Overview of Immune Checkpoint Inhibitors Therapy for Hepatocellular Carcinoma, and The ITA.LI.CA Cohort Derived Estimate of Amenability Rate to Immune Checkpoint Inhibitors in Clinical Practice

 ,
, 
 , , , ,
, , , ,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Cancer Immunotherapy
3. Rationale of Immune Checkpoint Blockade in Hepatocellular Carcinoma
4. Strategies for Patients Selection
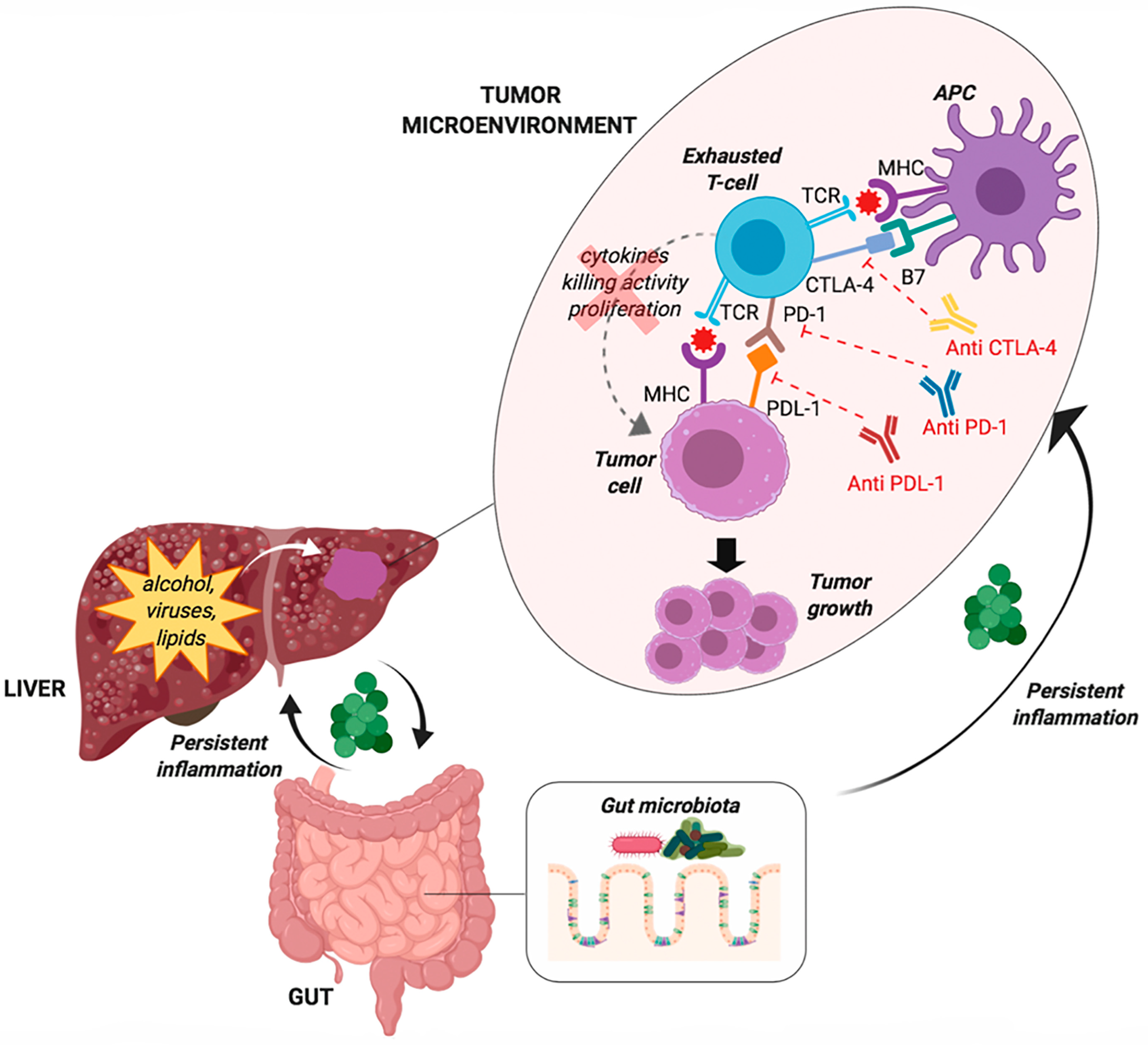
4.1. T-Cell Exhaustion
4.2. The Gut Microbiota
5. Outcome of Current Studies on Immunotherapy in Patients with HCC
5.1. Efficacy
5.2. Results of Monotherapy with Checkpoint Inhibitors in HCC
5.2.1. Tremelimumab
5.2.2. Nivolumab
5.2.3. Pembrolizumab
5.2.4. Camrelizumab
5.2.5. Tislelizumab
5.2.6. Durvalumab
5.3. Combination of Two Immune Checkpoint Inhibitors
5.3.1. Durvalumab plus Tremelimumab
5.3.2. Nivolumab plus Ipilimumab
5.4. Combination of Immune Checkpoint Inhibitors with Molecular-Targeted Agents
5.4.1. Atezolizumab plus Bevacizumab
5.4.2. Pembrolizumab plus Lenvatinib
5.4.3. Other Combinations
5.5. Immune Checkpoint Inhibitors as Neo-Adjuvant or Adjuvant Therapy, or in Combination with Local Treatments
6. Liver Involvement in Immune-Related-Adverse Events
7. Assessment of Treatment Response: The iRECIST Criteria
8. Rationale Underlying the Use of the ITA.LI.CA Database to Assess Real-Life Applicability of Checkpoint Inhibitors
8.1. First-Line Scenario
8.2. Second-Line Scenario
9. Conclusions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, D.L.; Kanwal, F.; Serag, H.B.E. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin. Gastroenterol. Hepatol. 2012, 10, 1342–1359. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology 2018, 155, 1828–1837. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Chronic inflammation due to the various underlying etiologies is a mainstay of HCC development. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Kimmoun, E.; Legrand, A.; Sauvanet, A.; Degott, C.; Lardeux, B.; Bernuau, D. Activation of NF-kappa B, AP-1 and STAT transcription factors is a frequent and early event in human hepatocellular carcinomas. J. Hepatol. 2002, 37, 63–71. [Google Scholar] [CrossRef]
- He, G.; Yu, G.Y.; Temkin, V.; Ogata, H.; Kuntzen, C.; Sakurai, T.; Sieghart, W.; Peck-Radosavljevic, M.; Leffert, H.L.; Karin, M. Hepatocyte IKKβ/NF-κB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 2010, 17, 286–297. [Google Scholar] [CrossRef]
- Kanda, T.; Goto, T.; Hirotsu, Y.; Moriyama, M.; Omata, M. Molecular mechanisms driving progression of liver cirrhosis towards hepatocellular carcinoma in chronic hepatitis B and C infections: A review. Int. J. Mol. Sci. 2019, 20, 1358. [Google Scholar] [CrossRef]
- Bitzer, M.; Horger, M.; Giannini, E.G.; Ganten, T.M.; Wörns, M.A.; Siveke, J.T.; Dollinger, M.M.; Gerken, G.; Scheulen, M.E.; Wege, H.; et al. Resminostat plus sorafenib as second-line therapy of advanced hepatocellular carcinoma—The SHELTER study. J. Hepatol. 2016, 65, 280–288. [Google Scholar] [CrossRef]
