The Metabolic Inhibitor CPI-613 Negates Treatment Enrichment of Ovarian Cancer Stem Cells



Abstract
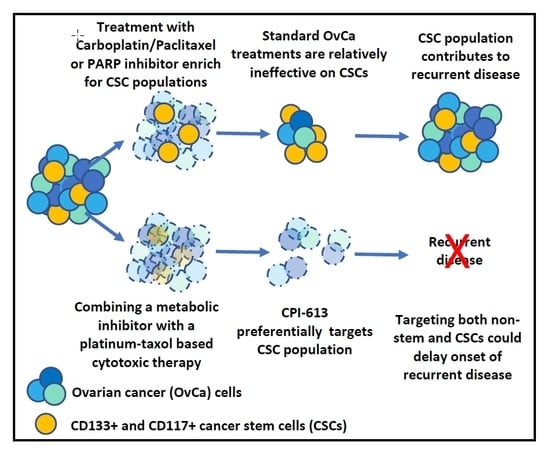
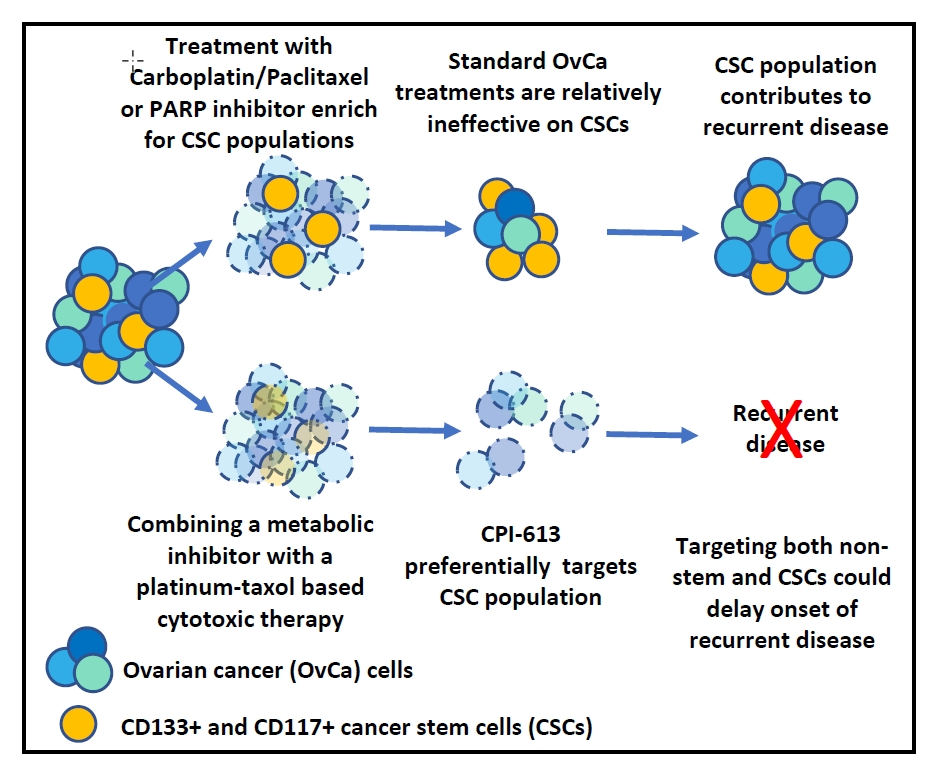{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. CD133 and CD117 Marker Expression in OvCa Cell Lines Identifies a Cell Population with Stem-Like Features
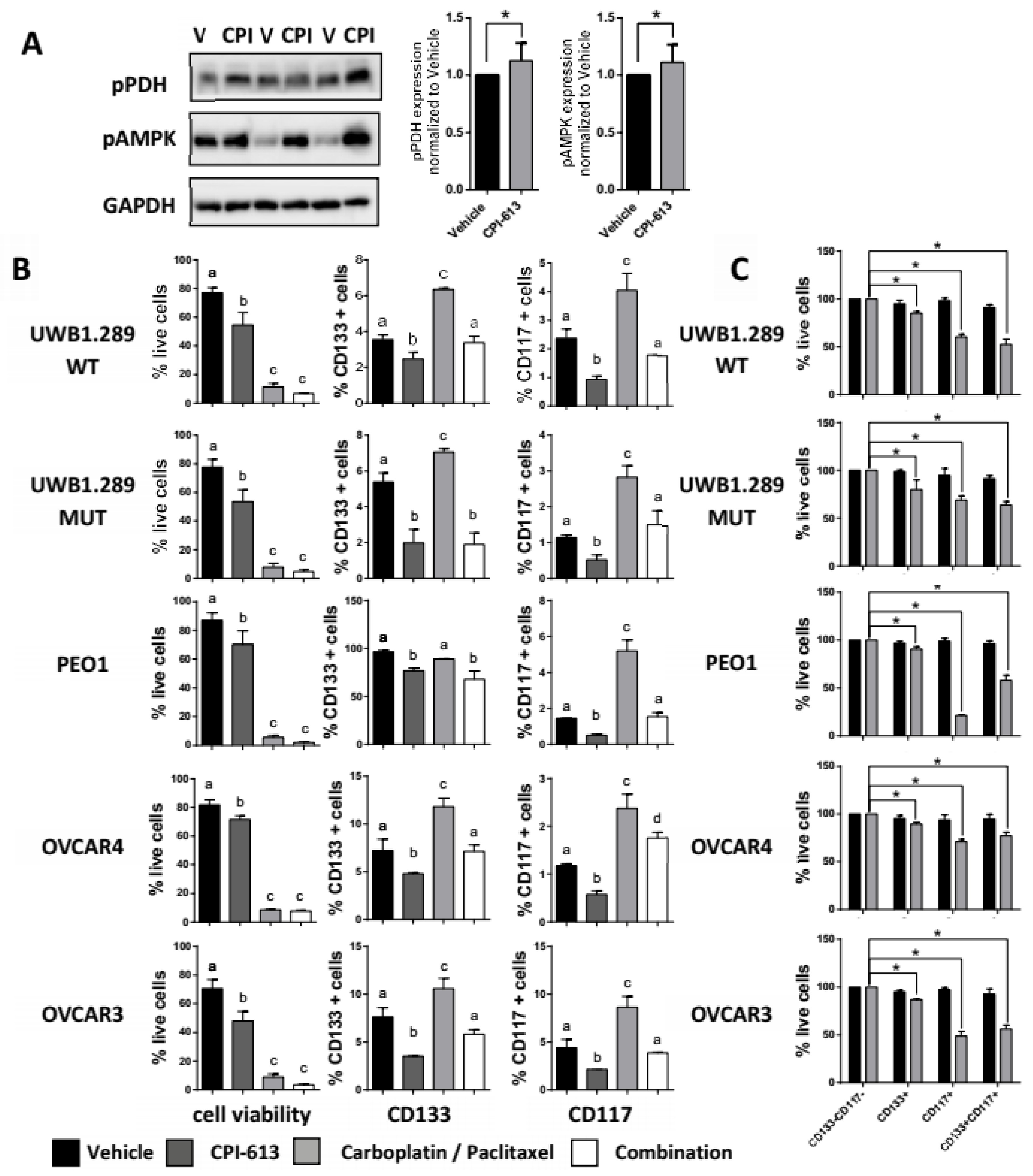
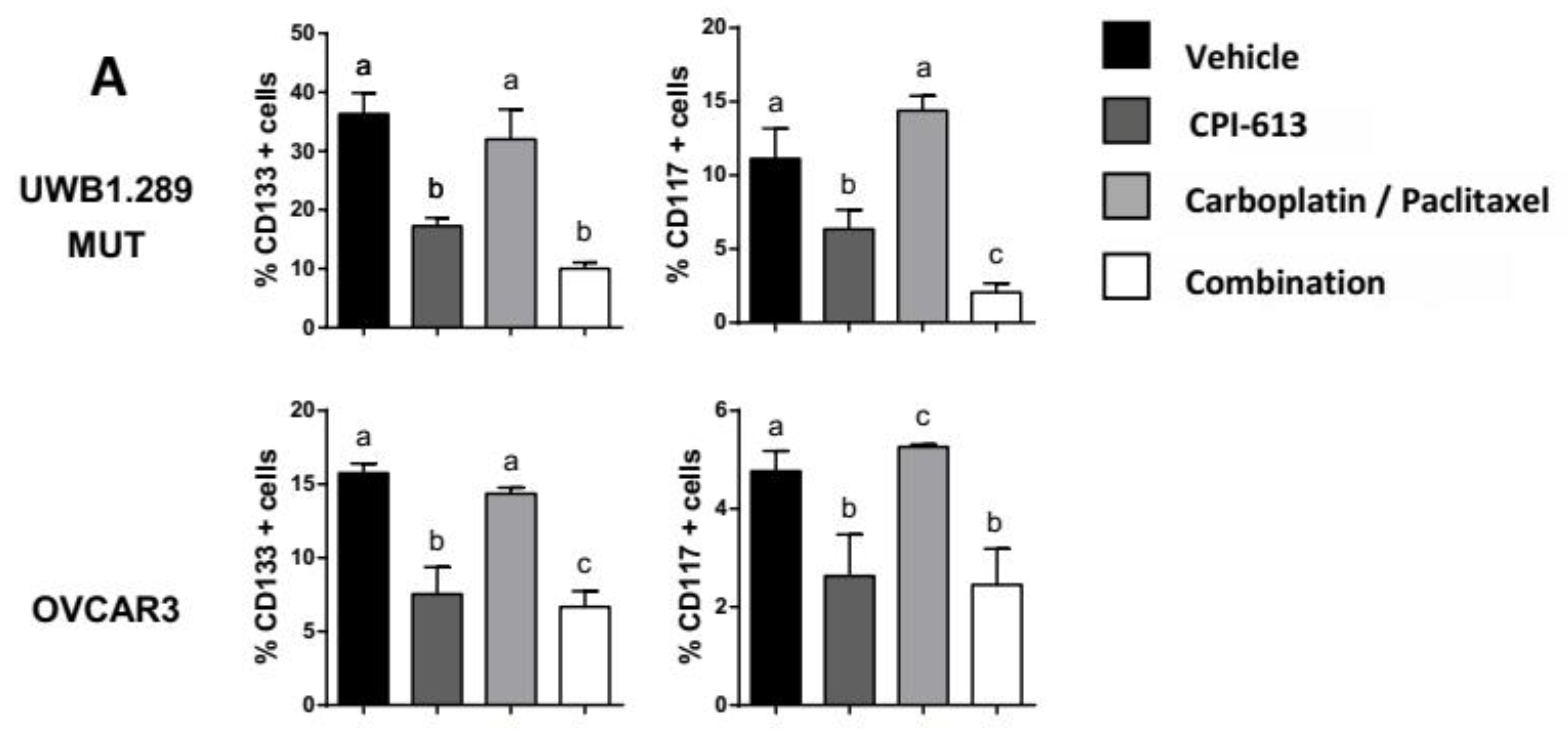
2.2. CPI-613 In Vitro Induces a Decrease in CD133 and CD117 Positive Cell Frequency
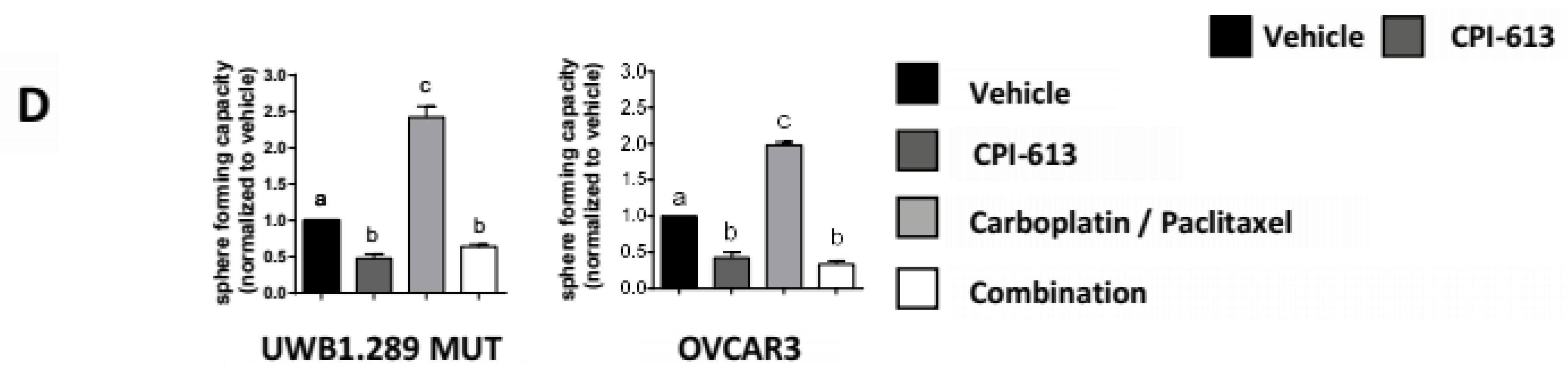
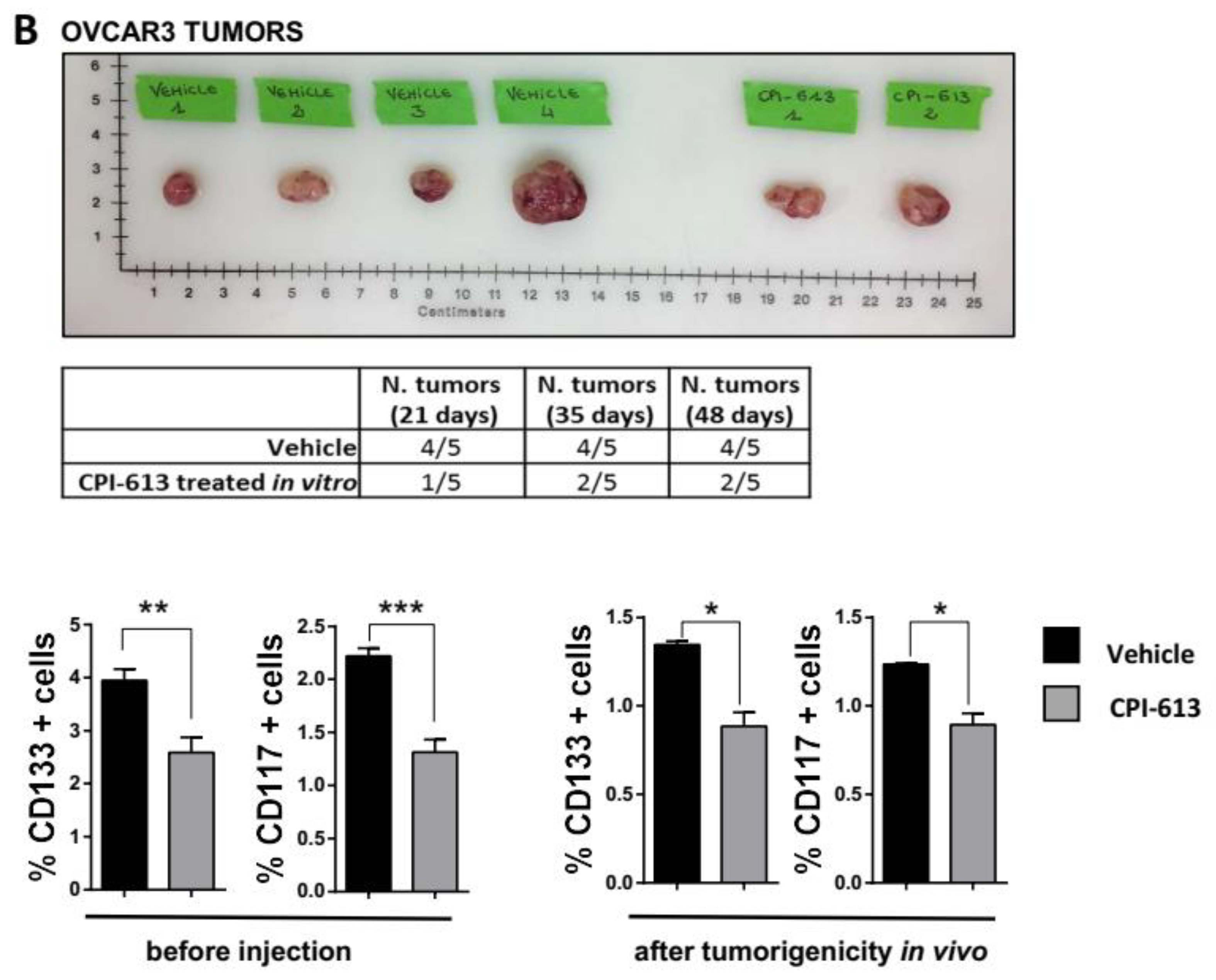
2.3. CPI-613 Treatment Negatively Impacts CSC-Rich Spheres and Results in a Decrease in Tumorigenicity In Vivo
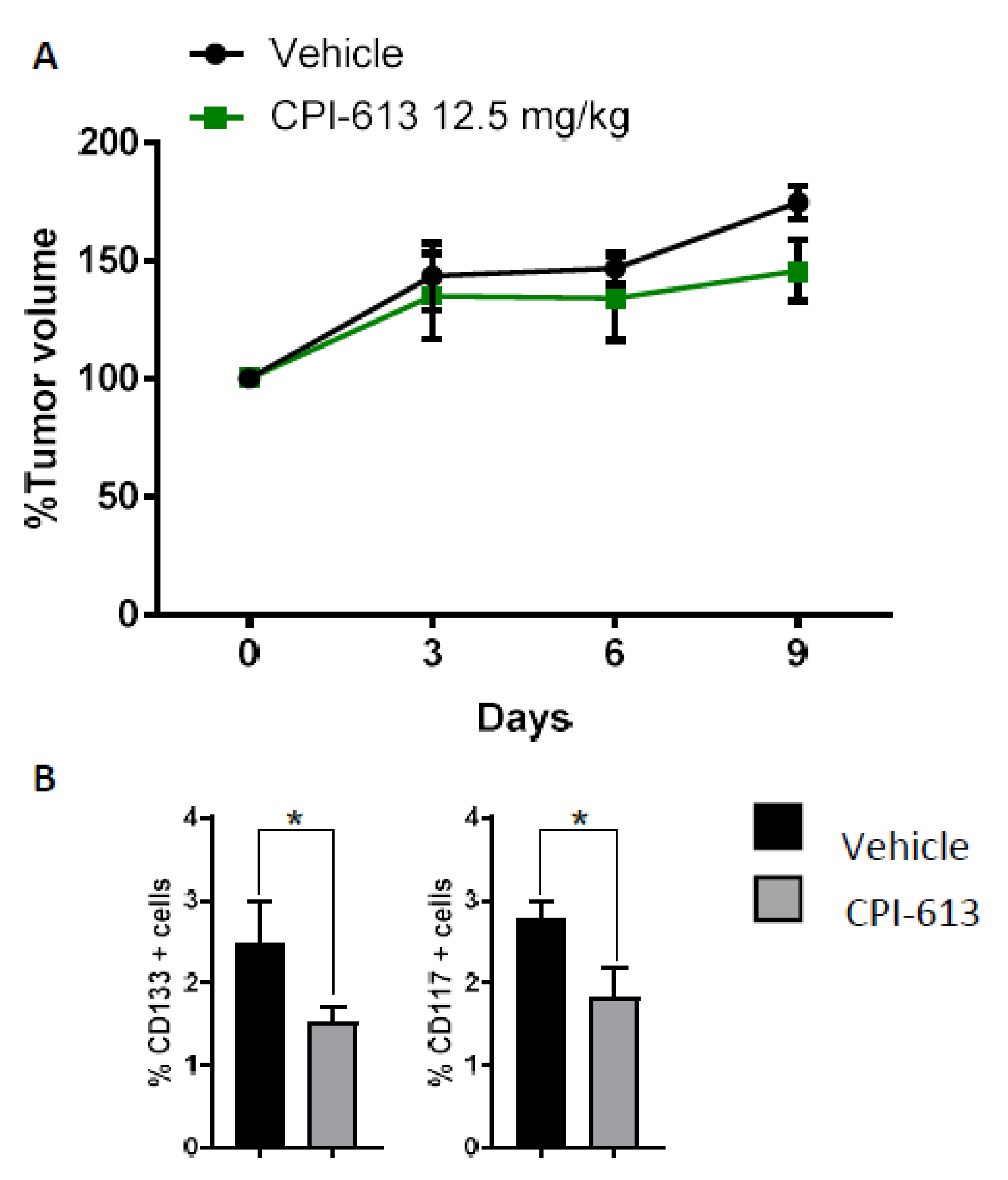
2.4. CPI-613 Treatment In Vivo Induces a Decrease in CD133+ and CD117+ Cell Frequency
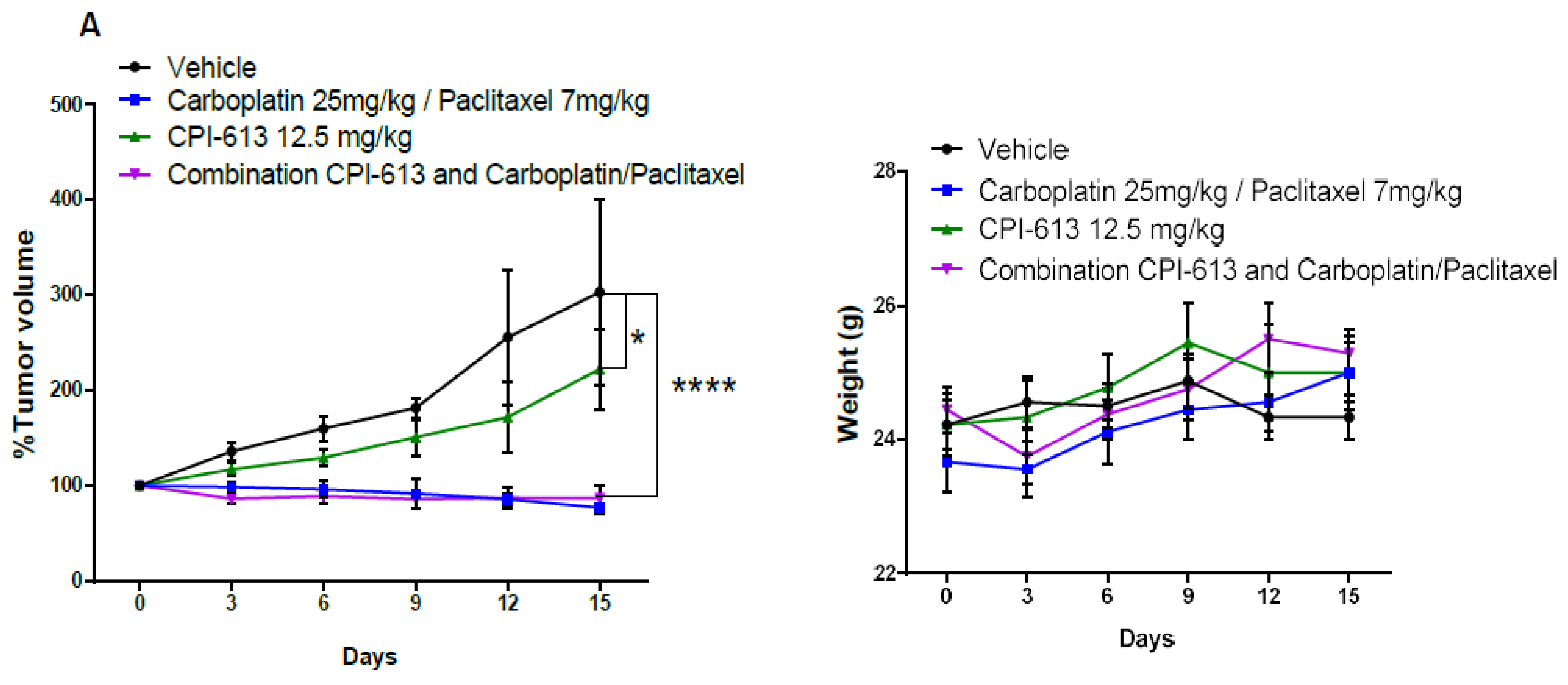
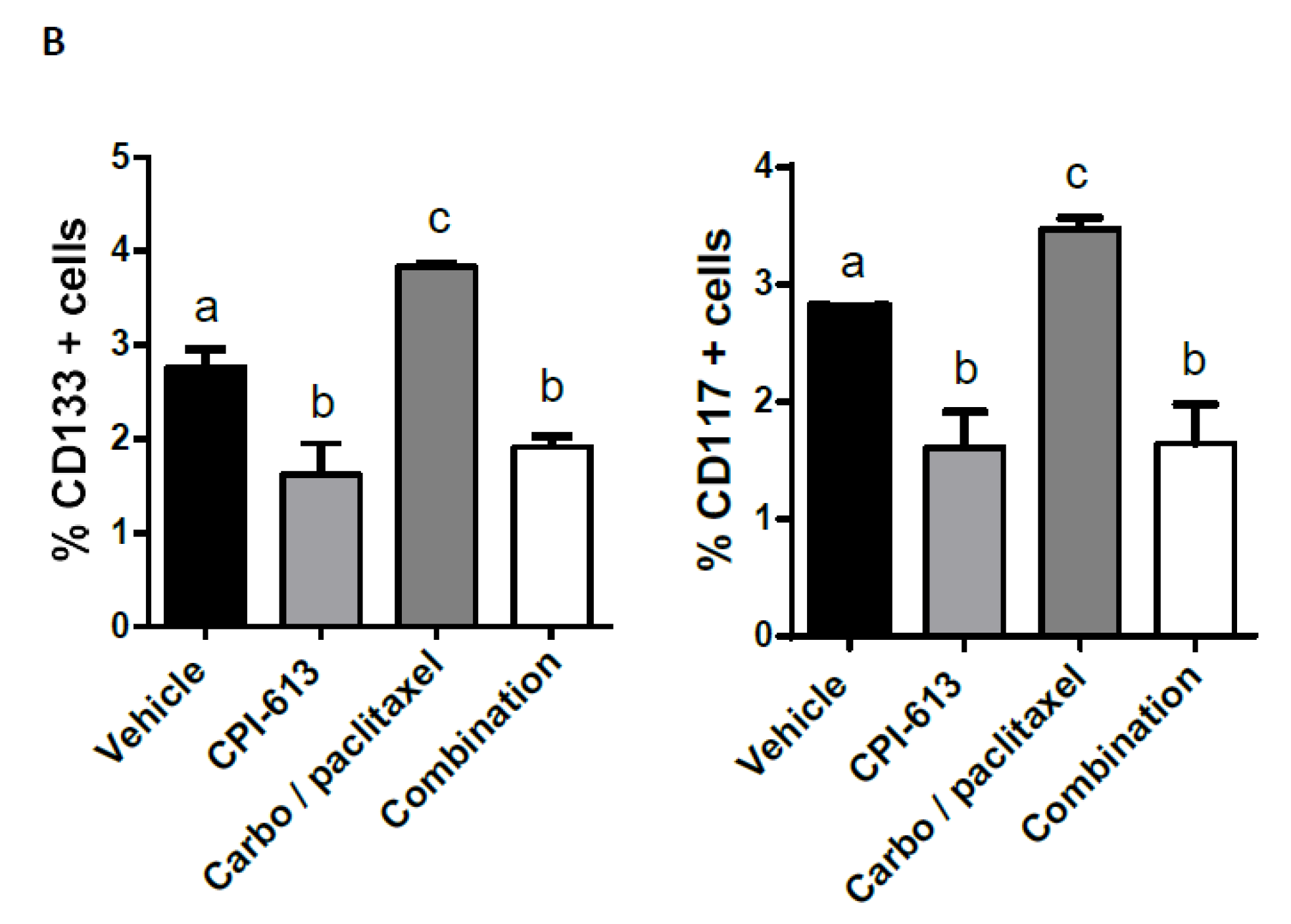
2.5. Combining CPI-613 and Carboplatin/Paclitaxel Treatment Impacts Tumor Growth Compared to CPI-613 Single Agent
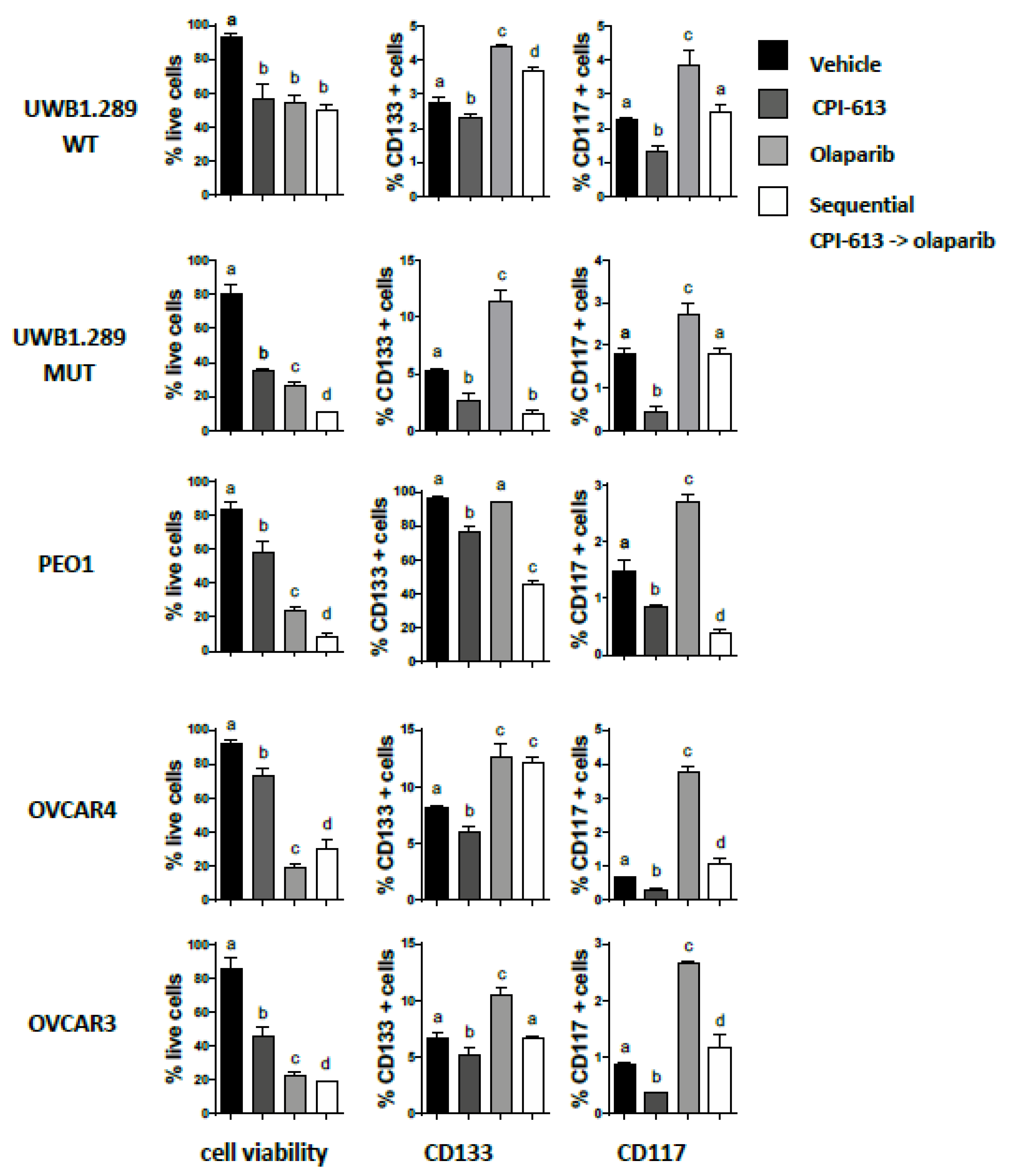
2.6. Pretreatment with CPI-613 Negates the Olaparib Induced an Increase in CD133 and CD117 Positive Cells
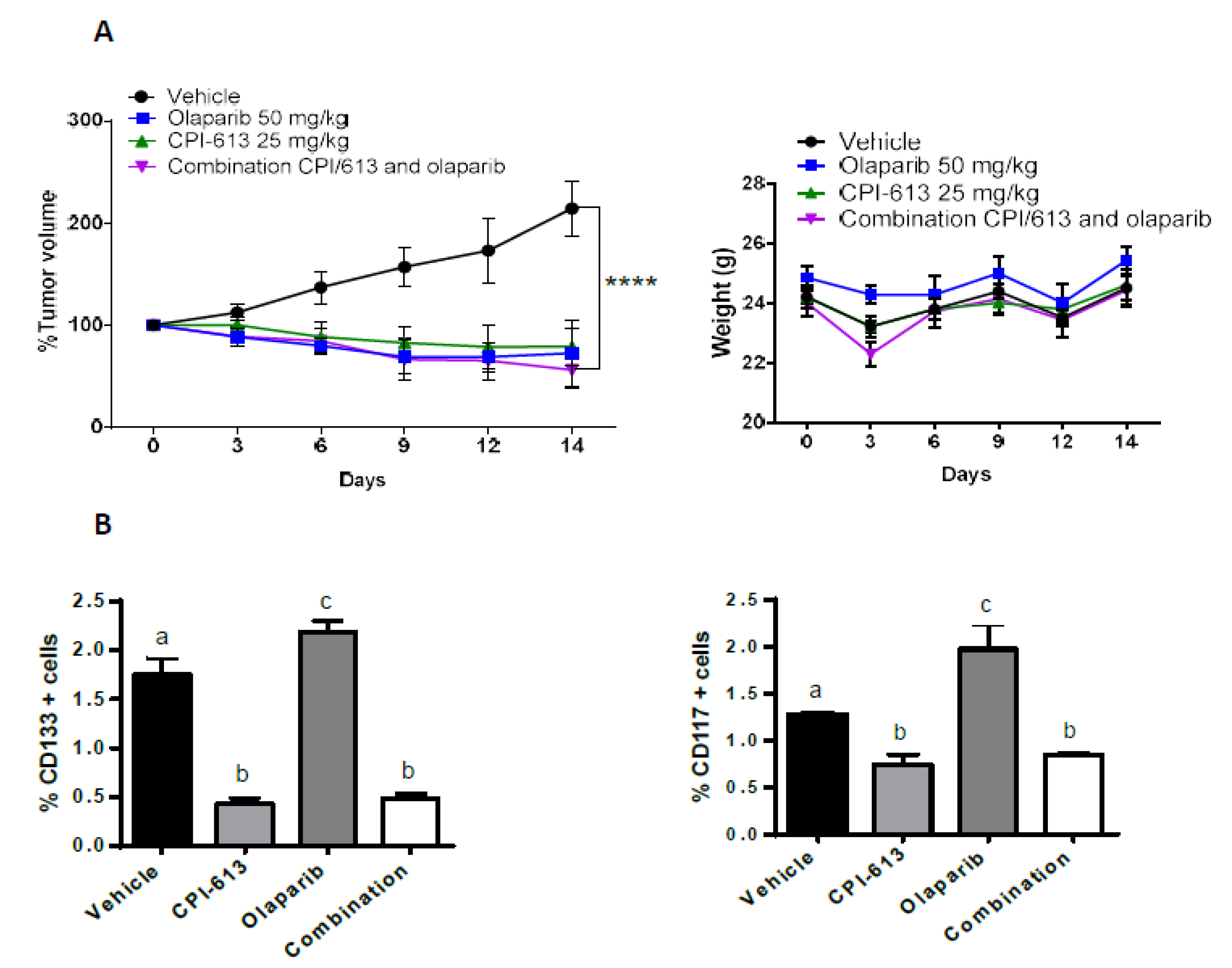
2.7. The Combination of CPI-613 and Olaparib Impacts Tumor Growth and Olaparib Induced Enrichment of CD133+ and CD117+ Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and In Vitro Culture
4.2. Drug Treatment (Carboplatin, Paclitaxel, Olaparib, and CPI-613)-MTT and Cell Counting
4.3. Flow Cytometry (CD133, CD117, Cell Viability)
4.4. Sphere Formation and ELDA
4.5. Tumorigenicity In Vivo Assay
4.6. In Vivo Experiment
4.7. Western Blotting
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| OvCa | ovarian cancer |
| EOC | epithelial ovarian cancer |
| CSCs | cancer stem cells |
| OXPHOS | oxidative phosphorylation |
| TCA | tricarboxylic acid |
| PDH | pyruvate dehydrogenase |
| KGDH | alpha-ketoglutarate dehydrogenase |
