamaguchi, R.; Saito, A.; Imoto, S.; et al. Molecular features of triple negative breast cancer cells by genome-wide gene expression profiling analysis. Int. J. Oncol. 2013, 42, 478–506. [Google Scholar] [CrossRef]
- Cui, Y.; Bai, X.; Niu, M.; Qin, Y.X.; Pang, D. Upregulated expression of AT-rich interactive domain-containing protein 1B predicts poor prognosis in patients with triple-negative breast cancer. Oncol. Lett. 2019, 17, 3289–3295. [Google Scholar] [CrossRef]
- Chen, B.; Wang, J.; Dai, D.; Zhou, Q.; Guo, X.; Tian, Z.; Huang, X.; Yang, L.; Tang, H.; Xie, X. AHNAK suppresses tumour proliferation and invasion by targeting multiple pathways in triple-negative breast cancer. J. Exp. Clin. Cancer Res. 2017, 36, 65. [Google Scholar] [CrossRef]
- Shiovitz, S.; Korde, L.A. Genetics of breast cancer: A topic in evolution. Ann. Oncol. 2015, 26, 1291–1299. [Google Scholar] [CrossRef]
- Kawazu, M.; Kojima, S.; Ueno, T.; Totoki, Y.; Nakamura, H.; Kunita, A.; Qu, W.; Yoshimura, J.; Soda, M.; Yasuda, T.; et al. Integrative analysis of genomic alterations in triple-negative breast cancer in association with homologous recombination deficienc. PLoS Genet. 2017, 13, e1006853. [Google Scholar] [CrossRef]
- Purrington, K.S.; Slager, S.; Eccles, D.; Yannoukakos, D.; Fasching, P.A.; Miron, P.; Carpenter, J.; Chang-Claude, J.; Martin, N.G.; Montgomery, G.W.; et al. Genome-wide association study identifies 25 known breast cancer susceptibility loci as risk factors for triple-negative breast cancer. Carcinogenesis 2014, 35, 1012–1019. [Google Scholar] [CrossRef]
- Stevens, K.N.; Vachon, C.M.; Couch, F.J. Genetic susceptibility to triple-negative breast cancer. Cancer Res. 2013, 73, 2025–2030. [Google Scholar] [CrossRef]
- Couch, F.J.; Hart, S.N.; Sharma, P.; Toland, A.E.; Wang, X.; Miron, P.; Olson, J.E.; Godwin, A.K.; Pankratz, V.S.; Olswold, C.; et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J. Clin. Oncol. 2015, 33, 304–311. [Google Scholar] [CrossRef]
- Stevens, K.N.; Vachon, C.M.; Lee, A.M.; Slager, S.; Lesnick, T.; Olswold, C.; Fasching, P.A.; Miron, P.; Eccles, D.; Carpenter, J.E.; et al. Common breast cancer susceptibility loci are associated with triple-negative breast cancer. Cancer Res. 2011, 71, 6240–6249. [Google Scholar] [CrossRef]
- Zhang, F.; Ren, C.; Zhao, H.; Yang, L.; Su, F.; Zhou, M.M.; Han, J.; Sobie, E.A.; Walsh, M.J. Identification of novel prognostic indicators for triple-negative breast cancer patients through integrative analysis of cancer genomics data and protein interactome data. Oncotarget 2016, 7, 71620–71634. [Google Scholar] [CrossRef][Green Version]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V.; et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell. 2018, 33, 690–705.e9. [Google Scholar] [CrossRef] [PubMed]