- Klener, P., Jr.; Otahal, P.; Lateckova, L.; Klener, P. Immunotherapy approaches in cancer treatment. Curr. Pharm. Biotechnol. 2015, 16, 771–781. [Google Scholar] [CrossRef]
- Ventola, C.L. Cancer Immunotherapy, Part 1: Current Strategies and Agents. Pharm. Ther. 2017, 42, 375–383. [Google Scholar]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–195. [Google Scholar] [CrossRef] [PubMed]
- Alatrash, G.; Jakher, H.; Stafford, P.D.; Mittendorf, E.A. Cancer immunotherapies, their safety and toxicity. Expert Opin. Drug Saf. 2013, 12, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Nault, J.C.; Roberts, L.R.; Zucman-Rossi, J. Genomic medicine and implications for hepatocellular carcinoma prevention and therapy. Gastroenterology 2019, 156, 492–509. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology 2015, 149, 1226–1239. [Google Scholar] [CrossRef]
- Nault, J.C.; Villanueva, A. Intratumor molecular and phenotypic diversity in hepatocellular carcinoma. Clin. Cancer Res. 2015, 21, 1786–1788. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. RESORCE investigators. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Sprengers, D.; Boor, P.P.C.; Doukas, M.; Schutz, H.; Mancham, S.; Pedroza-Gonzalez, A.; Polak, W.G.; de Jonge, J.; Gaspersz, M.; et al. Antibodies against immune checkpoint molecules restore functions of tumor-infiltrating T cells in hepatocellular carcinomas. Gastroenterology 2017, 153, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Shi, M.; Zeng, Z.; Qi, R.Z.; Liu, Z.W.; Zhang, J.Y.; Yang, Y.P.; Tien, P.; Wang, F.S. PD-1 and PD-L1 upregulation promotes CD8+ Tcell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int. J. Cancer 2011, 128, 887–896. [Google Scholar] [CrossRef]
- Kuang, D.M.; Zhao, Q.; Peng, C.; Xu, J.; Zhang, J.P.; Wu, C.; Zheng, L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med. 2009, 206, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Chen, Z.; Yang, Y.; Jiang, Z.; Gu, Y.; Liu, Y.; Lin, C.; Pan, Z.; Yu, Y.; Jiang, M.; et al. Human CD14+ CTLA-4+ regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine-2,3-dioxygenase production in hepatocellular carcinoma. Hepatology 2014, 59, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Kalathil, S.; Lugade, A.A.; Miller, A.; Iyer, R.; Thanavala, Y. Higher frequencies of GARP+ CTLA-4+ Foxp3+ T regulatory cells and myeloid-derived suppressor cells in hepatocellular carcinoma patients are associated with impaired T-cell functionality. Cancer Res. 2013, 73, 2435–2444. [Google Scholar] [CrossRef]
- Wu, X.; Gu, Z.; Chen, Y.; Chen, B.; Chen, W.; Weng, L.; Liu, X. Application of PD-1 blockade in cancer immunotherapy. Comput. Struct. Biotechnol. J. 2019, 17, 661–674. [Google Scholar] [CrossRef]
- Wu, K.; Kryczek, I.; Chen, L.; Zou, W.; Welling, T.H. Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7–H1/programmed death-1 interactions. Cancer Res. 2009, 69, 8067–8075. [Google Scholar] [CrossRef]