| FDA | Food and Drug Administration |
| AML | acute myeloid leukemia |
| MDS | myelodysplastic syndromes |
| PTCL | peripheral T-cell lymphoma |
| ROS | reactive oxygen species |
| PARPi | PARP inhibitors |
| ATP | PARP inhibitors |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Kaku, T.; Ogawa, S.; Kawano, Y.; Ohishi, Y.; Kobayashi, H.; Hirakawa, T.; Nakano, H. Histological classification of ovarian cancer. Med. Electron. Microsc. 2003, 36, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.W.; Ruiz, B.; Killeen, J.L.; Cote, T.R.; Wu, X.C.; Correa, C.N. Pathology and classification of ovarian tumors. Cancer 2003, 97, 2631–2642. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Lisio, M.A.; Fu, L.; Goyeneche, A.; Gao, Z.H.; Telleria, C. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int. J. Mol. Sci. 2019, 20, e952. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.K. Relapsed ovarian cancer: Challenges and management strategies for a chronic disease. Oncologist 2002, 7, 20–28. [Google Scholar] [CrossRef]
- Kim, S.; Han, Y.; Kim, S.I.; Kim, H.S.; Kim, S.J.; Song, Y.S. Tumor evolution and chemoresistance in ovarian cancer. NPJ Precis. Oncol. 2018, 2, e20. [Google Scholar] [CrossRef]
- Cojocaru, E.; Parkinson, C.A.; Brenton, J.D. Personalising Treatment for High-Grade Serous Ovarian Carcinoma. Clin. Oncol. R. Coll. Radiol. 2018, 30, 515–524. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Fu, X.; Bai, M.; Li, X.; Mei, Q.; Nie, J.; Wu, Z.; Han, W. Eliminating ovarian cancer stem cells: A potential therapeutic target for ovarian cancer chemoresistance. Curr. Protein Pept. Sci. 2015, 16, 270–278. [Google Scholar] [CrossRef]
- Markowska, A.; Sajdak, S.; Huczynski, A.; Rehlis, S.; Markowska, J. Ovarian cancer stem cells: A target for oncological therapy. Adv. Clin. Exp. Med. 2018, 27, 1017–1020. [Google Scholar] [CrossRef]