- Shilpi, A.; Bi, Y.; Jung, S.; Patra, S.K.; Davuluri, R.V. Identification of Genetic and Epigenetic Variants Associated with Breast Cancer Prognosis by integrative bioinformatics analysis. Cancer Inf. 2017, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, D.E.; Johnson, K.C.; Skinner, L.; Koestler, D.C.; Christensen, B.C. Epigenetic and genetic burden measures are associated with tumor characteristics in invasive breast carcinoma. Epigenetics 2016, 11, 344–353. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, B.P.; Apolónio, J.D.; Binnie, A.; Castelo-Branco, P. Roadmap of DNA methylation in breast cancer identifies novel prognostic biomarkers. BMC Cancer 2019, 19, 219. [Google Scholar] [CrossRef] [PubMed]
- Vel Szic, K.S.; Declerck, K.; Crans, R.A.; Diddens, J.; Scherf, D.B.; Gerhäuser, C.; Berghe, W.V. Epigenetic silencing of triple negative breast cancer hallmarks by Withaferin A. Oncotarget 2017, 8, 40434. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef]
- Scott, C.M.; Joo, J.E.; O’Callaghan, N.; Buchanan, D.D.; Clendenning, M.; Giles, G.G.; Hopper, J.L.; Wong, E.M.; Southey, M.C. Methylation of Breast Cancer Predisposition Genes in Early-Onset Breast Cancer: Australian Breast Cancer Family Registry. PLoS ONE 2016, 11, e0165436. [Google Scholar] [CrossRef]
- Gao, Y.; Widschwendter, M.; Teschendorff, A.E. DNA Methylation Patterns in Normal Tissue Correlate more Strongly with Breast Cancer Status than Copy-Number Variants. EBioMedicine 2018, 31, 243–252. [Google Scholar] [CrossRef]
- Catteau, A.; Morris, J.R. BRCA1 methylation: A significant role in tumour development? Semin Cancer Biol. 2002, 12, 359–371. [Google Scholar] [CrossRef]
- Shoemaker, R.; Deng, J.; Wang, W.; Zhang, K. Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res. 2010, 20, 883–889. [Google Scholar] [CrossRef]
- Lin, W.; Lin, H.D.; Guo, X.Y.; Lin, Y.; Su, F.X.; Jia, W.H.; Tang, L.Y.; Zheng, W.; Long, J.R.; Ren, Z.F. Allelic expression imbalance polymorphisms in susceptibility chromosome regions and the risk and survival of breast cancer. Mol. Carcinog. 2017, 56, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Shi, Y.; Wang, X.Y.L.; Guan, X. A novel BRCA1 germline mutation promotes triple-negative breast cancer cells progression and enhances sensitivity to DNA damage agents. Cancer Genet. 2019, 239, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; Chung, S.; Brough, R.; Vail, P.; Franco, J.; Lord, C.J.; Knudsen, E.S. Targeting the Vulnerability of RB Tumor Suppressor Loss in Triple-Negative Breast Cance. Cell Rep. 2018, 22, 1185–1199. [Google Scholar] [CrossRef]