- Wang, B.J.; Bao, J.J.; Wang, J.Z.; Wang, Y.; Jiang, M.; Xing, M.Y.; Zhang, W.G.; Qi, J.Y.; Roggendorf, M.; Lu, M.J.; et al. Immunostaining of PD-1/PD-Ls in liver tissues of patients with hepatitis and hepatocellular carcinoma. World J. Gastroenterol. 2011, 17, 3322–3329. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, X.Y.; Qiu, S.J.; Yamato, I.; Sho, M.; Nakajima, Y.; Zhou, J.; Li, B.Z.; Shi, Y.H.; Xiao, Y.S.; et al. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin. Cancer Res. 2009, 15, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Unitt, E.; Marshall, A.; Gelson, W.; Rushbrook, S.M.; Davies, S.; Vowler, S.L.; Morris, L.S.; Coleman, N.; Alexander, G.J. Tumour lymphocytic infiltrate and recurrence of hepatocellular carcinoma following liver transplantation. J. Hepatol. 2006, 45, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Nakashima, O.; Kutami, R.; Yamamoto, O.; Kojiro, M. Clinicopathological study on hepatocellular carcinoma with lymphocytic infiltration. Hepatology 1998, 27, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Flecken, T.; Schmidt, N.; Hild, S.; Gostick, E.; Drognitz, O.; Zeiser, R.; Schemmer, P.; Bruns, H.; Eiermann, T.; Price, D.A.; et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma. Hepatology 2014, 59, 1415–1426. [Google Scholar] [CrossRef] [PubMed]
- Kurebayashi, Y.; Ojima, H.; Tsujikawa, H.; Kubota, N.; Maehara, J.; Abe, Y.; Kitago, M.; Shinoda, M.; Kitagawa, Y.; Sakamoto, M. Landscape of immune microenvironment in hepatocellular carcinoma and its additional impact on histological and molecular classification. Hepatology 2018, 68, 1025–1041. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zheng, L.; Yoo, J.K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 2017, 169, 1342–1356. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Bald, T.; Landsberg, J.; Lopez-Ramos, D.; Renn, M.; Glodde, N.; Jansen, P.; Gaffal, E.; Steitz, J.; Tolba, R.; Kalinke, U.; et al. Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov. 2014, 4, 674–687. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Long, G.V.; Scolyer, R.A.; Teng, M.W.; Smyth, M.J. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 2017, 52, 71–81. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; de Moura, M.C.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an immune-specific class of hepatocellular carcinoma, based on molecular features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.J.; Zhu, A.X.; Bauer, T.M.; Choueiri, T.K.; Drilon, A.; Voss, M.H.; Fuchs, C.S.; Abou-Alfa, G.K.; Wijayawardana, S.R.; Wang, X.A.; et al. A Phase 1b/2 study of ramucirumab in combination with emibetuzumab in patients with advanced cancer. Clin. Cancer Res. 2019, 25, 2116–2126. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Meyer, T.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Sangro, B.; Crocenzi, T.S.; Welling, T.H.; Iñarrairaegui, M.; Prieto, J.; Fuertes, C.; Feely, W.; Anderson, J.; Grasela, D.M.; Wigginton, J.M.; et al. Phase I dose escalation study of nivolumab (anti-PD-1; BMS-936558; ONO-4538) in patients (pts) with advanced hepatocellular carcinoma (HCC) with or without chronic viral hepatitis. J. Clin. Oncol. 2013, 31 (Suppl. 15). [Google Scholar] [CrossRef]