- Al-Alem, L.F.; Pandya, U.M.; Baker, A.T.; Bellio, C.; Zarrella, B.D.; Clark, J.; DiGloria, C.M.; Rueda, B.R. Ovarian cancer stem cells: What progress have we made? Int. J. Biochem. Cell Biol. 2019, 107, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.N. Cancer stem cells: Understanding tumor hierarchy and heterogeneity. Medicine 2016, 95, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Fulawka, L.; Donizy, P.; Halon, A. Cancer stem cells-The current status of an old concept: Literature review and clinical approaches. Biol. Res. 2014, 47, e66. [Google Scholar] [CrossRef] [PubMed]
- Gkountela, S.; Aceto, N. Stem-like features of cancer cells on their way to metastasis. Biol. Direct 2016, 11, e33. [Google Scholar] [CrossRef]
- Maugeri-Sacca, M.; Bartucci, M.; De Maria, R. DNA damage repair pathways in cancer stem cells. Mol. Cancer Ther. 2012, 11, 1627–1636. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, e5416923. [Google Scholar] [CrossRef]
- Chow, E.K. Implication of cancer stem cells in cancer drug development and drug delivery. J. Lab. Autom. 2013, 18, 6–11. [Google Scholar] [CrossRef]
- Abdullah, L.N.; Chow, E.K. Mechanisms of chemoresistance in cancer stem cells. Clin. Transl. Med. 2013, 2, e3. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Deheeger, M.; Lesniak, M.S.; Ahmed, A.U. Cellular plasticity regulated cancer stem cell niche: A possible new mechanism of chemoresistance. Cancer Cell Microenviron. 2014, 1, e295. [Google Scholar]
- Doherty, M.R.; Smigiel, J.M.; Junk, D.J.; Jackson, M.W. Cancer Stem Cell Plasticity Drives Therapeutic Resistance. Cancers 2016, 8, e8. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.; Curry, E.; Magnani, L.; Wilhelm-Benartzi, C.S.; Borley, J. Poised epigenetic states and acquired drug resistance in cancer. Nat. Rev. Cancer 2014, 14, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Huang, Y.H.; Chen, J.L. Understanding and targeting cancer stem cells: Therapeutic implications and challenges. Acta Pharmacol. Sin. 2013, 34, 732–740. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Fadaka, A.; Basiru, A.; Ojo, O.; Adewale, O.; Olayide, I.; Emuowhochere, R. Biology of glucose metabolism in cancer cells. J. Oncol. Sci. 2017, 3, 45–51. [Google Scholar]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Zhou, M.; Zhao, Y.; Ding, Y.; Liu, H.; Liu, Z.; Fodstad, O.; Riker, A.I.; Kamarajugadda, S.; Lu, J.; Owen, L.B.; et al. Warburg effect in chemosensitivity: Targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol. Cancer 2010, 9, e33. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, H.; Liu, Z.; Ding, Y.; Ledoux, S.P.; Wilson, G.L.; Voellmy, G.L.; Lin, Y.; Lin, W.; Nahta, R.; et al. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res. 2011, 71, 4585–4597. [Google Scholar] [CrossRef]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13, e89. [Google Scholar] [CrossRef]
- Abaza, M.; Luqmani, Y.A. The influence of pH and hypoxia on tumor metastasis. Expert Rev. Anticancer Ther. 2013, 13, 1229–1242. [Google Scholar] [CrossRef]
- Rahman, M.; Hasan, M.R. Cancer Metabolism and Drug Resistance. Metabolites 2015, 5, 571–600. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y. Cancer Metabolism: A Hope for Curing Cancer. Biomol. Ther. 2018, 26, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; Herst, P.M.; Tan, A.S. Metabolic flexibility and cell hierarchy in metastatic cancer. Mitochondrion 2010, 10, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Peiris-Pages, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, e55. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, J.; Lima, J. Metabolic traits of cancer stem cells. Dis. Model. Mech. 2018, 11, dmm033464. [Google Scholar] [CrossRef]
- Snyder, V.; Reed-Newman, T.C.; Arnold, L.; Thomas, S.M.; Anant, S. Cancer Stem Cell Metabolism and Potential Therapeutic Targets. Front. Oncol. 2018, 8, e203. [Google Scholar] [CrossRef]
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting Metabolism for Cancer Therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar] [CrossRef]
