Role of Focal Adhesion Kinase in Small-Cell Lung Cancer and Its Potential as a Therapeutic Target


Abstract
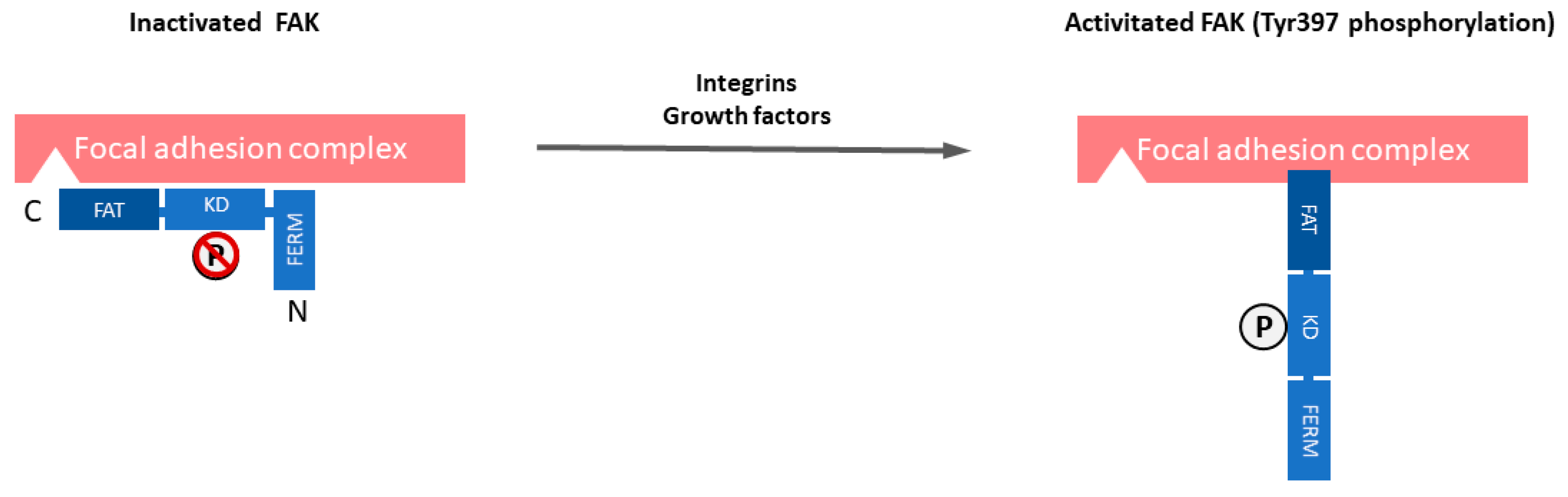
1. Introduction
2. FAK Overexpression and/or Activation in Human Cancers, Its Frequency and Mechanisms
3. FAK Role in Proliferation, Cell Cycle, and Survival
4. FAK Role in Adhesion, Migration, and Invasion
5. FAK in Epithelial to Mesenchymal Transition
6. FAK-Mediated Angiogenesis and Vascular Permeability
7. FAK and DNA Damage Repair
8. FAK and Radioresistance
9. Regulation of Cancer Stem Cells
10. FAK in Tumor Immune Escape
11. Prognostic and Predictive Value of FAK Alterations
12. Conclusions and Therapeutic Perspectives
Funding
Acknowledgments
Conflicts of Interest
Appendix A

References
- Govindan, R.; Page, N.; Morgensztern, D.; Read, W.; Tierney, R.; Vlahiotis, A.; Spitznagel, E.L.; Piccirillo, J. Changing Epidemiology of Small-Cell Lung Cancer in the United States Over the Last 30 Years: Analysis of the Surveillance, Epidemiologic, and End Results Database. J. Clin. Oncol. 2006, 24, 4539–4544. [Google Scholar] [CrossRef] [PubMed]
- Recondo, G.; Facchinetti, F.; Olaussen, K.A.; Besse, B.; Friboulet, L. Making the first move in EGFR-driven or ALK-driven NSCLC: First-generation or next-generation TKI? Nat. Rev. Clin. Oncol. 2018, 15, 694–708. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crino, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet Lond. Engl. 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef]
- Wang, J.C.; Sone, S.; Feng, L.; Yang, Z.G.; Takashima, S.; Maruyama, Y.; Hasegawa, M.; Kawakami, S.; Honda, T.; Yamanda, T. Rapidly growing small peripheral lung cancers detected by screening CT: Correlation between radiological appearance and pathological features. Br. J. Radiol. 2000, 73, 930–937. [Google Scholar] [CrossRef]
- Thomas, A.; Pattanayak, P.; Szabo, E.; Pinsky, P. Characteristics and Outcomes of Small Cell Lung Cancer Detected by CT Screening. Chest 2018, 154, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Turrisi, A.T.; Kim, K.; Blum, R.; Sause, W.T.; Livingston, R.B.; Komaki, R.; Wagner, H.; Aisner, S.; Johnson, D.H. Twice-daily compared with once-daily thoracic radiotherapy in limited small-cell lung cancer treated concurrently with cisplatin and etoposide. N. Engl. J. Med. 1999, 340, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Kubota, K.; Hida, T.; Ishikura, S.; Mizusawa, J.; Nishio, M.; Kawahara, M.; Yokoyama, A.; Imamura, F.; Takeda, K.; Negoro, S.; et al. Etoposide and cisplatin versus irinotecan and cisplatin in patients with limited-stage small-cell lung cancer treated with etoposide and cisplatin plus concurrent accelerated hyperfractionated thoracic radiotherapy (JCOG0202): A randomised phase 3 study. Lancet Oncol. 2014, 15, 106–113. [Google Scholar] [CrossRef]
- Pujol, J.L.; Daures, J.P.; Riviere, A.; Quoix, E.; Westeel, V.; Quantin, X.; Breton, J.L.; Lemarie, E.; Poudenx, M.; Milleron, B.; et al. Etoposide plus cisplatin with or without the combination of 4’-epidoxorubicin plus cyclophosphamide in treatment of extensive small-cell lung cancer: A French Federation of Cancer Institutes multicenter phase III randomized study. J. Natl. Cancer Inst. 2001, 93, 300–308. [Google Scholar] [CrossRef]
- Saunders, L.R.; Bankovich, A.J.; Anderson, W.C.; Aujay, M.A.; Bheddah, S.; Black, K.; Desai, R.; Escarpe, P.A.; Hampl, J.; Laysang, A.; et al. A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci. Transl. Med. 2015, 7, 302ra136. [Google Scholar] [CrossRef]
- Rudin, C.M.; Pietanza, M.C.; Bauer, T.M.; Ready, N.; Morgensztern, D.; Glisson, B.S.; Byers, L.A.; Johnson, M.L.; Burris, H.A.; Robert, F.; et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: A first-in-human, first-in-class, open-label, phase 1 study. Lancet. Oncol. 2017, 18, 42–51. [Google Scholar] [CrossRef]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef]
- Stinchcombe, T.E. Current Treatments for Surgically Resectable, Limited-Stage, and Extensive-Stage Small Cell Lung Cancer. Oncologist 2017, 22, 1510–1517. [Google Scholar] [CrossRef]
- Povsic, M.; Enstone, A.; Wyn, R.; Kornalska, K.; Penrod, J.R.; Yuan, Y. Real-world effectiveness and tolerability of small-cell lung cancer (SCLC) treatments: A systematic literature review (SLR). PLoS ONE 2019, 14, e0219622. [Google Scholar] [CrossRef]
- Carelli, S.; Zadra, G.; Vaira, V.; Falleni, M.; Bottiglieri, L.; Nosotti, M.; Di Giulio, A.M.; Gorio, A.; Bosari, S. Up-regulation of focal adhesion kinase in non-small cell lung cancer. Lung Cancer 2006, 53, 263–271. [Google Scholar] [CrossRef]
- Dy, G.K.; Ylagan, L.; Pokharel, S.; Miller, A.; Brese, E.; Bshara, W.; Morrison, C.; Cance, W.G.; Golubovskaya, V.M. The Prognostic Significance of Focal Adhesion Kinase Expression in Stage I Non-Small-Cell Lung Cancer. J. Thorac. Oncol. 2014, 9, 1278–1284. [Google Scholar] [CrossRef]
- Wang, C.; Yang, R.; Yue, D.; Zhang, Z. Expression of FAK and PTEN in Bronchioloalveolar Carcinoma and Lung Adenocarcinoma. Lung 2009, 187, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, M.; Nishimura, M.; Takeuchi, S.; Murase, M.; Hamaguchi, M. Role of tyrosine specific phosphorylation of cellular proteins, especially EGF receptor and p125FAK in human lung cancer cells. Lung Cancer 1997, 17, 69–84. [Google Scholar] [CrossRef]
- Ocak, S.; Chen, H.; Callison, C.; Gonzalez, A.L.; Massion, P.P. Expression of focal adhesion kinase in small-cell lung carcinoma. Cancer 2012, 118, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Hsu, N.Y.; Chen, C.Y.; Hsu, C.P.; Lin, T.Y.; Chou, M.C.; Chiou, S.H.; Chow, K.C. Prognostic significance of expression of nm23-H1 and focal adhesion kinase in non-small cell lung cancer. Oncol. Rep. 2007, 18, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.F.; Pang, D.; Fu, S.B.; Jin, Y.; Yao, L.; Qi, J.P.; Bai, J. Overexpression of focal adhesion kinase correlates with increased lymph node metastasis and poor prognosis in non-small-cell lung cancer. J. Cancer Res. Clin. Oncol. 2013, 139, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Owens, L.V.; Xu, L.; Craven, R.J.; Dent, G.A.; Weiner, T.M.; Kornberg, L.; Liu, E.T.; Cance, W.G. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res. 1995, 55, 2752–2755. [Google Scholar]
- Ocak, S.; Yamashita, H.; Udyavar, A.R.; Miller, A.N.; Gonzalez, A.L.; Zou, Y.; Jiang, A.; Yi, Y.; Shyr, Y.; Estrada, L.; et al. DNA copy number aberrations in small-cell lung cancer reveal activation of the focal adhesion pathway. Oncogene 2010, 29, 6331–6342. [Google Scholar] [CrossRef]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355. [Google Scholar] [CrossRef]
- Schaller, M.D.; Borgman, C.A.; Parsons, J.T. Autonomous expression of a noncatalytic domain of the focal adhesion-associated protein tyrosine kinase pp125FAK. Mol. Cell. Biol. 1993, 13, 785–791. [Google Scholar] [CrossRef]
- Richardson, A.; Parsons, T. A mechanism for regulation of the adhesion-associated proteintyrosine kinase pp125FAK. Nature 1996, 380, 538–540. [Google Scholar] [CrossRef] [PubMed]