- Knolle, P.A.; Thimme, R. Hepatic immune regulation and its involvement in viral hepatitis infection. Gastroenterology 2014, 146, 1193–1207. [Google Scholar] [CrossRef]
- Sangro, B.; Gomez-Martin, C.; de la Mata, M.; Iñarrairaegui, M.; Garralda, E.; Barrera, P.; Riezu-Boj, J.I.; Larrea, E.; Alfaro, C.; Sarobe, P.; et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 2013, 59, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; ElGindi, M.; et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.S.; Cox, M.A.; Zajac, A.J. T-cell exhaustion: Characteristics, causes and conversion. Immunology 2010, 129, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.D.; Song, G.W.; Park, S.; Jung, M.K.; Kim, M.H.; Kang, H.J.; Yoo, C.; Yi, K.; Kim, K.H.; Eo, S.; et al. Association between expression level of PD1 by tumor-infiltrating CD8+ T cells and features of hepatocellular carcinoma. Gastroenterology 2018, 155, 1936–1950. [Google Scholar]
- Feun, L.G.; Li, Y.Y.; Wu, C.; Wangpaichitr, M.; Jones, P.D.; Richman, S.P.; Madrazo, B.; Kwon, D.; Garcia-Buitrago, M.; Martin, P.; et al. Phase 2 study of pembrolizumab and circulating biomarkers to predict anticancer response in advanced, unresectable hepatocellular carcinoma. Cancer 2019, 125, 3603–3614. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Koeppen, H.; Astarita, J.L.; Cubas, R.; Jhunjhunwala, S.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Blander, J.M.; Longman, R.S.; Iliev, I.D.; Sonnenberg, G.F.; Artis, D. Regulation of inflammation by microbiota interactions with the host. Nat. Immunol. 2017, 18, 851–860. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef]
- Routy, B.; le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef]
- Li, J.; Sung, C.Y.; Lee, N.; Ni, Y.; Pihlajamäki, J.; Panagiotou, G.; El-Nezami, H. Probiotics modulated gut microbiota suppresses hepatocellular carcinoma growth in mice. Proc. Natl. Acad. Sci. USA 2016, 113, E1306–E1315. [Google Scholar] [CrossRef]
- Yu, L.X.; Yan, H.X.; Liu, Q.; Yang, W.; Wu, H.P.; Dong, W.; Tang, L.; Lin, Y.; He, Y.Q.; Zou, S.S.; et al. Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology 2010, 52, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Dapito, D.H.; Mencin, A.; Gwak, G.Y.; Pradere, J.P.; Jang, M.K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Yu, L.X.; Yang, W.; Tang, L.; Lin, Y.; Wu, H.; Zhai, B.; Tan, Y.X.; Shan, L.; Liu, Q.; et al. Profound impact of gut homeostasis on chemically-induced pro-tumorigenic inflammation and hepatocarcinogenesis in rats. J. Hepatol. 2012, 57, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; del Chierico, F.; Sanguinetti, M.; Morelli, D.; Paroni Sterbini, F.; Petito, V.; et al. Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology 2019, 69, 107–120. [Google Scholar] [CrossRef]