- Wong, T.L.; Che, N.; Ma, S. Reprogramming of central carbon metabolism in cancer stem cells. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1728–1738. [Google Scholar] [CrossRef]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef]
- Stuart, S.D.; Schauble, A.; Gupta, S.; Kennedy, A.D.; Keppler, B.R.; Bingham, P.M.; Zachara, Z. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab. 2014, 2, e4. [Google Scholar] [CrossRef] [PubMed]
- Zachar, Z.; Marecek, J.; Maturo, C.; Gupta, S.; Stuart, S.D.; Howell, K.; Schauble, A.; Lem, J.; Piramzadian, A.; Karnik, S.; et al. Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J. Mol. Med. 2011, 89, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Alistar, A.; Morris, B.B.; Desnoyer, R.; Klepin, H.D.; Hosseinzadeh, K.; Clark, C.; Cameron, A.; Layendecker, J.; D’Agostino, R., Jr.; Topaloglu, U.; et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: A single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2017, 18, 770–778. [Google Scholar] [CrossRef]
- Pardee, T.S.; Lee, K.; Luddy, J.; Maturo, C.; Rodriguez, R.; Isom, S.; Miller, L.D.; Stadelman, K.M.; Levitan, D.; Hurd, D.; et al. A phase I study of the first-in-class antimitochondrial metabolism agent, CPI-613, in patients with advanced hematologic malignancies. Clin. Cancer Res. 2014, 20, 5255–5264. [Google Scholar] [CrossRef] [PubMed]
- Ke, C.; Li, A.; Hou, Y.; Sun, M.; Yang, K.; Cheng, J.; Wang, J.; Ge, T.; Zhang, F.; Li, Q.; et al. Metabolic phenotyping for monitoring ovarian cancer patients. Sci. Rep. 2016, 6, e23334. [Google Scholar] [CrossRef] [PubMed]
- Dar, S.; Chhina, J.; Mert, I.; Chitale, D.; Buekers, T.; Kaur, H.; Giri, S.; Munkarah, A.; Rattan, R. Bioenergetic Adaptations in Chemoresistant Ovarian Cancer Cells. Sci. Rep. 2017, 7, e8760. [Google Scholar] [CrossRef]