- Kleinschmidt, E.G.; Schlaepfer, D.D. Focal adhesion kinase signaling in unexpected places. Curr. Opin. Cell Biol. 2017, 45, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Almeida, E.A.; Ilic, D.; Han, Q.; Hauck, C.R.; Jin, F.; Kawakatsu, H.; Schlaepfer, D.D.; Damsky, C.H. Matrix survival signaling: From fibronectin via focal adhesion kinase to c-Jun NH(2)-terminal kinase. J. Cell Biol. 2000, 149, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Mak, G.; Soria, J.C.; Blagden, S.P.; Plummer, R.; Fleming, R.A.; Nebot, N.; Zhang, J.; Mazumdar, J.; Rogan, D.; Gazzah, A.; et al. A phase Ib dose-finding, pharmacokinetic study of the focal adhesion kinase inhibitor GSK2256098 and trametinib in patients with advanced solid tumours. Br. J. Cancer 2019, 120, 975–981. [Google Scholar] [CrossRef]
- Soria, J.C.; Gan, H.K.; Blagden, S.P.; Plummer, R.; Arkenau, H.T.; Ranson, M.; Evans, T.R.; Zalcman, G.; Bahleda, R.; Hollebecque, A.; et al. A phase I, pharmacokinetic and pharmacodynamic study of GSK2256098, a focal adhesion kinase inhibitor, in patients with advanced solid tumors. Ann. Oncol. 2016, 27, 2268–2274. [Google Scholar] [CrossRef]
- Brown, N.F.; Williams, M.; Arkenau, H.T.; Fleming, R.A.; Tolson, J.; Yan, L.; Zhang, J.; Swartz, L.; Singh, R.; Auger, K.R.; et al. A study of the focal adhesion kinase inhibitor GSK2256098 in patients with recurrent glioblastoma with evaluation of tumor penetration of [11C] GSK2256098. Neuro Oncol. 2018, 20, 1634–1642. [Google Scholar] [CrossRef]
- Infante, J.R.; Camidge, D.R.; Mileshkin, L.R.; Chen, E.X.; Hicks, R.J.; Rischin, D.; Fingert, H.; Pierce, K.J.; Xu, H.; Roberts, W.G.; et al. Safety, pharmacokinetic, and pharmacodynamic phase I dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J. Clin. Oncol. 2012, 30, 1527–1533. [Google Scholar] [CrossRef]
- Jones, S.F.; Siu, L.L.; Bendell, J.C.; Cleary, J.M.; Razak, A.R.; Infante, J.R.; Pandya, S.S.; Bedard, P.L.; Pierce, K.J.; Houk, B.; et al. A phase I study of VS-6063, a second-generation focal adhesion kinase inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 1100–1107. [Google Scholar] [CrossRef]
- Patel, M.R.; Infante, J.R.; Moore, K.N.; Keegan, M.; Poli, A.; Padval, M.; Jones, S.F.; Horobin, J.; Burris, H.A. Phase 1/1b study of the FAK inhibitor defactinib (VS-6063) in combination with weekly paclitaxel for advanced ovarian cancer. J. Clin. Oncol. 2014, 32, 5521. [Google Scholar] [CrossRef]
- Shimizu, T.; Fukuoka, K.; Takeda, M.; Iwasa, T.; Yoshida, T.; Horobin, J.; Keegan, M.; Vaickus, L.; Chavan, A.; Padval, M.; et al. A first-in-Asian phase 1 study to evaluate safety, pharmacokinetics and clinical activity of VS-6063, a focal adhesion kinase (FAK) inhibitor in Japanese patients with advanced solid tumors. Cancer Chemother. Pharm. 2016, 77, 997–1003. [Google Scholar] [CrossRef]
- Doi, T.; Yang, J.C.; Shitara, K.; Naito, Y.; Cheng, A.L.; Sarashina, A.; Pronk, L.C.; Takeuchi, Y.; Lin, C.C. Phase I Study of the Focal Adhesion Kinase Inhibitor BI 853520 in Japanese and Taiwanese Patients with Advanced or Metastatic Solid Tumors. Target Oncol. 2019, 14, 57–65. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, M.J.A.; Steeghs, N.; Lolkema, M.P.; Hotte, S.J.; Hirte, H.W.; van der Biessen, D.A.J.; Abdul Razak, A.R.; De Vos, F.; Verheijen, R.B.; Schnell, D.; et al. Phase I Study of BI 853520, an Inhibitor of Focal Adhesion Kinase, in Patients with Advanced or Metastatic Nonhematologic Malignancies. Target Oncol. 2019, 14, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Verheijen, R.B.; van der Biessen, D.A.J.; Hotte, S.J.; Siu, L.L.; Spreafico, A.; de Jonge, M.J.A.; Pronk, L.C.; De Vos, F.; Schnell, D.; Hirte, H.W.; et al. Randomized, Open-Label, Crossover Studies Evaluating the Effect of Food and Liquid Formulation on the Pharmacokinetics of the Novel Focal Adhesion Kinase (FAK) Inhibitor BI 853520. Target Oncol. 2019, 14, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Aung, K.L.; McWhirter, E.; Welch, S.; Wang, L.; Lovell, S.; Stayner, L.-A.; Ali, S.; Malpage, A.; Makepeace, B.; Ramachandran, M.; et al. A phase II trial of GSK2256098 and trametinib in patients with advanced pancreatic ductal adenocarcinoma (PDAC) (MOBILITY-002 Trial, NCT02428270). J. Clin. Oncol. 2018, 36, 409. [Google Scholar] [CrossRef]
- Fennell, D.A.; Baas, P.; Taylor, P.; Nowak, A.K.; Gilligan, D.; Nakano, T.; Pachter, J.A.; Weaver, D.T.; Scherpereel, A.; Pavlakis, N.; et al. Maintenance Defactinib Versus Placebo After First-Line Chemotherapy in Patients With Merlin-Stratified Pleural Mesothelioma: COMMAND—A Double-Blind, Randomized, Phase II Study. J. Clin. Oncol. 2019, 37, 790–798. [Google Scholar] [CrossRef]
- Bueno, R.; Gill, R.R.; Lizotte, P.H.; Sprott, K.; Jackman, D.M.; Barlow, J.; Sharma, S.; Yeap, B.Y.; Chirieac, L.R.; Lebenthal, A.; et al. Effect of FAK inhibitor defactinib on tumor immune changes and tumor reductions in a phase II window of opportunity study in malignant pleural mesothelioma (MPM). J. Clin. Oncol. 2017, 35, 8555. [Google Scholar] [CrossRef]
- Liu, T.-J.; LaFortune, T.; Honda, T.; Ohmori, O.; Hatakeyama, S.; Meyer, T.; Jackson, D.; de Groot, J.; Yung, W.K.A. Inhibition of both focal adhesion kinase and insulin-like growth factor-I receptor kinase suppresses glioma proliferation in vitro and in vivo. Mol. Cancer Ther. 2007, 6, 1357–1367. [Google Scholar] [CrossRef]
- Slack-Davis, J.K.; Martin, K.H.; Tilghman, R.W.; Iwanicki, M.; Ung, E.J.; Autry, C.; Luzzio, M.J.; Cooper, B.; Kath, J.C.; Roberts, W.G.; et al. Cellular Characterization of a Novel Focal Adhesion Kinase Inhibitor. J. Biol. Chem. 2007, 282, 14845–14852. [Google Scholar] [CrossRef]
- Auger, K.R.; Smitheman, K.N.; Korenchuk, S.; McHugh, C.; Kruger, R.; Van Aller, G.S.; Smallwood, A.; Gontarek, R.R.; Faitg, T.; Johnson, N. 387 The Focal Adhesion Kinase Inhibitor GSK2256098: A Potent and Selective Inhibitor for the Treatment of Cancer. Eur. J. Cancer 2012, 48, 118. [Google Scholar] [CrossRef]
- Weis, S.M.; Lim, S.T.; Lutu-Fuga, K.M.; Barnes, L.A.; Chen, X.L.; Gothert, J.R.; Shen, T.L.; Guan, J.L.; Schlaepfer, D.D.; Cheresh, D.A. Compensatory role for Pyk2 during angiogenesis in adult mice lacking endothelial cell FAK. J. Cell Biol. 2008, 181, 43–50. [Google Scholar] [CrossRef]
- Walsh, C.; Tanjoni, I.; Uryu, S.; Tomar, A.; Nam, J.O.; Luo, H.; Phillips, A.; Patel, N.; Kwok, C.; McMahon, G.; et al. Oral delivery of PND-1186 FAK inhibitor decreases tumor growth and spontaneous breast to lung metastasis in pre-clinical models. Cancer Biol. 2010, 9, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Roberts, W.G.; Ung, E.; Whalen, P.; Cooper, B.; Hulford, C.; Autry, C.; Richter, D.; Emerson, E.; Lin, J.; Kath, J.; et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008, 68, 1935–1944. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Hu, W.; Ivan, C.; Dalton, H.J.; Miyake, T.; Pecot, C.V.; Zand, B.; Liu, T.; Huang, J.; Jennings, N.B.; et al. Role of focal adhesion kinase in regulating YB-1-mediated paclitaxel resistance in ovarian cancer. J. Natl. Cancer Inst. 2013, 105, 1485–1495. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, T.; Seenisamy, J.; Emmanuvel, L.; Kulkarni, S.S.; Bomke, J.; Rohdich, F.; Greiner, H.; Esdar, C.; Krier, M.; Grädler, U.; et al. Fragment-Based Discovery of New Highly Substituted 1H-Pyrrolo[2,3-b]- and 3H-Imidazolo[4,5-b]-Pyridines as Focal Adhesion Kinase Inhibitors. J. Med. Chem. 2013, 56, 1160–1170. [Google Scholar] [CrossRef] [PubMed]
- Kurenova, E.; Liao, J.; He, D.H.; Hunt, D.; Yemma, M.; Bshara, W.; Seshadri, M.; Cance, W.G. The FAK scaffold inhibitor C4 disrupts FAK-VEGFR-3 signaling and inhibits pancreatic cancer growth. Oncotarget 2013, 4, 1632–1646. [Google Scholar] [CrossRef] [PubMed]