- Okusaka, T.; Ikeda, M. Immunotherapy for hepatocellular carcinoma: Current status and future perspectives. ESMO Open 2018, 3 (Suppl. 1), e000455. [Google Scholar] [CrossRef]
- Sangro, B.; Park, J.W.; Cruz, C.M.D.; Anderson, J.; Lang, L.; Neely, J.; Shaw, J.W.; Cheng, A.L. A randomized, multicenter, phase 3 study of nivolumab vs. sorafenib as first-line treatment in patients (pts) with advanced hepatocellular carcinoma (HCC): CheckMate-459. J. Clin. Oncol. 2016, 34, TPS4147. [Google Scholar] [CrossRef]
- Bristol-Myers Squibb Announces Results from CheckMate-459 Study Evaluating Opdivo (Nivolumab) as a First-Line Treatment for Patients with Unresectable Hepatocellular Carcinoma. Available online: https://news.bms.com/press-release/bmy/bristol-myers-squibb-announces-results-checkmate-459-study-evaluating-opdivo-nivol (accessed on 13 August 2019).
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Finn, R.S.; Chan, S.L.; Zhu, A.X.; Knox, J.J.; Cheng, A.L.; Siegel, A.B.; Bautista, O.; Kudo, M. Phase 3, randomized study of pembrolizumab (pembro) vs best supportive care (BSC) for second-line advanced hepatocellular carcinoma (HCC): KEYNOTE-240. J. Clin. Oncol. 2017, 35 (Suppl. 15), TPS4143. [Google Scholar] [CrossRef]
- Qin, S.K.; Ren, Z.G.; Meng, Z.Q.; Chen, Z.D.; Chai, X.L.; Xiong, J.P.; Bai, Y.X.; Yang, L.; Zhu, H.; Fang, W.J.; et al. A randomized multicentered phase II study to evaluate SHR-1210 (PD-1 antibody) in subjects with advanced hepatocellular carcinoma (HCC) who failed or intolerable to prior systemic treatment. Ann. Oncol. 2018, 29 (Suppl. 8). [Google Scholar] [CrossRef]
- Qin, S.; Finn, R.S.; Kudo, M.; Meyer, T.; Vogel, A.; Ducreux, M.; Macarulla, T.M.; Tomasello, G.; Boisserie, F.; Hou, J.; et al. Rationale 301 study: Tislelizumab versus sorafenib as first-line treatment for unresectable hepatocellular carcinoma. Future Oncol. 2019, 15. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Segal, N.H.; Jaeger, D.; Lee, K.H.; Marshall, J.; Joseph, A.S.; Butler, M.; Sandborn, R.E.; Nemunaitis, J.J.; Carlson, C.A.; et al. Safety and clinical activity of durvalumab monotherapy in patients with hepatocellular carcinoma (HCC). J. Clin. Oncol. 2017, 35 (Suppl. 15), 4071. [Google Scholar] [CrossRef]
- Kelley, R.K.; Abou-Alfa, G.K.; Bendell, J.C.; Kim, T.Y.; Borad, M.J.; Yong, W.P.; Morse, M.; Kang, Y.K.; Rebelatto, M.; Makowsky, M.; et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. J. Clin. Oncol. 2017, 35 (Suppl. 15), 4073. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Chan, S.L.; Furuse, J.; Galle, P.R.; Kelley, R.K.; Qin, A.; Armstrong, J.; Darilay, A.; Vlahovic, G.; Negro, A.; et al. A randomized, multi center phase 3 study of durvalumab (D) and tremelimumab (T) as first-line treatment in patients with unresectable hepatocellular carcinoma (HCC): HIMALAYA study. J. Clin. Oncol. 2018, 36 (Suppl. 15), TPS4144. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.M.; Matilla, A.; et al. Nivolumab (NIVO) + ipilimumab (IPI) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Results from CheckMate 040. J. Clin. Oncol. 2019, 37 (Suppl. 15), 4012. [Google Scholar] [CrossRef]