- Liao, J.; Qian, F.; Tchabo, N.; Mhawech-Fauceglia, P.; Beck, A.; Qian, Z.; Wang, X.; Huss, W.J.; Lele, S.B.; Morrison, C.D.; et al. Ovarian cancer spheroid cells with stem cell-like properties contribute to tumor generation, metastasis and chemotherapy resistance through hypoxia-resistant metabolism. PLoS ONE 2014, 9, e84941. [Google Scholar] [CrossRef]
- Anderson, A.S.; Roberts, P.C.; Frisard, M.I.; Hulver, M.W.; Schmelz, E.M. Ovarian tumor-initiating cells display a flexible metabolism. Exp. Cell Res. 2014, 328, 44–57. [Google Scholar] [CrossRef]
- Pasto, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef]
- Bellio, C.; DiGloria, C.; Foster, R.; James, K.; Konstantinopoulos, P.A.; Growdon, W.B.; Rueda, B.R. PARP Inhibition Induces Enrichment of DNA Repair-Proficient CD133 and CD117 Positive Ovarian Cancer Stem Cells. Mol. Cancer Res. 2019, 17, 431–445. [Google Scholar] [CrossRef]
- Parte, S.C.; Batra, S.K.; Kakar, S.S. Characterization of stem cell and cancer stem cell populations in ovary and ovarian tumors. J. Ovarian Res. 2018, 11, e69. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Zeng, J.; Liang, B.; Zhao, Z.; Sun, L.; Cao, D.; Yang, J.; Shen, K. Ovarian cancer cells with the CD117 phenotype are highly tumorigenic and are related to chemotherapy outcome. Exp. Mol. Pathol. 2011, 91, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, M.; D’Alterio, C.; Camerlingo, R.; Tirino, V.; Consales, C.; Riccio, A.; Ierano, C.; Cecere, S.C.; Losito, N.S.; Greggi, S.; et al. Identification of a distinct population of CD133(+)CXCR4(+) cancer stem cells in ovarian cancer. Sci. Rep. 2015, 5, e10357. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Wicha, M.S. Metabolic plasticity of cancer stem cells. Oncotarget 2015, 6, 35141–35142. [Google Scholar] [CrossRef] [PubMed]
- Chiodi, I.; Belgiovine, C.; Dona, F.; Scovassi, A.I.; Mondello, C. Drug treatment of cancer cell lines: A way to select for cancer stem cells? Cancers 2011, 3, 1111–1128. [Google Scholar] [CrossRef]
- Holness, M.J.; Sugden, M.C. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem. Soc. Trans. 2003, 31, 1143–1151. [Google Scholar] [CrossRef]