- Kurenova, E.V.; Hunt, D.L.; He, D.; Magis, A.T.; Ostrov, D.A.; Cance, W.G. Small molecule chloropyramine hydrochloride (C4) targets the binding site of focal adhesion kinase and vascular endothelial growth factor receptor 3 and suppresses breast cancer growth in vivo. J. Med. Chem. 2009, 52, 4716–4724. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.E.; Ma, X.; Megison, M.; Nabers, H.; Cance, W.G.; Kurenova, E.V.; Beierle, E.A. Inhibition of FAK and VEGFR-3 binding decreases tumorigenicity in neuroblastoma. Mol. Carcinog. 2015, 54, 9–23. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Ho, B.; Zheng, M.; Magis, A.; Ostrov, D.; Morrison, C.; Cance, W.G. Disruption of focal adhesion kinase and p53 interaction with small molecule compound R2 reactivated p53 and blocked tumor growth. BMC Cancer 2013, 13, 342. [Google Scholar] [CrossRef]
- Ho, B.; Olson, G.; Figel, S.; Gelman, I.; Cance, W.G.; Golubovskaya, V.M. Nanog increases focal adhesion kinase (FAK) promoter activity and expression and directly binds to FAK protein to be phosphorylated. J. Biol. Chem. 2012, 287, 18656–18673. [Google Scholar] [CrossRef]
- Tiede, S.; Meyer-Schaller, N.; Kalathur, R.K.R.; Ivanek, R.; Fagiani, E.; Schmassmann, P.; Stillhard, P.; Hafliger, S.; Kraut, N.; Schweifer, N.; et al. The FAK inhibitor BI 853520 exerts anti-tumor effects in breast cancer. Oncogenesis 2018, 7, 73. [Google Scholar] [CrossRef]
- Laszlo, V.; Valko, Z.; Ozsvar, J.; Kovacs, I.; Garay, T.; Hoda, M.A.; Klikovits, T.; Stockhammer, P.; Aigner, C.; Groger, M.; et al. The FAK inhibitor BI 853520 inhibits spheroid formation and orthotopic tumor growth in malignant pleural mesothelioma. J. Mol. Med. 2019, 97, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Hjelmeland, A.B.; Keir, S.T.; Song, L.; Wickman, S.; Jackson, D.; Ohmori, O.; Bigner, D.D.; Friedman, H.S.; Rich, J.N. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Mol. Carcinog. 2007, 46, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M. Focal adhesion kinase as a cancer therapy target. Anti-Cancer Agents Med. Chem. 2010, 10, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Gabarra-Niecko, V.; Schaller, M.D.; Dunty, J.M. FAK regulates biological processes important for the pathogenesis of cancer. Cancer Metastasis Rev. 2003, 22, 359–374. [Google Scholar] [CrossRef]
- Van Nimwegen, M.J.; van de Water, B. Focal adhesion kinase: A potential target in cancer therapy. Biochem. Pharm. 2007, 73, 597–609. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Cance, W.G. Focal adhesion kinase and p53 signaling in cancer cells. Int. Rev. Cytol. 2007, 263, 103–153. [Google Scholar] [CrossRef]
- Aboubakar Nana, F.; Hoton, D.; Ambroise, J.; Lecocq, M.; Vanderputten, M.; Sibille, Y.; Vanaudenaerde, B.; Pilette, C.; Bouzin, C.; Ocak, S. Increased Expression and Activation of FAK in Small-Cell Lung Cancer Compared to Non-Small-Cell Lung Cancer. Cancers 2019, 11, 1526. [Google Scholar] [CrossRef]
- Aboubakar Nana, F.; Lecocq, M.; Ladjemi, M.Z.; Detry, B.; Dupasquier, S.; Feron, O.; Massion, P.P.; Sibille, Y.; Pilette, C.; Ocak, S. Therapeutic Potential of Focal Adhesion Kinase Inhibition in Small Cell Lung Cancer. Mol. Cancer 2019, 18, 17–27. [Google Scholar] [CrossRef]
- Cance, W.G.; Harris, J.E.; Iacocca, M.V.; Roche, E.; Yang, X.; Chang, J.; Simkins, S.; Xu, L. Immunohistochemical analyses of focal adhesion kinase expression in benign and malignant human breast and colon tissues: Correlation with preinvasive and invasive phenotypes. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 2417–2423. [Google Scholar]
- Ayaki, M.; Komatsu, K.; Mukai, M.; Murata, K.; Kameyama, M.; Ishiguro, S.; Miyoshi, J.; Tatsuta, M.; Nakamura, H. Reduced Expression of Focal Adhesion Kinase in Liver Metastases Compared with Matched Primary Human Colorectal Adenocarcinomas. Clin. Cancer Res. 2001, 7, 3106–3112. [Google Scholar]
- Rovin, J.D.; Frierson, H.F.; Ledinh, W.; Parsons, J.T.; Adams, R.B. Expression of focal adhesion kinase in normal and pathologic human prostate tissues. Prostate 2002, 53, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.B.; Kurago, Z.; Zaharias, R.; Gruman, L.M.; Schaller, M.D.; Hendrix, M.J. Elevated focal adhesion kinase expression facilitates oral tumor cell invasion. Cancer 2002, 95, 2508–2515. [Google Scholar] [CrossRef] [PubMed]
- Aronsohn, M.S.; Brown, H.M.; Hauptman, G.; Kornberg, L.J. Expression of focal adhesion kinase and phosphorylated focal adhesion kinase in squamous cell carcinoma of the larynx. Laryngoscope 2003, 113, 1944–1948. [Google Scholar] [CrossRef] [PubMed]
- Lark, A.L.; Livasy, C.A.; Calvo, B.; Caskey, L.; Moore, D.T.; Yang, X.; Cance, W.G. Overexpression of Focal Adhesion Kinase in Primary Colorectal Carcinomas and Colorectal Liver Metastases. Clin. Cancer Res. 2003, 9, 215–222. [Google Scholar] [PubMed]
- Oktay, M.H.; Oktay, K.; Hamele-Bena, D.; Buyuk, A.; Koss, L.G. Focal adhesion kinase as a marker of malignant phenotype in breast and cervical carcinomas. Hum. Pathol. 2003, 34, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, S.E.; Kouraklis, G.P.; Kakisis, J.D.; Kanelli, H.G.; Apostolakou, F.E.; Karatzas, G.M.; Koutselinis, A.S. Focal adhesion kinase expression is not a prognostic predictor in colon adenocarcinoma patients. Eur. J. Surg. Oncol. 2003, 29, 571–574. [Google Scholar] [CrossRef]
- Itoh, S.; Maeda, T.; Shimada, M.; Aishima, S.; Shirabe, K.; Tanaka, S.; Maehara, Y. Role of expression of focal adhesion kinase in progression of hepatocellular carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 2812–2817. [Google Scholar] [CrossRef]
- Kim, S.J.; Park, J.W.; Yoon, J.S.; Mok, J.O.; Kim, Y.J.; Park, H.K.; Kim, C.H.; Byun, D.W.; Lee, Y.J.; Jin, S.Y.; et al. Increased expression of focal adhesion kinase in thyroid cancer: Immunohistochemical study. J. Korean Med. Sci. 2004, 19, 710–715. [Google Scholar] [CrossRef]
- Lightfoot, H.M.; Lark, A.; Livasy, C.A.; Moore, D.T.; Cowan, D.; Dressler, L.; Craven, R.J.; Cance, W.G. Upregulation of focal adhesion kinase (FAK) expression in ductal carcinoma in situ (DCIS) is an early event in breast tumorigenesis. Breast Cancer Res. Treat. 2004, 88, 109–116. [Google Scholar] [CrossRef]
- Lark, A.L.; Livasy, C.A.; Dressler, L.; Moore, D.T.; Millikan, R.C.; Geradts, J.; Iacocca, M.; Cowan, D.; Little, D.; Craven, R.J.; et al. High focal adhesion kinase expression in invasive breast carcinomas is associated with an aggressive phenotype. Mod. Pathol. 2005, 18, 1289–1294. [Google Scholar] [CrossRef]
- Canel, M.; Secades, P.; Rodrigo, J.-P.; Cabanillas, R.; Herrero, A.; Suarez, C.; Chiara, M.-D. Overexpression of Focal Adhesion Kinase in Head and Neck Squamous Cell Carcinoma Is Independent of ak Gene Copy Number. Clin. Cancer Res. 2006, 12, 3272–3279. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, J.P.; Dominguez, F.; Suárez, V.; Canel, M.; Secades, P.; Chiara, M.D. Focal Adhesion Kinase and E-Cadherin as Markers for Nodal Metastasis in Laryngeal Cancer. Arch. Otolaryngol. Head Neck Surg. 2007, 133, 145–150. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Beierle, E.A.; Massoll, N.A.; Hartwich, J.; Kurenova, E.V.; Golubovskaya, V.M.; Cance, W.G.; McGrady, P.; London, W.B. Focal adhesion kinase expression in human neuroblastoma: Immunohistochemical and real-time PCR analyses. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 3299–3305. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Naomoto, Y.; Yamatsuji, T.; Okawa, T.; Shirakawa, Y.; Gunduz, M.; Nobuhisa, T.; Takaoka, M.; Sirmali, M.; Nakajima, M.; et al. Localization of FAK is related with colorectal carcinogenesis. Int. J. Oncol. 2008, 32, 791–796. [Google Scholar]