- Finn, R.S.; Ducreux, M.; Qin, S.; Galle, P.R.; Zhu, A.X.; Ikeda, M.; Kim, T.Y.; Xu, D.Z.; Verret, W.; Liu, J.; et al. IMbrave150: A randomized phase III study of atezolizumab plus bevacizumab vs sorafenib in locally advanced or metastatic hepatocellular carcinoma. J. Clin. Oncol. 2018, 36 (Suppl. 15), TPS4141. [Google Scholar] [CrossRef]
- Roche’s Tecentriq in Combination with Avastin Increased Overall Survival and Progression-Free Survival in People with Unresectable Hepatocellular Carcinoma. Available online: https://www.roche.com/media/releases/med-cor-2019-10-21.htm (accessed on 28 October 2019).
- Ikeda, M.; Sung, M.W.; Kudo, M.; Kobayashi, M.; Baron, A.D.; Finn, R.S.; Kanek, S.; Zhu, A.X.; Kubota, T.; Kraljevic, S.; et al. A phase 1b trial of lenvatinib (LEN) plus pembrolizumab (PEM) in patients (pts) with unresectable hepatocellular carcinoma (uHCC). J. Clin. Oncol. 2018, 36 (Suppl. 15), 4076. [Google Scholar] [CrossRef]
- Weber, J.; Mandala, M.; del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant nivolumab versus ipilimumab in resected Stage III or IV melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef]
- Haanen, J.B.A.G.; Carbonnel, F.; Robert, C.; Kerr, K.M.; Peters, S.; Larkin, J.; Jordan, K. ESMO Guidelines Committee Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv264–iv266. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- O’Day, S.J.; Maio, M.; Chiarion-Sileni, V.; Gajewski, T.F.; Pehamberger, H.; Bondarenko, I.N.; Queirolo, P.; Lundgren, L.; Mikhailov, S.; Roman, L.; et al. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: A multicenter single-arm phase II study. Ann. Oncol. 2010, 21, 1712–1717. [Google Scholar] [CrossRef]
- Daly, L.E.; Power, D.G.; O’Reilly, Á.; Donnellan, P.; Cushen, S.J.; O’Sullivan, K.; Twomey, M.; Woodlock, D.P.; Redmond, H.P.; Ryan, A.M. The impact of body composition parameters on ipilimumab toxicity and survival in patients with metastatic melanoma. Br. J. Cancer 2017, 116, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.M.; Rowland, A.; Kichenadasse, G.; Wiese, M.D.; Gurney, H.; McKinnon, R.A.; Karapetis, C.S.; Sorich, M.J. Predicting response and toxicity to immune checkpoint inhibitors using routinely available blood and clinical markers. Br. J. Cancer 2017, 117, 913–920. [Google Scholar] [CrossRef] [PubMed]
- De Martin, E.; Michot, J.M.; Papouin, B.; Champiat, S.; Mateus, C.; Lambotte, O.; Roche, B.; Antonini, T.M.; Coilly, A.; Laghouati, S.; et al. Characterization of liver injury induced by cancer immunotherapy using immune checkpoint inhibitors. J. Hepatol. 2018, 68, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.J.; Duckworth, A.M.; Davies, S.E.; Mells, G.F.; Brais, R.; Harden, S.V.; Parkinson, C.A.; Corrie, P.G. Severe steroid-resistant anti-PD1 T-cell checkpoint inhibitor-induced hepatotoxicity driven by biliary injury. ESMO Open 2017, 2, e000268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seymour, L.; Bogaerts, J.; Perrone, A.; Ford, R.; Schwartz, L.H.; Mandrekar, S.; Lin, N.U.; Litière, S.; Dancey, J.; Chen, A.; et al. iRECIST: Guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017, 18, e143–e152. [Google Scholar] [CrossRef]