- Mehta, P.; Novak, C.; Raghavan, S.; Ward, M.; Mehta, G. Self-Renewal and CSCs In Vitro Enrichment: Growth as Floating Spheres. Methods Mol. Biol. 2018, 1692, 61–75. [Google Scholar]
- Richichi, C.; Osti, D.; Del Bene, M.; Fornasari, L.; Patane, M.; Pollo, B.; DiMeco, F.; Pelicci, G. Tumor-initiating cell frequency is relevant for glioblastoma aggressiveness. Oncotarget 2016, 7, 71491–71503. [Google Scholar] [CrossRef][Green Version]
- Desale, S.S.; Soni, K.S.; Romanova, S.; Cohen, S.M.; Bronich, T.K. Targeted delivery of platinum-taxane combination therapy in ovarian cancer. J. Control. Release 2015, 220, 651–659. [Google Scholar] [CrossRef]
- Lee, K.C.; Maturo, C.; Perera, C.N.; Luddy, J.; Rodriguez, R.; Shorr, R. Translational assessment of mitochondrial dysfunction of pancreatic cancer from in vitro gene microarray and animal efficacy studies, to early clinical studies, via the novel tumor-specific anti-mitochondrial agent, CPI-613. Ann. Transl. Med. 2014, 2, e91. [Google Scholar]
- Chu, E. Sequential versus combination therapy: Where are we? Clin. Colorectal Cancer. 2008, 7, e295. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Bedard, P.L.; Winer, E.P.; Pagani, O.; Senkus-Konefka, E.; Fallowfield, L.J.; Kyriakides, S.; Costa, A.; Cufer, T.; Albain, K.S. International guidelines for management of metastatic breast cancer: Combination vs sequential single-agent chemotherapy. J. Natl. Cancer Inst. 2009, 101, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni-Datar, K.; Orsulic, S.; Foster, R.; Rueda, B.R. Ovarian tumor initiating cell populations persist following paclitaxel and carboplatin chemotherapy treatment in vivo. Cancer Lett. 2013, 339, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Steg, A.D.; Bevis, K.S.; Katre, A.A.; Ziebarth, A.; Dobbin, Z.C.; Alvarez, R.D.; Zhang, K.; Conner, M.; Landen, C.N. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin. Cancer Res. 2012, 18, 869–881. [Google Scholar] [CrossRef]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef]