- Cai, L.; Han, J.; Zhuo, X.; Xiong, Y.; Dong, J.; Li, X. Overexpression and significance of focal adhesion kinase in hepatocellular carcinoma and its relationship with HBV infection. Med. Oncol. 2009, 26, 409–414. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Conway-Dorsey, K.; Edmiston, S.N.; Tse, C.K.; Lark, A.A.; Livasy, C.A.; Moore, D.; Millikan, R.C.; Cance, W.G. FAK overexpression and p53 mutations are highly correlated in human breast cancer. Int. J. Cancer. J. Int. Du Cancer 2009, 125, 1735–1738. [Google Scholar] [CrossRef]
- Lau, G.M.; Lau, G.M.; Yu, G.L.; Gelman, I.H.; Gutowski, A.; Hangauer, D.; Fang, J.W. Expression of Src and FAK in hepatocellular carcinoma and the effect of Src inhibitors on hepatocellular carcinoma in vitro. Dig. Dis. Sci. 2009, 54, 1465–1474. [Google Scholar] [CrossRef]
- Theocharis, S.E.; Klijanienko, J.T.; Padoy, E.; Athanassiou, S.; Sastre-Garau, X.X. Focal adhesion kinase (FAK) immunocytochemical expression in breast ductal invasive carcinoma (DIC): Correlation with clinicopathological parameters and tumor proliferative capacity. Med. Sci. Monit. Int. Med J. Exp. Clin. Res. 2009, 15, 221–226. [Google Scholar]
- Lai, I.R.; Chu, P.Y.; Lin, H.S.; Liou, J.Y.; Jan, Y.J.; Lee, J.C.; Shen, T.L. Phosphorylation of focal adhesion kinase at Tyr397 in gastric carcinomas and its clinical significance. Am. J. Pathol. 2010, 177, 1629–1637. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, B.L.; Yoon, J.; Kim, J.; Kim, M.A.; Yang, H.K.; Kim, W.H. Focal adhesion kinase (FAK) gene amplification and its clinical implications in gastric cancer. Hum. Pathol. 2010, 41, 1664–1673. [Google Scholar] [CrossRef]
- Yuan, Z.; Zheng, Q.; Fan, J.; Ai, K.X.; Chen, J.; Huang, X.Y. Expression and prognostic significance of focal adhesion kinase in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2010, 136, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Yom, C.K.; Noh, D.Y.; Kim, W.H.; Kim, H.S. Clinical significance of high focal adhesion kinase gene copy number and overexpression in invasive breast cancer. Breast Cancer Res. Treat 2011, 128, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xia, X.; Wang, S.; Wu, X.; Zhang, J.; Zhou, Y.; Tan, Y.; He, S.; Qiang, F.; Li, A.; et al. High FAK combined with low JWA expression: Clinical prognostic and predictive role for adjuvant fluorouracil-leucovorin-oxaliplatin treatment in resectable gastric cancer patients. J. Gastroenterol. 2013, 48, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- De Vicente, J.C.; Rosado, P.; Lequerica-Fernandez, P.; Allonca, E.; Villallain, L.; Hernandez-Vallejo, G. Focal adhesion kinase overexpression: Correlation with lymph node metastasis and shorter survival in oral squamous cell carcinoma. Head Neck 2013, 35, 826–830. [Google Scholar] [CrossRef]
- Faingold, D.; Filho, V.B.; Fernandes, B.; Jagan, L.; de Barros, A.M.; Orellana, M.E.; Antecka, E.; Burnier, M.N. Expression of focal adhesion kinase in uveal melanoma and the effects of Hsp90 inhibition by 17-AAG. Pathol. Res. Pract. 2014, 210, 739–745. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Ylagan, L.; Miller, A.; Hughes, M.; Wilson, J.; Wang, D.; Brese, E.; Bshara, W.; Edge, S.; Morrison, C.; et al. High focal adhesion kinase expression in breast carcinoma is associated with lymphovascular invasion and triple-negative phenotype. BMC Cancer 2014, 14, 769. [Google Scholar] [CrossRef]
- Stone, R.L.; Baggerly, K.A.; Armaiz-Pena, G.N.; Kang, Y.; Sanguino, A.M.; Thanapprapasr, D.; Dalton, H.J.; Bottsford-Miller, J.; Zand, B.; Akbani, R.; et al. Focal adhesion kinase: An alternative focus for anti-angiogenesis therapy in ovarian cancer. Cancer Biol. Ther. 2014, 15, 919–929. [Google Scholar] [CrossRef][Green Version]
- Li, M.; Hong, L.I.; Liao, M.; Guo, G. Expression and clinical significance of focal adhesion kinase and adrenomedullin in epithelial ovarian cancer. Oncol. Lett. 2015, 10, 1003–1007. [Google Scholar] [CrossRef]
- Ren, K.; Lu, X.; Yao, N.; Chen, Y.; Yang, A.; Chen, H.; Zhang, J.; Wu, S.; Shi, X.; Wang, C.; et al. Focal adhesion kinase overexpression and its impact on human osteosarcoma. Oncotarget 2015, 6, 31085–31103. [Google Scholar] [CrossRef]
- Gomez Del Pulgar, T.; Cebrian, A.; Fernandez-Acenero, M.J.; Borrero-Palacios, A.; Del Puerto-Nevado, L.; Martinez-Useros, J.; Marin-Arango, J.P.; Carames, C.; Vega-Bravo, R.; Rodriguez-Remirez, M.; et al. Focal adhesion kinase: Predictor of tumour response and risk factor for recurrence after neoadjuvant chemoradiation in rectal cancer. J. Cell. Mol. Med. 2016, 20, 1729–1736. [Google Scholar] [CrossRef]
- Omura, G.; Ando, M.; Saito, Y.; Kobayashi, K.; Yoshida, M.; Ebihara, Y.; Kanaya, K.; Fujimoto, C.; Sakamoto, T.; Kondo, K.; et al. Association of the upregulated expression of focal adhesion kinase with poor prognosis and tumor dissemination in hypopharyngeal cancer. Head Neck 2016, 38, 1164–1169. [Google Scholar] [CrossRef] [PubMed]
- Almstedt, K.; Sicking, I.; Battista, M.J.; Huangfu, S.; Heimes, A.S.; Weyer-Elberich, V.; Hasenburg, A.; Schmidt, M. Prognostic Significance of Focal Adhesion Kinase in Node-Negative Breast Cancer. Breast Care 2017, 12, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Thanapprapasr, K.; Nartthanarung, A.; Thanapprapasr, D.; Jinawath, A. pFAK-Y397 overexpression as both a prognostic and a predictive biomarker for patients with metastatic osteosarcoma. PLoS ONE 2017, 12, e0182989. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.J.; Zhou, B. Focal adhesion kinase promotes progression and predicts poor clinical outcomes in patients with osteosarcoma. Oncol. Lett. 2018, 15, 6225–6232. [Google Scholar] [CrossRef]
- Munguia-Calzada, P.; Fernandez-Vega, I.; Martinez-Camblor, P.; Diaz-Coto, S.; Garcia-Pedrero, J.M.; Vivanco, B.; Osuna, C.G.; Vazquez-Lopez, F.; Rodrigo, J.P.; Santos-Juanes, J. Correlation of focal adhesion kinase expression with nodal metastasis in patients with head and neck cutaneous squamous cell carcinoma. Head Neck 2018, 41, 1291–1296. [Google Scholar] [CrossRef]
- Schmitz, K.J.; Grabellus, F.; Callies, R.; Otterbach, F.; Wohlschlaeger, J.; Levkau, B.; Kimmig, R.; Schmid, K.W.; Baba, H.A. High expression of focal adhesion kinase (p125FAK) in node-negative breast cancer is related to overexpression of HER-2/neu and activated Akt kinase but does not predict outcome. Breast Cancer Res. BCR 2005, 7, 194–203. [Google Scholar] [CrossRef]
- Fujii, T.; Koshikawa, K.; Nomoto, S.; Okochi, O.; Kaneko, T.; Inoue, S.; Yatabe, Y.; Takeda, S.; Nakao, A. Focal adhesion kinase is overexpressed in hepatocellular carcinoma and can be served as an independent prognostic factor. J. Hepatol. 2004, 41, 104–111. [Google Scholar] [CrossRef]
- Sun, C.K.; Ng, K.T.; Sun, B.S.; Ho, J.W.Y.; Lee, T.K.; Ng, I.; Poon, R.T.P.; Lo, C.M.; Liu, C.L.; Man, K.; et al. The significance of proline-rich tyrosine kinase2 (Pyk2) on hepatocellular carcinoma progression and recurrence. Br. J. Cancer 2007, 97, 50. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851. [Google Scholar] [CrossRef]
- In The Cancer Genome Atlas (TCGA) Database. Available online: http://www.cbioportal.org/ (accessed on 20 May 2019).