- Synnott, N.C.; Bauer, M.R.; Madden, S.; Murray, A.; Klinger, R.; O’Donovan, N.; O’Connor, D.; Gallagher, W.M.; Crown, J.; Fersht, A.R.; et al. Mutant p53 as a therapeutic target for the treatment of triple-negative breast cancer: Preclinical investigation with the anti-p53 drug, PK11007. Cancer Lett. 2018, 414, 99–106. [Google Scholar] [CrossRef]
- Ademuyiwa, F.O.; Tao, Y.; Luo, J.; Weilbaecher, K.; Ma, C.X. Differences in the mutational landscape of triple-negative breast cancer in African Americans and Caucasians. Breast Cancer Res Treat. 2017, 161, 491–499. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Type | TNBC | ||
|---|---|---|---|
| Genes or Probes | Tumor Samples | Control Samples | |
| Gene expression | 60,484 Probes | 110 | 113 |
| Methylation | 485,578 probes | 83 | 83 |
| Somatic | 7659 genes | 110 | 113 |
| GWAS * | 825 genes | >300,000 | >300,000 |
| Gene_Symbol | Chromosome | Methylation | RNAseq | |
|---|---|---|---|---|
| Event | Adjust p-Value | Adjust p-Value | ||
| RP1 | 8q11.23 | 992 | 3.96 × 10−24 | 8.94 × 10−5 |
| PTPRN2 | 7q36.3 | 917 | 7.85 × 10−24 | 4.24 × 10−16 |
| PRDM16 | 1p36.32 | 434 | 1.78 × 10−22 | 3.42 × 10−17 |
| TNXB | 6p21.33 | 364 | 3.11 × 10−22 | 1.29 × 10−24 |
| MAD1L1 | 7p22.3 | 317 | 3.89 × 10−23 | 2.29 × 10−7 |
| DIP2C | 10p15.3 | 298 | 4.71 × 10−23 | 1.24 × 10−9 |
| PCDHGA2 | 5q31.3 | 291 | 3.57 × 10−24 | 1.30 × 10−19 |
| SNHG14 | 15q11.2 | 283 | 1.56 × 10−20 | 2.15 × 10−13 |
| PCDHGA3 | 5q31.3 | 277 | 3.57 × 10−24 | 2.05 × 10−21 |
| ERICH1 | 8p23.3 | 270 | 3.25 × 10−23 | 1.47 × 10−2 |
| ADARB2 | 10p15.3 | 257 | 5.96 × 10−24 | 6.18 × 10−10 |
| PCDHGA4 | 5q31 | 253 | 3.57 × 10−24 | 3.25 × 10−15 |
| PCDHGB2 | 5q31 | 239 | 3.57 × 10−24 | 4.51 × 10−8 |
| PCDHGA5 | 5q31 | 229 | 3.57 × 10−24 | 6.38 × 10−15 |
| EIF2B5 | 3q27.1 | 217 | 2.57 × 10−23 | 2.26 × 10−4 |
| PCDHGB3 | 5q31 | 213 | 3.57 × 10−24 | 2.64 × 10−8 |
| TBCD | 17q25.3 | 204 | 1.30 × 10−23 | 8.35 × 10−7 |
| HDAC4 | 2q37.3 | 202 | 6.13 × 10−24 | 3.71 × 10−8 |
| MCF2L | 13q34 | 202 | 6.15 × 10−24 | 9.10 × 10−5 |
| PCDHGA6 | 5q31 | 202 | 3.57 × 10−24 | 3.20 × 10−14 |
| SDK1 | 7p22.2 | 202 | 4.35 × 10−22 | 1.13 × 10−8 |
| INPP5A | 10q26.3 | 190 | 3.55 × 10−22 | 4.39 × 10−13 |
| PCDHGA7 | 5q31 | 188 | 3.57 × 10−24 | 1.69 × 10−13 |
| ATP11A | 13q34 | 177 | 6.26 × 10−23 | 2.83 × 10−2 |
| PCDHGB4 | 5q31 | 175 | 3.57 × 10−24 | 6.45 × 10−10 |
| KCNQ1 | 11p15.5 | 174 | 2.29 × 10−22 | 2.51 × 10−3 |
| HOXA3 | 7p15.2 | 168 | 1.26 × 10−23 | 3.43 × 10−11 |
| PCDHGA8 | 5q31.3 | 166 | 3.57 × 10−24 | 3.74 × 10−3 |
| C7orf50 | 7p22.3 | 163 | 4.59 × 10−23 | 1.54 × 10−7 |
| AGAP1 | 2q37.2 | 160 | 5.28 × 10−21 | 2.25 × 10−3 |
| Gene | Chromosome | Methylation | RNAseq | Somatic Events | |
|---|---|---|---|---|---|
| DM_Sites | Adjust p-Value | Adjust p-Value | |||