- Vlashi, E.; Pajonk, F. The metabolic state of cancer stem cells-a valid target for cancer therapy? Free Radic. Biol. Med. 2015, 79, 264–268. [Google Scholar] [CrossRef]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef]
- Ahmed, N.; Escalona, R.; Leung, D.; Chan, E.; Kannourakis, G. Tumour microenvironment and metabolic plasticity in cancer and cancer stem cells: Perspectives on metabolic and immune regulatory signatures in chemoresistant ovarian cancer stem cells. Semin. Cancer Biol. 2018, 53, 265–281. [Google Scholar] [CrossRef]
- Curley, M.D.; Therrien, V.A.; Cummings, C.L.; Sergent, P.A.; Koulouris, C.R.; Friel, A.M.; Roberts, D.J.; Seiden, M.V.; Scadden, D.T.; Rueda, B.R.; et al. CD133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells 2009, 27, 2875–2883. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellio, C.; DiGloria, C.; Spriggs, D.R.; Foster, R.; Growdon, W.B.; Rueda, B.R. The Metabolic Inhibitor CPI-613 Negates Treatment Enrichment of Ovarian Cancer Stem Cells. Cancers 2019, 11, 1678. https://doi.org/10.3390/cancers11111678
Bellio C, DiGloria C, Spriggs DR, Foster R, Growdon WB, Rueda BR. The Metabolic Inhibitor CPI-613 Negates Treatment Enrichment of Ovarian Cancer Stem Cells. Cancers. 2019; 11(11):1678. https://doi.org/10.3390/cancers11111678
Chicago/Turabian StyleBellio, Chiara, Celeste DiGloria, David R. Spriggs, Rosemary Foster, Whitfield B. Growdon, and Bo R. Rueda. 2019. "The Metabolic Inhibitor CPI-613 Negates Treatment Enrichment of Ovarian Cancer Stem Cells" Cancers 11, no. 11: 1678. https://doi.org/10.3390/cancers11111678
APA StyleBellio, C., DiGloria, C., Spriggs, D. R., Foster, R., Growdon, W. B., & Rueda, B. R. (2019). The Metabolic Inhibitor CPI-613 Negates Treatment Enrichment of Ovarian Cancer Stem Cells. Cancers, 11(11), 1678. https://doi.org/10.3390/cancers11111678