- Agochiya, M.; Brunton, V.G.; Owens, D.W.; Parkinson, E.K.; Paraskeva, C.; Keith, W.N.; Frame, M.C. Increased dosage and amplification of the focal adhesion kinase gene in human cancer cells. Oncogene 1999, 18, 5646–5653. [Google Scholar] [CrossRef]
- Damstrup, L.; Rygaard, K.; Spang-Thomsen, M.; Skovgaard Poulsen, H. Expression of transforming growth factor beta (TGF beta) receptors and expression of TGF beta 1, TGF beta 2 and TGF beta 3 in human small cell lung cancer cell lines. Br. J. Cancer 1993, 67, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Dowell, J.E.; Amirkhan, R.H.; Lai, W.S.; Frawley, W.H.; Minna, J.D. Survival in small cell lung cancer is independent of tumor expression of VEGF and COX-2. Anticancer Res. 2004, 24, 2367–2373. [Google Scholar] [PubMed]
- Ma, P.C.; Tretiakova, M.S.; Nallasura, V.; Jagadeeswaran, R.; Husain, A.N.; Salgia, R. Downstream signalling and specific inhibition of c-MET/HGF pathway in small cell lung cancer: Implications for tumour invasion. Br. J. Cancer 2007, 97, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, H.; Badzio, A.; Boyle, T.A.; Schildhaus, H.-U.; Lu, X.; Dziadziuszko, R.; Jassem, J.; Varella-Garcia, M.; Heasley, L.E.; et al. Fibroblast Growth Factor Receptor 1 and Related Ligands in Small-Cell Lung Cancer. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2015, 10, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Lee, J.H.; Abdullaev, Z.; Park, K.S.; Pineda, M.; Saidkhodjaeva, L.; Miettinen, M.; Wang, Y.; Pack, S.D.; Giaccone, G. Characterization of fibroblast growth factor receptor 1 in small-cell lung cancer. J. Thorac. Oncol. 2014, 9, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Lucchi, M.; Mussi, A.; Fontanini, G.; Faviana, P.; Ribechini, A.; Angeletti, C.A. Small cell lung carcinoma (SCLC): The angiogenic phenomenon. Eur. J. Cardio Thorac. Surg. Off. J. Eur. Assoc. Cardio Thorac. Surg. 2002, 21, 1105–1110. [Google Scholar] [CrossRef]
- Beviglia, L.; Kramer, R.H. HGF induces FAK activation and integrin-mediated adhesion in MTLn3 breast carcinoma cells. Int. J. Cancer. J. Int. Du Cancer 1999, 83, 640–649. [Google Scholar] [CrossRef]
- Rodríguez-Fernández, J.L.; Rozengurt, E. Bombesin, Vasopressin, Lysophosphatidic Acid, and Sphingosylphosphorylcholine Induce Focal Adhesion Kinase Activation in Intact Swiss 3T3 Cells. J. Biol. Chem. 1998, 273, 19321–19328. [Google Scholar] [CrossRef]
- Leyton, J.; Garcia-Marin, L.J.; Tapia, J.A.; Jensen, R.T.; Moody, T.W. Bombesin and gastrin releasing peptide increase tyrosine phosphorylation of focal adhesion kinase and paxillin in non-small cell lung cancer cells. Cancer Lett. 2001, 162, 87–95. [Google Scholar] [CrossRef]
- Zhou, B.; Wang, G.-Z.; Zhou, G.-B.; Wen, Z.-S.; Zhou, Y.-C.; Huang, Y.-C.; Chen, Y. Somatic Mutations and Splicing Variants of Focal Adhesion Kinase in Non–Small Cell Lung Cancer. JNCI J. Natl. Cancer Inst. 2017, 110, 195–204. [Google Scholar] [CrossRef]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.Q.; Li, N.; Ma, L.L.; Tseng, Y.J.; Zhao, N.Q.; Chen, S.Y. Prognostic Value of Focal Adhesion Kinase (FAK) in Human Solid Carcinomas: A Meta-Analysis. PLoS ONE 2016, 11, e0162666. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.; Kaur, A.; Cance, W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: Nuclear factor kappa B and p53 binding sites. Biochim. Biophys. Acta 2004, 1678, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Li, Y.; Han, Z.G. Argonaute2 promotes tumor metastasis by way of up-regulating focal adhesion kinase expression in hepatocellular carcinoma. Hepatology 2013, 57, 1906–1918. [Google Scholar] [CrossRef]
- Golubovskaya, V.M. FAK and Nanog cross talk with p53 in cancer stem cells. Anticancer Agents Med. Chem. 2013, 13, 576–580. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretic, L.; Kong, G.; Leenders, F.; Lu, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]
- Tallett, A.; Chilvers, E.R.; MacKinnon, A.C.; Haslett, C.; Sethi, T. Neuropeptides stimulate tyrosine phosphorylation and tyrosine kinase activity in small cell lung cancer cell lines. Peptides 1996, 17, 665–673. [Google Scholar] [CrossRef]
- Frisch, S.M.; Vuori, K.; Ruoslahti, E.; Chan-Hui, P.Y. Control of adhesion-dependent cell survival by focal adhesion kinase. J. Cell Biol. 1996, 134, 793–799. [Google Scholar] [CrossRef]
- Kurenova, E.; Xu, L.H.; Yang, X.; Baldwin, A.S.; Craven, R.J.; Hanks, S.K.; Liu, Z.G.; Cance, W.G. Focal adhesion kinase suppresses apoptosis by binding to the death domain of receptor-interacting protein. Mol. Cell. Biol. 2004, 24, 4361–4371. [Google Scholar] [CrossRef]
- Schlaepfer, D.D.; Hanks, S.K.; Hunter, T.; van der Geer, P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature 1994, 372, 786–791. [Google Scholar] [CrossRef]
- Oktay, M.; Wary, K.K.; Dans, M.; Birge, R.B.; Giancotti, F.G. Integrin-mediated activation of focal adhesion kinase is required for signaling to Jun NH2-terminal kinase and progression through the G1 phase of the cell cycle. J. Cell Biol. 1999, 145, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bian, Z.C.; Yee, K.; Chen, B.P.; Chien, S.; Guan, J.L. Identification of transcription factor KLF8 as a downstream target of focal adhesion kinase in its regulation of cyclin D1 and cell cycle progression. Mol. Cell 2003, 11, 1503–1515. [Google Scholar] [CrossRef]
- Zhao, J.; Pestell, R.; Guan, J.L. Transcriptional activation of cyclin D1 promoter by FAK contributes to cell cycle progression. Mol. Biol. Cell 2001, 12, 4066–4077. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.H.; Reiske, H.; Guan, J.L. Regulation of the cell cycle by focal adhesion kinase. J. Cell Biol. 1998, 143, 1997–2008. [Google Scholar] [CrossRef]
- Bond, M.; Sala-Newby, G.B.; Newby, A.C. Focal adhesion kinase (FAK)-dependent regulation of S-phase kinase-associated protein-2 (Skp-2) stability. A novel mechanism regulating smooth muscle cell proliferation. J. Biol. Chem. 2004, 279, 37304–37310. [Google Scholar] [CrossRef]
- Bryant, P.; Zheng, Q.; Pumiglia, K. Focal adhesion kinase controls cellular levels of p27/Kip1 and p21/Cip1 through Skp2-dependent and -independent mechanisms. Mol. Cell. Biol. 2006, 26, 4201–4213. [Google Scholar] [CrossRef]
- Carrano, A.C.; Eytan, E.; Hershko, A.; Pagano, M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol. 1999, 1, 193–199. [Google Scholar] [CrossRef]
- Ding, Q.; Grammer, J.R.; Nelson, M.A.; Guan, J.L.; Stewart, J.E.; Gladson, C.L. p27Kip1 and cyclin D1 are necessary for focal adhesion kinase regulation of cell cycle progression in glioblastoma cells propagated in vitro and in vivo in the scid mouse brain. J. Biol. Chem. 2005, 280, 6802–6815. [Google Scholar] [CrossRef]
- Walker, J.L.; Fournier, A.K.; Assoian, R.K. Regulation of growth factor signaling and cell cycle progression by cell adhesion and adhesion-dependent changes in cellular tension. Cytokine Growth Factor Rev. 2005, 16, 395–405. [Google Scholar] [CrossRef]
- Haskell, H.; Natarajan, M.; Hecker, T.P.; Ding, Q.; Stewart, J.; Grammer, J.R.; Gladson, C.L. Focal adhesion kinase is expressed in the angiogenic blood vessels of malignant astrocytic tumors in vivo and promotes capillary tube formation of brain microvascular endothelial cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 2157–2165. [Google Scholar]
- Hauck, C.R.; Hsia, D.A.; Ilic, D.; Schlaepfer, D.D. v-Src SH3-enhanced interaction with focal adhesion kinase at beta 1 integrin-containing invadopodia promotes cell invasion. J. Biol. Chem. 2002, 277, 12487–12490. [Google Scholar] [CrossRef] [PubMed]
- Hsia, D.A.; Mitra, S.K.; Hauck, C.R.; Streblow, D.N.; Nelson, J.A.; Ilic, D.; Huang, S.; Li, E.; Nemerow, G.R.; Leng, J.; et al. Differential regulation of cell motility and invasion by FAK. J. Cell Biol. 2003, 160, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, L.J. Focal adhesion kinase and its potential involvement in tumor invasion and metastasis. Head Neck 1998, 20, 745–752. [Google Scholar] [CrossRef]
- Burridge, K.; Turner, C.E.; Romer, L.H. Tyrosine phosphorylation of paxillin and pp125FAK accompanies cell adhesion to extracellular matrix: A role in cytoskeletal assembly. J. Cell Biol. 1992, 119, 893–903. [Google Scholar] [CrossRef]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef]