| TTN * | 2q31.2 | 20 | 1.53 × 10−13 | 8.34 × 10−10 | 27 |
| MUC4 * | 3q29 | 28 | 3.91 × 10−19 | 7.60 × 10−4 | 13 |
| FAT3 | 11q14.3 | 23 | 9.50 × 10−15 | 1.67 × 10−9 | 12 |
| USH2A | 1q41 | 12 | 1.53 × 10−11 | 1.73 × 10−6 | 12 |
| SYNE1 * | 6q25.2 | 29 | 1.04 × 10−18 | 1.66 × 10−20 | 9 |
| FCGBP | 19q13.2 | 14 | 1.60 × 10−17 | 3.48 × 10−2 | 9 |
| SPTA1 | 1q23.1 | 3 | 2.00 × 10−14 | 8.63 × 10−5 | 9 |
| DNAH17 | 17q25.3 | 76 | 3.52 × 10−21 | 3.74 × 10−4 | 8 |
| DST | 6p12.1 | 41 | 1.76 × 10−18 | 1.14 × 10−21 | 8 |
| MUC5B | 11p15.5 | 40 | 9.44 × 10−16 | 1.71 × 10−8 | 8 |
| PIK3CA * | 3q26.32 | 7 | 1.11 × 10−16 | 3.26 × 10−6 | 8 |
| PLEC | 8q24.3 | 103 | 1.36 × 10−21 | 1.28 × 10−4 | 7 |
| CSMD2 | 1p35.1 | 47 | 1.81 × 10−19 | 8.84 × 10−12 | 7 |
| CREBBP | 16p13.3 | 38 | 1.40 × 10−21 | 1.85 × 10−3 | 7 |
| FLG | 1q21.3 | 33 | 3.22 × 10−17 | 4.48 × 10−9 | 7 |
| KMT2D | 12q13.12 | 17 | 3.06 × 10−17 | 6.93 × 10−4 | 7 |
| AHCTF1 | 1q44 | 10 | 1.20 × 10−7 | 3.48 × 10−4 | 7 |
| ASPM * | 1q31.3 | 10 | 1.17 × 10−12 | 5.39 × 10−24 | 7 |
| MYO18B | 22q12.1 | 4 | 4.93 × 10−16 | 3.73 × 10−6 | 7 |
| USP34 | 2p15 | 4 | 2.38 × 10−3 | 1.36 × 10−2 | 7 |
| KIF26B | 1q44 | 79 | 8.82 × 10−15 | 7.07 × 10−16 | 6 |
| SPTBN1 | 2p16.2 | 54 | 4.56 × 10−24 | 8.06 × 10−21 | 6 |
| LRP1 | 12q13.3 | 52 | 5.94 × 10−23 | 1.08 × 10−17 | 6 |
| COL18A1 | 21q22.3 | 47 | 1.17 × 10−21 | 1.41 × 10−3 | 6 |
| ARID1B * | 6q25.3 | 43 | 1.71 × 10−16 | 5.95 × 10−3 | 6 |
| ZNF512B | 20q13.33 | 42 | 3.57 × 10−24 | 4.92 × 10−5 | 6 |
| AHNAK * | 11q12.3 | 30 | 8.81 × 10−23 | 1.94 × 10−24 | 6 |
| CACNA1B | 9q34.3 | 24 | 6.13 × 10−15 | 1.31 × 10−7 | 6 |
| STAB1 | 3p21.1 | 19 | 1.17 × 10−17 | 2.65 × 10−4 | 6 |
| LAMA3 | 18q11.2 | 18 | 1.13 × 10−22 | 7.72 × 10−19 | 6 |
| Gene Symbol | Chromosome | Methylation | RNAseq | Somatic Events | GWAS | |||
|---|---|---|---|---|---|---|---|---|
| DM Sites | Adjust p-Value | Adjust p-Value | SNP | p-Value | Event | |||
| BRCA1 | 17q21.31 | 12 | 2.01 × 10−5 | 2.78 × 10−3 | 5 | rs1799950 | 2.00 × 10−4 | 2 |
| FHOD3 | 18q12.2 | 11 | 8.83 × 10−14 | 9.05 × 10−11 | 5 | rs9956546 | 2.90 × 10−6 | 2 |
| MYO10 | 5p15.1 | 45 | 1.86 × 10−19 | 2.29 × 10−11 | 4 | rs2562343 | 9.20 × 10−3 | 2 |
| CNTNAP2 | 7q35 | 30 | 8.36 × 10−17 | 1.83 × 10−3 | 4 | rs10487920 | 3.90 × 10−4 | 2 |
| RELN | 7q22.1 | 12 | 1.92 × 10−13 | 2.94 × 10−18 | 4 | rs17157903 | 1.00 × 10−2 | 2 |
| MSH3 | 5q14.1 | 10 | 2.46 × 10−10 | 7.38 × 10−15 | 4 | rs6151904 | 1.24 × 10−2 | 2 |
| ATM | 11q22.3 | 20 | 9.88 × 10−7 | 1.47 × 10−5 | 3 | rs1801516 | 2.00 × 10−4 | 2 |
| MTHFR | 1p36.22 | 9 | 7.33 × 10−12 | 1.33 × 10−6 | 3 | rs180113 | 4.10 × 10−2 | 2 |
| PALB2 | 16p12.2 | 3 | 1.54 × 10−2 | 1.87 × 10−3 | 3 | deletion | 4.00 × 10−4 | 2 |