- Giannini, E.G.; Bucci, L.; Garuti, F.; Brunacci, M.; Lenzi, B.; Valente, M.; Caturelli, E.; Cabibbo, G.; Piscaglia, F.; Virdone, R.; et al. Patients with advanced hepatocellular carcinoma need a personalized management: A lesson from clinical practice. Hepatology 2018, 67, 1784–1796. [Google Scholar] [CrossRef]
- Bucci, L.; Garuti, F.; Lenzi, B.; Pecorelli, A.; Farinati, F.; Giannini, E.G.; Granito, A.; Ciccarese, F.; Rapaccini, G.L.; di Marco, M.; et al. The evolutionary scenario of hepatocellular carcinoma in Italy: An update. Liver Int. 2017, 37, 259–270. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| NCT | Phase | Drug | Procedure | Line of Treatment | Primary End-Point | Estimated Study Completion Date | Company Conducting the Trial |
|---|---|---|---|---|---|---|---|
| NCT03298451 | III | Tremelilumab (+Durvalumab) vs Sorafenib | - | 1 | OS | 06/2021 | Astra Zeneca |
| NCT02576509 | III | Nivolumab vs Sorafenib | - | 1 | OS | 07/2020 | BMS |
| NCT03412773 | III | Tislelizumab vs Sorafenib | - | 1 | OS | 05/2022 | BeiGene |
| NCT03062358 | III | Pembrolizumab vs placebo | - | 2 | OS | 01/2022 | MSD |
| NCT02702401 | III | Pembrolizumab vs placebo | - | 2 | OS, PFS | 06/2020 | MSD |
| NCT02702414 | II | Pembrolizumab | - | 1-2 | ORR | 05/2021 | MSD |
| NCT02519348 | II | Tremelilumab (+Durvalumab) | - | 2 | Safety, DLT | 04/2021 | MedImmune LLC |
| NCT03163992 | II | Pembrolizumab | - | 2 | ORR | 12/2020 | Samsung Medical Center |
| NCT02658019 | II | Pembrolizumab | - | >2 | DCR, Safety | 11/2020 | Lynn Feun |
| NCT03389126 | II | Avelumab | - | >2 | ORR | 03/2020 | Seoul National University Hospital |
| NCT03419897 | II | Tislelizumab | - | >2 | ORR | 09/2021 | BeiGene |
| NCT03033446 | II | Nivolumab | SIRT | Any | ORR | 12/2019 | National Cancer Centre, Singapore |
| ign="middle" style="border-bottom:solid thin">1 | ORR | 11/2020 | Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins | ||||
| NCT02821754 | II | Tremelilumab | Local ablation | 1 | PFS | 04/2021 | National Cancer Institute (NCI) |
| NCT03630640 | II | Nivolumab | Electroporation | 1 | RFS | 09/2020 | Assistance Publique—Hôpitaux de Paris |
| NCT03482102 | II | Tremelilumab (+Durvalumab) | BRT | 2 | ORR | 10/2025 | Massachusetts General Hospital |
| NCT03316872 | II | Pembrolizumab | SBRT | 2 | ORR | 04/2022 | University Health Network, Toronto |
| NCT01658878 | IB/II | Nivolumab vs Sorafenib | - | 1 | ORR | 12/2019 | BMS |
| NCT02423343 | IB/II | Nivolumab + Galunisertib | - | 2 | MTD, Safety | 12/2019 | Eli Lilly and Company |
| NCT01658878 | IB/II | Nivolumab + Ipilimumab | - | >2 | ORR | 12/2019 | BMS |
| NCT02940496 | I/II | Pembrolizumab | - | 2 | Biomarkers | 12/2019 | M.D. Anderson Cancer Center |