- Sheetz, M.P.; Felsenfeld, D.P.; Galbraith, C.G. Cell migration: Regulation of force on extracellular-matrix-integrin complexes. Trends Cell. Biol. 1998, 8, 51–54. [Google Scholar] [CrossRef]
- Pelham, R.J.; Wang, Y. High resolution detection of mechanical forces exerted by locomoting fibroblasts on the substrate. Mol. Biol. Cell. 1999, 10, 935–945. [Google Scholar] [CrossRef]
- Katsumi, A.; Orr, A.W.; Tzima, E.; Schwartz, M.A. Integrins in mechanotransduction. J. Biol. Chem. 2004, 279, 12001–12004. [Google Scholar] [CrossRef]
- Hanks, S.K.; Ryzhova, L.; Shin, N.Y.; Brabek, J. Focal adhesion kinase signaling activities and their implications in the control of cell survival and motility. Front. Biosci. A J. Virtual Libr. 2003, 8, 982–996. [Google Scholar] [CrossRef]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell. Biol. 2005, 6, 56–68. [Google Scholar] [CrossRef]
- Parsons, J.T. Focal adhesion kinase: The first ten years. J. Cell Sci. 2003, 116, 1409–1416. [Google Scholar] [CrossRef] [PubMed]
- Siesser, P.M.; Hanks, S.K. The signaling and biological implications of FAK overexpression in cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 3233–3237. [Google Scholar] [CrossRef] [PubMed]
- Webb, D.J.; Donais, K.; Whitmore, L.A.; Thomas, S.M.; Turner, C.E.; Parsons, J.T.; Horwitz, A.F. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 2004, 6, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Tilghman, R.W.; Slack-Davis, J.K.; Sergina, N.; Martin, K.H.; Iwanicki, M.; Hershey, E.D.; Beggs, H.E.; Reichardt, L.F.; Parsons, J.T. Focal adhesion kinase is required for the spatial organization of the leading edge in migrating cells. J. Cell Sci. 2005, 118, 2613–2623. [Google Scholar] [CrossRef] [PubMed]
- Klemke, R.L.; Leng, J.; Molander, R.; Brooks, P.C.; Vuori, K.; Cheresh, D.A. CAS/Crk coupling serves as a “molecular switch” for induction of cell migration. J. Cell Biol. 1998, 140, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Cary, L.A.; Chang, J.F.; Guan, J.L. Stimulation of cell migration by overexpression of focal adhesion kinase and its association with Src and Fyn. J. Cell Sci. 1996, 109, 1787–1794. [Google Scholar] [PubMed]
- Cary, L.A.; Han, D.C.; Polte, T.R.; Hanks, S.K.; Guan, J.L. Identification of p130Cas as a mediator of focal adhesion kinase-promoted cell migration. J. Cell Biol. 1998, 140, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Owen, J.D.; Ruest, P.J.; Fry, D.W.; Hanks, S.K. Induced focal adhesion kinase (FAK) expression in FAK-null cells enhances cell spreading and migration requiring both auto- and activation loop phosphorylation sites and inhibits adhesion-dependent tyrosine phosphorylation of Pyk2. Mol. Cell. Biol. 1999, 19, 4806–4818. [Google Scholar] [CrossRef]
- Sieg, D.J.; Hauck, C.R.; Schlaepfer, D.D. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J. Cell Sci. 1999, 112, 2677–2691. [Google Scholar]
- Han, D.C.; Guan, J.L. Association of focal adhesion kinase with Grb7 and its role in cell migration. J. Biol. Chem. 1999, 274, 24425–24430. [Google Scholar] [CrossRef]
- Han, D.C.; Shen, T.L.; Guan, J.L. Role of Grb7 targeting to focal contacts and its phosphorylation by focal adhesion kinase in regulation of cell migration. J. Biol. Chem. 2000, 275, 28911–28917. [Google Scholar] [CrossRef] [PubMed]
- Irby, R.B.; Yeatman, T.J. Increased Src activity disrupts cadherin/catenin-mediated homotypic adhesion in human colon cancer and transformed rodent cells. Cancer Res. 2002, 62, 2669–2674. [Google Scholar] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell. Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Avizienyte, E.; Frame, M.C. Src and FAK signalling controls adhesion fate and the epithelial-to-mesenchymal transition. Curr. Opin. Cell Biol. 2005, 17, 542–547. [Google Scholar] [CrossRef] [PubMed]
- McLean, G.W.; Carragher, N.O.; Avizienyte, E.; Evans, J.; Brunton, V.G.; Frame, M.C. The role of focal-adhesion kinase in cancer—A new therapeutic opportunity. Nat. Rev. Cancer 2005, 5, 505–515. [Google Scholar] [CrossRef]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef]
- Canadas, I.; Taus, A.; Gonzalez, I.; Villanueva, X.; Gimeno, J.; Pijuan, L.; Domine, M.; Sanchez-Font, A.; Vollmer, I.; Menendez, S.; et al. High circulating hepatocyte growth factor levels associate with epithelial to mesenchymal transition and poor outcome in small cell lung cancer patients. Oncotarget 2014, 5, 5246–5256. [Google Scholar] [CrossRef]
- Ito, T.; Kudoh, S.; Ichimura, T.; Fujino, K.; Hassan, W.A.; Udaka, N. Small cell lung cancer, an epithelial to mesenchymal transition (EMT)-like cancer: Significance of inactive Notch signaling and expression of achaete-scute complex homologue 1. Hum. Cell 2017, 30, 1–10. [Google Scholar] [CrossRef]
- Zhao, L.; Li, J.; Liu, Y.; Zhou, W.; Shan, Y.; Fan, X.; Zhou, X.; Shan, B.; Song, Y.; Zhan, Q. Flotillin1 promotes EMT of human small cell lung cancer via TGF-beta signaling pathway. Cancer Biol. Med. 2018, 15, 400–414. [Google Scholar] [CrossRef]
- Cicchini, C.; Laudadio, I.; Citarella, F.; Corazzari, M.; Steindler, C.; Conigliaro, A.; Fantoni, A.; Amicone, L.; Tripodi, M. TGFbeta-induced EMT requires focal adhesion kinase (FAK) signaling. Exp. Cell Res. 2008, 314, 143–152. [Google Scholar] [CrossRef]
- Fan, H.; Zhao, X.; Sun, S.; Luo, M.; Guan, J.L. Function of focal adhesion kinase scaffolding to mediate endophilin A2 phosphorylation promotes epithelial-mesenchymal transition and mammary cancer stem cell activities in vivo. J. Biol. Chem. 2013, 288, 3322–3333. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Zhou, X.; Rowe, R.G.; Hu, Y.; Schlaepfer, D.D.; Ilic, D.; Dressler, G.; Park, A.; Guan, J.L.; Weiss, S.J. Snail1 controls epithelial-mesenchymal lineage commitment in focal adhesion kinase-null embryonic cells. J. Cell Biol. 2011, 195, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Urvalek, A.M.; Liu, J.; Zhao, J. Activation of KLF8 transcription by focal adhesion kinase in human ovarian epithelial and cancer cells. J. Biol. Chem. 2008, 283, 13934–13942. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zheng, M.; Liu, G.; Xia, W.; McKeown-Longo, P.J.; Hung, M.C.; Zhao, J. Kruppel-like factor 8 induces epithelial to mesenchymal transition and epithelial cell invasion. Cancer Res. 2007, 67, 7184–7193. [Google Scholar] [CrossRef]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; De Herreros, A.G. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84. [Google Scholar] [CrossRef]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor Snail controls epithelial—Mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Liu, S.Q.; Xu, C.Y.; Wu, W.H.; Fu, Z.H.; He, S.W.; Qin, M.B.; Huang, J.A. Sphingosine kinase 1 promotes the metastasis of colorectal cancer by inducing the epithelialmesenchymal transition mediated by the FAK/AKT/MMPs axis. Int. J. Oncol. 2019, 54, 41–52. [Google Scholar] [CrossRef]
- Xu, C.Y.; Liu, S.Q.; Qin, M.B.; Zhuge, C.F.; Qin, L.; Qin, N.; Lai, M.Y.; Huang, J.A. SphK1 modulates cell migration and EMT-related marker expression by regulating the expression of p-FAK in colorectal cancer cells. Int. J. Mol. Med. 2017, 39, 1277–1284. [Google Scholar] [CrossRef]
- Taliaferro-Smith, L.; Oberlick, E.; Liu, T.; McGlothen, T.; Alcaide, T.; Tobin, R.; Donnelly, S.; Commander, R.; Kline, E.; Nagaraju, G.P.; et al. FAK activation is required for IGF1R-mediated regulation of EMT, migration, and invasion in mesenchymal triple negative breast cancer cells. Oncotarget 2015, 6, 4757. [Google Scholar] [CrossRef]
- Wilson, C.; Nicholes, K.; Bustos, D.; Lin, E.; Song, Q.; Stephan, J.P.; Kirkpatrick, D.S.; Settleman, J. Overcoming EMT-associated resistance to anti-cancer drugs via Src/FAK pathway inhibition. Oncotarget 2014, 5, 7328–7341. [Google Scholar] [CrossRef]
- Azzi, S.; Hebda, J.; Gavard, J. Vascular Permeability and Drug Delivery in Cancers. Front. Oncol. 2013, 3, 211. [Google Scholar] [CrossRef] [PubMed]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef] [PubMed]
- Reymond, N.; d’Água, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858. [Google Scholar] [CrossRef]