| FBXL7 | 5p15.1 | 35 | 2.85 × 10−19 | 9.65 × 10−12 | 2 | rs12652447 | 5.60 × 10−4 | 2 |
| NUMA1 | 11q13.4 | 28 | 7.97 × 10−20 | 1.17 × 10−7 | 2 | rs3750913 | 1.00 × 10−2 | 2 |
| RB1 | 13q14.2 | 24 | 2.24 × 10−21 | 9.05 × 10−11 | 2 | rs2854344 | 7.00 × 10−3 | 2 |
| AACS | 12q24.31 | 19 | 1.04 × 10−15 | 1.12 × 10−3 | 2 | rs7307700 | 2.00 × 10−2 | 2 |
| WRN | 8p12 | 15 | 3.30 × 10−16 | 3.19 × 10−4 | 2 | rs1346044 | 2.00 × 10−2 | 2 |
| GRIN3A | 9q31.1 | 12 | 7.03 × 10−11 | 8.98 × 10−5 | 2 | rs10512287 | 2.30 × 10−4 | 2 |
| BID | 22q11.21 | 11 | 3.11 × 10−22 | 3.39 × 10−10 | 2 | rs8190315 | 1.00 × 10−2 | 2 |
| DMBT1 | 10q26.13 | 11 | 3.79 × 10−18 | 1.85 × 10−7 | 2 | rs11523871 | 2.00 × 10−3 | 2 |
| FOXM1 | 12p13.33 | 8 | 1.67 × 10−6 | 2.57 × 10−24 | 2 | rs2074985 | 3.40 × 10−2 | 2 |
| MSH6 | 2p16.3 | 8 | 1.11 × 10−11 | 5.51 × 10−10 | 2 | rs3136337 | 3.39 × 10−2 | 2 |
| MTR | 1q43 | 4 | 7.78 × 10−10 | 7.42 × 10−3 | 2 | rs1805087 | 2.00 × 10−2 | 2 |
| DSEL | 18q22.1 | 2 | 2.37 × 10−14 | 4.19 × 10−19 | 2 | rs17827708 | 9.00 × 10−3 | 2 |
| FANCG | 9p13.3 | 2 | 1.85 × 10−16 | 8.14 × 10−14 | 2 | rs4986940 | 2.79 × 10−2 | 2 |
| EHMT2 | 6p21.33 | 99 | 6.80 × 10−20 | 4.19 × 10−12 | 1 | rs535586 | 1.00 × 10−2 | 2 |
| MCC | 5q22.2 | 45 | 3.79 × 10−18 | 1.41 × 10−17 | 1 | rs6890833 | 3.40 × 10−2 | 2 |
| PRDM2 | 1p36.21 | 31 | 4.55 × 10−18 | 1.41 × 10−8 | 1 | rs2235515 | 2.00 × 10−2 | 2 |
| POR | 7q11.23 | 20 | 5.42 × 10−21 | 2.25 × 10−13 | 1 | rs10262966 | 3.00 × 10−2 | 2 |
| KCNJ6 | 21q22.13 | 17 | 8.09 × 10−18 | 8.25 × 10−15 | 1 | rs4817896 | 2.40 × 10−2 | 2 |
| SORBS1 | 10q24.1 | 17 | 8.00 × 10−9 | 5.09 × 10−23 | 1 | rs10450393 | 1.00 × 10−2 | 2 |
| SHBG | 17p13.1 | 14 | 3.97 × 10−12 | 5.12 × 10−5 | 1 | rs858524 | 3.00 × 10−2 | 2 |
| VDR | 12q13.11 | 14 | 1.39 × 10−18 | 6.75 × 10−3 | 1 | rs731236 | 3.00 × 10−2 | 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Mamidi, T.K.K.; Zhang, L.; Hicks, C. Deconvolution of the Genomic and Epigenomic Interaction Landscape of Triple-Negative Breast Cancer. Cancers 2019, 11, 1692. https://doi.org/10.3390/cancers11111692
Wu J, Mamidi TKK, Zhang L, Hicks C. Deconvolution of the Genomic and Epigenomic Interaction Landscape of Triple-Negative Breast Cancer. Cancers. 2019; 11(11):1692. https://doi.org/10.3390/cancers11111692
Chicago/Turabian StyleWu, Jiande, Tarun Karthik Kumar Mamidi, Lu Zhang, and Chindo Hicks. 2019. "Deconvolution of the Genomic and Epigenomic Interaction Landscape of Triple-Negative Breast Cancer" Cancers 11, no. 11: 1692. https://doi.org/10.3390/cancers11111692
APA StyleWu, J., Mamidi, T. K. K., Zhang, L., & Hicks, C. (2019). Deconvolution of the Genomic and Epigenomic Interaction Landscape of Triple-Negative Breast Cancer. Cancers, 11(11), 1692. https://doi.org/10.3390/cancers11111692