| NCT03397654 | IB | Pembrolizumab | TACE | 1 | Safety | 12/2020 | Imperial College London |
| NCT02837029 | I | Nivolumab | SIRT | Any | MTD | 07/2020 | Northwestern University |
| NCT03099564 | I | Pembrolizumab | SIRT | 1 | PFS | 01/2020 | Autumn McRee, MD |
| NCT03143270 | I | Nivolumab | debTACE | 1 | Safety | 04/2020 | Memorial Sloan Kettering Cancer Center |
| NCT03203304 | I | Nivolumab/Ipilimumab | SBRT | 1 | Safety | 08/2020 | University of Chicago |
| NCT01853618 | I | Tremelilumab | Local Ablation | 1 | Safety | 12/2020 | National Cancer Institute (NCI) |
| ITA.LI.CA Database | Number of HCCs = 11,483 (including recurrences) | |
| (A) First-step removal | 1. HCC diagnosis before 01/01/2008 = 3144 | |
| 2. HCC recurrence = 4453 | ||
| Number of patients = 3886 (01/01/2008-31/12/2016) | ||
| (B) Second step removal | Missing data = 1403 | |
| Examined population = 2483 (100.0%) | ||
| (C) Third step removal | Nivolumab | Pembrolizumab |
|
| |
| Final population = 525/2483 (21.1%) | Final population = 268/2483 (10.8%) | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giannini, E.G.; Aglitti, A.; Borzio, M.; Gambato, M.; Guarino, M.; Iavarone, M.; Lai, Q.; Levi Sandri, G.B.; Melandro, F.; Morisco, F.; et al. Overview of Immune Checkpoint Inhibitors Therapy for Hepatocellular Carcinoma, and The ITA.LI.CA Cohort Derived Estimate of Amenability Rate to Immune Checkpoint Inhibitors in Clinical Practice. Cancers 2019, 11, 1689. https://doi.org/10.3390/cancers11111689
Giannini EG, Aglitti A, Borzio M, Gambato M, Guarino M, Iavarone M, Lai Q, Levi Sandri GB, Melandro F, Morisco F, et al. Overview of Immune Checkpoint Inhibitors Therapy for Hepatocellular Carcinoma, and The ITA.LI.CA Cohort Derived Estimate of Amenability Rate to Immune Checkpoint Inhibitors in Clinical Practice. Cancers. 2019; 11(11):1689. https://doi.org/10.3390/cancers11111689
Chicago/Turabian StyleGiannini, Edoardo G., Andrea Aglitti, Mauro Borzio, Martina Gambato, Maria Guarino, Massimo Iavarone, Quirino Lai, Giovanni Battista Levi Sandri, Fabio Melandro, Filomena Morisco, and et al. 2019. "Overview of Immune Checkpoint Inhibitors Therapy for Hepatocellular Carcinoma, and The ITA.LI.CA Cohort Derived Estimate of Amenability Rate to Immune Checkpoint Inhibitors in Clinical Practice" Cancers 11, no. 11: 1689. https://doi.org/10.3390/cancers11111689
APA StyleGiannini, E. G., Aglitti, A., Borzio, M., Gambato, M., Guarino, M., Iavarone, M., Lai, Q., Levi Sandri, G. B., Melandro, F., Morisco, F., Ponziani, F. R., Rendina, M., Russo, F. P., Sacco, R., Viganò, M., Vitale, A., Trevisani, F., & on behalf of the Associazione Italiana per lo Studio del Fegato (AISF) HCC Special Interest Group. (2019). Overview of Immune Checkpoint Inhibitors Therapy for Hepatocellular Carcinoma, and The ITA.LI.CA Cohort Derived Estimate of Amenability Rate to Immune Checkpoint Inhibitors in Clinical Practice. Cancers, 11(11), 1689. https://doi.org/10.3390/cancers11111689