- Williamson, S.C.; Metcalf, R.L.; Trapani, F.; Mohan, S.; Antonello, J.; Abbott, B.; Leong, H.S.; Chester, C.P.E.; Simms, N.; Polanski, R.; et al. Vasculogenic mimicry in small cell lung cancer. Nat. Commun. 2016, 7, 13322. [Google Scholar] [CrossRef] [PubMed]
- Yancopoulos, G.D.; Davis, S.; Gale, N.W.; Rudge, J.S.; Wiegand, S.J.; Holash, J. Vascular-specific growth factors and blood vessel formation. Nature 2000, 407, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249. [Google Scholar] [CrossRef]
- Salven, P.; Ruotsalainen, T.; Mattson, K.; Joensuu, H. High pre-treatment serum level of vascular endothelial growth factor (VEGF) is associated with poor outcome in small-cell lung cancer. Int. J. Cancer 1998, 79, 144–146. [Google Scholar] [CrossRef]
- Hodgkinson, C.L.; Morrow, C.J.; Li, Y.; Metcalf, R.L.; Rothwell, D.G.; Trapani, F.; Polanski, R.; Burt, D.J.; Simpson, K.L.; Morris, K.; et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat. Med. 2014, 20, 897–903. [Google Scholar] [CrossRef]
- Gaspar, L.E.; McNamara, E.J.; Gay, E.G.; Putnam, J.B.; Crawford, J.; Herbst, R.S.; Bonner, J.A. Small-Cell Lung Cancer: Prognostic Factors and Changing Treatment Over 15 Years. Clin. Lung Cancer 2012, 13, 115–122. [Google Scholar] [CrossRef]
- Hamilton, G.; Moser, D.; Hochmair, M. Metastasis: Circulating Tumor Cells in Small Cell Lung Cancer. Trends Cancer 2016, 2, 159–160. [Google Scholar] [CrossRef]
- Sun, J.-M.; Lee, K.H.; Kim, B.-S.; Kim, H.-G.; Min, Y.J.; Yi, S.Y.; Yun, H.J.; Jung, S.-H.; Lee, S.-H.; Ahn, J.S.; et al. Pazopanib maintenance after first-line etoposide and platinum chemotherapy in patients with extensive disease small-cell lung cancer: A multicentre, randomised, placebo-controlled Phase II study (KCSG-LU12-07). Br. J. Cancer 2018, 118, 648. [Google Scholar] [CrossRef] [PubMed]
- Marcello, T.; Boni, L.; Ambrosio, F.; Camerini, A.; Baldini, E.; Cinieri, S.; Brighenti, M.; Zanelli, F.; Defraia, E.; Chiari, R.; et al. Italian, Multicenter, Phase III, Randomized Study of Cisplatin Plus Etoposide With or Without Bevacizumab as First-Line Treatment in Extensive-Disease Small-Cell Lung Cancer: The GOIRC-AIFA FARM6PMFJM Trial. J. Clin. Oncol. 2017, 35, 1281–1287. [Google Scholar] [CrossRef]
- Spigel, D.R.; Townley, P.M.; Waterhouse, D.M.; Fang, L.; Adiguzel, I.; Huang, J.E.; Karlin, D.A.; Faoro, L.; Scappaticci, F.A.; Socinski, M.A. Randomized Phase II Study of Bevacizumab in Combination with Chemotherapy in Previously Untreated Extensive-Stage Small-Cell Lung Cancer: Results From the SALUTE Trial. J. Clin. Oncol. 2011, 29, 2215–2222. [Google Scholar] [CrossRef] [PubMed]
- Ready, N.E.; Pang, H.H.; Gu, L.; Otterson, G.A.; Thomas, S.P.; Miller, A.A.; Baggstrom, M.; Masters, G.A.; Graziano, S.L.; Crawford, J.; et al. Chemotherapy With or Without Maintenance Sunitinib for Untreated Extensive-Stage Small-Cell Lung Cancer: A Randomized, Double-Blind, Placebo-Controlled Phase II Study-CALGB 30504 (Alliance). J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 1660–1665. [Google Scholar] [CrossRef] [PubMed]
- Ilic, D.; Kovacic, B.; McDonagh, S.; Jin, F.; Baumbusch, C.; Gardner David, G.; Damsky Caroline, H. Focal Adhesion Kinase Is Required for Blood Vessel Morphogenesis. Circ. Res. 2003, 92, 300–307. [Google Scholar] [CrossRef]
- Ueda, H.; Nagy, T.; Shen, T.-L.; Peng, X.; Guan, J.-L.; Stokol, T.; Zhou, H.; Vassalli, J.-D.; Alcaraz, A. Overexpression of focal adhesion kinase in vascular endothelial cells promotes angiogenesis in transgenic mice. Cardiovasc. Res. 2004, 64, 421–430. [Google Scholar] [CrossRef]
- Chen, X.L.; Nam, J.-O.; Jean, C.; Lawson, C.; Walsh, C.T.; Goka, E.; Lim, S.-T.; Tomar, A.; Tancioni, I.; Uryu, S.; et al. VEGF-induced vascular permeability is mediated by FAK. Dev. Cell 2012, 22, 146–157. [Google Scholar] [CrossRef]
- Jean, C.; Chen, X.L.; Nam, J.-O.; Tancioni, I.; Uryu, S.; Lawson, C.; Ward, K.K.; Walsh, C.T.; Miller, N.L.G.; Ghassemian, M.; et al. Inhibition of endothelial FAK activity prevents tumor metastasis by enhancing barrier function. J. Cell Biol. 2014, 204, 247–263. [Google Scholar] [CrossRef]
- Stokes, J.B.; Adair, S.J.; Slack-Davis, J.K.; Walters, D.M.; Tilghman, R.W.; Hershey, E.D.; Lowrey, B.; Thomas, K.S.; Bouton, A.H.; Hwang, R.F.; et al. Inhibition of Focal Adhesion Kinase by PF-562,271 Inhibits the Growth and Metastasis of Pancreatic Cancer Concomitant with Altering the Tumor Microenvironment. Mol. Cancer Ther. 2011, 10, 2135–2145. [Google Scholar] [CrossRef]
- Ward, K.K.; Tancioni, I.; Lawson, C.; Miller, N.L.G.; Jean, C.; Chen, X.L.; Uryu, S.; Kim, J.; Tarin, D.; Stupack, D.G.; et al. Inhibition of focal adhesion kinase (FAK) activity prevents anchorage-independent ovarian carcinoma cell growth and tumor progression. Clin. Exp. Metastasis 2013, 30, 579–594. [Google Scholar] [CrossRef]
- Serrels, B.; McGivern, N.; Canel, M.; Byron, A.; Johnson, S.C.; McSorley, H.J.; Quinn, N.; Taggart, D.; Von Kreigsheim, A.; Anderton, S.M.; et al. IL-33 and ST2 mediate FAK-dependent antitumor immune evasion through transcriptional networks. Sci. Signal. 2017, 10, aan8355. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, M.; Bottsford-Miller, J.; Pradeep, S.; Taylor, M.L.; Choi, H.J.; Hansen, J.M.; Dalton, H.J.; Stone, R.L.; Cho, M.S.; Nick, A.M.; et al. FAK regulates platelet extravasation and tumor growth after antiangiogenic therapy withdrawal. J. Clin. Investig. 2016, 126, 1885–1896. [Google Scholar] [CrossRef] [PubMed]
- Borsig, L. The role of platelet activation in tumor metastasis. Expert Rev. Anticancer Ther. 2008, 8, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R. Direct Signaling between Platelets and Cancer Cells Induces an Epithelial-Mesenchymal-Like Transition and Promotes Metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef]
- Cho, M.S.; Bottsford-Miller, J.; Vasquez, H.G.; Stone, R.; Zand, B.; Kroll, M.H.; Sood, A.K.; Afshar-Kharghan, V. Platelets increase the proliferation of ovarian cancer cells. Blood 2012, 120, 4869–4872. [Google Scholar] [CrossRef]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2015, 16, 20. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H.J. DNA Damage, Aging, and Cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- Sabari, J.K.; Lok, B.H.; Laird, J.H.; Poirier, J.T.; Rudin, C.M. Unravelling the biology of SCLC: Implications for therapy. Nat. Rev. Clin. Oncol. 2017, 14, 549–561. [Google Scholar] [CrossRef]
- Behera, M.; Ragin, C.; Kim, S.; Pillai, R.N.; Chen, Z.; Steuer, C.E.; Saba, N.F.; Belani, C.P.; Khuri, F.R.; Ramalingam, S.S.; et al. Trends, predictors, and impact of systemic chemotherapy in small cell lung cancer patients between 1985 and 2005. Cancer 2015, 122, 50–60. [Google Scholar] [CrossRef]
- Faivre-Finn, C.; Snee, M.; Ashcroft, L.; Appel, W.; Barlesi, F.; Bhatnagar, A.; Bezjak, A.; Cardenal, F.; Fournel, P.; Harden, S.; et al. Concurrent once-daily versus twice-daily chemoradiotherapy in patients with limited-stage small-cell lung cancer (CONVERT): An open-label, phase 3, randomised, superiority trial. Lancet Oncol. 2017, 18, 1116–1125. [Google Scholar] [CrossRef]
- Byers, L.A.; Wang, J.; Nilsson, M.B.; Fujimoto, J.; Saintigny, P.; Yordy, J.; Giri, U.; Peyton, M.; Fan, Y.H.; Diao, L.; et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2012, 2, 798–811. [Google Scholar] [CrossRef]
- Pietanza, M.C.; Waqar, S.N.; Krug, L.M.; Dowlati, A.; Hann, C.L.; Chiappori, A.; Owonikoko, T.K.; Woo, K.M.; Cardnell, R.J.; Fujimoto, J.; et al. Randomized, Double-Blind, Phase II Study of Temozolomide in Combination With Either Veliparib or Placebo in Patients With Relapsed-Sensitive or Refractory Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 2386–2394. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Vilimas, R.; Trindade, C.; Erwin-Cohen, R.; Roper, N.; Xi, L.; Krishnasamy, V.; Levy, E.; Mammen, A.; Nichols, S.; et al. Durvalumab in Combination with Olaparib in Patients with Relapsed SCLC: Results from a Phase II Study. J. Thorac. Oncol. 2019, 14, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Constanzo, J.D.; Tang, K.-j.; Rindhe, S.; Melegari, M.; Liu, H.; Tang, X.; Rodriguez-Canales, J.; Wistuba, I.; Scaglioni, P.P. PIAS1-FAK Interaction Promotes the Survival and Progression of Non-Small Cell Lung Cancer. Neoplasia 2016, 18, 282–293. [Google Scholar] [CrossRef]
- Tang, K.-J.; Constanzo, J.D.; Venkateswaran, N.; Melegari, M.; Ilcheva, M.; Morales, J.C.; Skoulidis, F.; Heymach, J.V.; Boothman, D.A.; Scaglioni, P.P. Focal Adhesion Kinase Regulates the DNA Damage Response and Its Inhibition Radiosensitizes Mutant KRAS Lung Cancer. Clin. Cancer Res. 2016, 22, 5851–5863. [Google Scholar] [CrossRef] [PubMed]
- Beinke, C.; Van Beuningen, D.; Cordes, N. Ionizing radiation modules of the expression and tyrosine phosphorylation of the focal adhesion-associated proteins focal adhesion kinase (FAK) and its substrates p130cas and paxillin in A549 human lung carcinoma cells in vitro. Int. J. Radiat. Biol. 2003, 79, 721–731. [Google Scholar] [CrossRef]
- Williams, K.E.; Bundred, N.J.; Landberg, G.; Clarke, R.B.; Farnie, G. Focal adhesion kinase and Wnt signaling regulate human ductal carcinoma in situ stem cell activity and response to radiotherapy. Stem Cells 2015, 33, 327–341. [Google Scholar] [CrossRef]
- Tavora, B.; Reynolds, L.E.; Batista, S.; Demircioglu, F.; Fernandez, I.; Lechertier, T.; Lees, D.M.; Wong, P.P.; Alexopoulou, A.; Elia, G.; et al. Endothelial-cell FAK targeting sensitizes tumours to DNA-damaging therapy. Nature 2014, 514, 112–116. [Google Scholar] [CrossRef]
- Cordes, N.; Frick, S.; Brunner, T.B.; Pilarsky, C.; Grutzmann, R.; Sipos, B.; Kloppel, G.; McKenna, W.G.; Bernhard, E.J. Human pancreatic tumor cells are sensitized to ionizing radiation by knockdown of caveolin-1. Oncogene 2007, 26, 6851–6862. [Google Scholar] [CrossRef]
- Eke, I.; Sandfort, V.; Mischkus, A.; Baumann, M.; Cordes, N. Antiproliferative effects of EGFR tyrosine kinase inhibition and radiation-induced genotoxic injury are attenuated by adhesion to fibronectin. Radiother. Oncol. 2006, 80, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Estrugo, D.; Fischer, A.; Hess, F.; Scherthan, H.; Belka, C.; Cordes, N. Ligand bound beta1 integrins inhibit procaspase-8 for mediating cell adhesion-mediated drug and radiation resistance in human leukemia cells. PLoS ONE 2007, 2, e269. [Google Scholar] [CrossRef] [PubMed]
- Fuks, Z.; Vlodavsky, I.; Andreeff, M.; McLoughlin, M.; Haimovitz-Friedman, A. Effects of extracellular matrix on the response of endothelial cells to radiation in vitro. Eur. J. Cancer 1992, 28, 725–731. [Google Scholar] [CrossRef]
- Li, L.; Dong, X.; Peng, F.; Shen, L. Integrin beta1 regulates the invasion and radioresistance of laryngeal cancer cells by targeting CD147. Cancer Cell Int. 2018, 18, 80. [Google Scholar] [CrossRef]
- Brodin, O.; Arnberg, H.; Bergh, J.; Nilsson, S. Increased radioresistance of an in vitro transformed human small cell lung cancer cell line. Lung Cancer 1995, 12, 183–198. [Google Scholar] [CrossRef]
- Kraus, A.C.; Ferber, I.; Bachmann, S.O.; Specht, H.; Wimmel, A.; Gross, M.W.; Schlegel, J.; Suske, G.; Schuermann, M. In vitro chemo- and radio-resistance in small cell lung cancer correlates with cell adhesion and constitutive activation of AKT and MAP kinase pathways. Oncogene 2002, 21, 8683–8695. [Google Scholar] [CrossRef]
- Kasahara, T.; Koguchi, E.; Funakoshi, M.; Aizu-Yokota, E.; Sonoda, Y. Antiapoptotic action of focal adhesion kinase (FAK) against ionizing radiation. Antioxid. Redox Signal. 2002, 4, 491–499. [Google Scholar] [CrossRef]
- Skinner, H.D.; Giri, U.; Yang, L.; Woo, S.H.; Story, M.D.; Pickering, C.R.; Byers, L.A.; Williams, M.D.; El-Naggar, A.; Wang, J.; et al. Proteomic Profiling Identifies PTK2/FAK as a Driver of Radioresistance in HPV-negative Head and Neck Cancer. Clin. Cancer Res. 2016, 22, 4643–4650. [Google Scholar] [CrossRef]
- Nguemgo Kouam, P.; Buhler, H.; Hero, T.; Adamietz, I.A. The increased adhesion of tumor cells to endothelial cells after irradiation can be reduced by FAK-inhibition. Radiat. Oncol. 2019, 14, 25. [Google Scholar] [CrossRef]
- Hehlgans, S.; Lange, I.; Eke, I.; Cordes, N. 3D cell cultures of human head and neck squamous cell carcinoma cells are radiosensitized by the focal adhesion kinase inhibitor TAE226. Radiother. Oncol. 2009, 92, 371–378. [Google Scholar] [CrossRef]
- Hehlgans, S.; Eke, I.; Cordes, N. Targeting FAK radiosensitizes 3-dimensional grown human HNSCC cells through reduced Akt1 and MEK1/2 signaling. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Eke, I.; Deuse, Y.; Hehlgans, S.; Gurtner, K.; Krause, M.; Baumann, M.; Shevchenko, A.; Sandfort, V.; Cordes, N. beta(1)Integrin/FAK/cortactin signaling is essential for human head and neck cancer resistance to radiotherapy. J. Clin. Investig. 2012, 122, 1529–1540. [Google Scholar] [CrossRef] [PubMed]
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer stem cells: An old idea—A paradigm shift. Cancer Res. 2006, 66, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Afify, S.M.; Seno, M. Conversion of Stem Cells to Cancer Stem Cells: Undercurrent of Cancer Initiation. Cancers 2019, 11, 345. [Google Scholar] [CrossRef]
- Suresh, R.; Ali, S.; Ahmad, A.; Philip, P.A.; Sarkar, F.H. The Role of Cancer Stem Cells in Recurrent and Drug-Resistant Lung Cancer. Adv. Exp. Med. Biol. 2016, 890, 57–74. [Google Scholar] [CrossRef]
- Salcido, C.D.; Larochelle, A.; Taylor, B.J.; Dunbar, C.E.; Varticovski, L. Molecular characterisation of side population cells with cancer stem cell-like characteristics in small-cell lung cancer. Br. J. Cancer 2010, 102, 1636–1644. [Google Scholar] [CrossRef]
- Wang, P.; Gao, Q.; Suo, Z.; Munthe, E.; Solberg, S.; Ma, L.; Wang, M.; Westerdaal, N.A.; Kvalheim, G.; Gaudernack, G. Identification and characterization of cells with cancer stem cell properties in human primary lung cancer cell lines. PLoS ONE 2013, 8, e57020. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, Y.; Qian, H.; Shao, G.; Lu, X.; Chen, Q.; Sun, X.; Chen, D.; Yin, R.; Zhu, H.; et al. Stemness and inducing differentiation of small cell lung cancer NCI-H446 cells. Cell Death Dis. 2013, 4, e633. [Google Scholar] [CrossRef]
- Sarvi, S.; Mackinnon, A.C.; Avlonitis, N.; Bradley, M.; Rintoul, R.C.; Rassl, D.M.; Wang, W.; Forbes, S.J.; Gregory, C.D.; Sethi, T. CD133+ cancer stem-like cells in small cell lung cancer are highly tumorigenic and chemoresistant but sensitive to a novel neuropeptide antagonist. Cancer Res. 2014, 74, 1554–1565. [Google Scholar] [CrossRef]
- Qiu, X.; Wang, Z.; Li, Y.; Miao, Y.; Ren, Y.; Luan, Y. Characterization of sphere-forming cells with stem-like properties from the small cell lung cancer cell line H446. Cancer Lett. 2012, 323, 161–170. [Google Scholar] [CrossRef]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Gutova, M.; Najbauer, J.; Gevorgyan, A.; Metz, M.Z.; Weng, Y.; Shih, C.C.; Aboody, K.S. Identification of uPAR-positive chemoresistant cells in small cell lung cancer. PLoS ONE 2007, 2, e243. [Google Scholar] [CrossRef] [PubMed]
- Codony-Servat, J.; Verlicchi, A.; Rosell, R. Cancer stem cells in small cell lung cancer. Transl. Lung Cancer Res. 2016, 5, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Morise, M.; Hishida, T.; Takahashi, A.; Yoshida, J.; Ohe, Y.; Nagai, K.; Ishii, G. Clinicopathological significance of cancer stem-like cell markers in high-grade neuroendocrine carcinoma of the lung. J. Cancer Res. Clin. Oncol. 2015, 141, 2121–2130. [Google Scholar] [CrossRef]
- Sodja, E.; Rijavec, M.; Koren, A.; Sadikov, A.; Korosec, P.; Cufer, T. The prognostic value of whole blood SOX2, NANOG and OCT4 mRNA expression in advanced small-cell lung cancer. Radiol. Oncol. 2016, 50, 188–196. [Google Scholar] [CrossRef]
- Yang, F.; Gao, Y.; Geng, J.; Qu, D.; Han, Q.; Qi, J.; Chen, G. Elevated expression of SOX2 and FGFR1 in correlation with poor prognosis in patients with small cell lung cancer. Int. J. Clin. Exp. Pathol. 2013, 6, 2846–2854. [Google Scholar]
- Kobayashi, K.; Takahashi, H.; Inoue, A.; Harada, H.; Toshimori, S.; Kobayashi, Y.; Goto, K.; Sugimoto, K.; Yano, H.; Ohnishi, T.; et al. Oct-3/4 promotes migration and invasion of glioblastoma cells. J. Cell Biochem. 2012, 113, 508–517. [Google Scholar] [CrossRef]
- Liu, C.; Li, Y.; Xing, Y.; Cao, B.; Yang, F.; Yang, T.; Ai, Z.; Wei, Y.; Jiang, J. The Interaction between Cancer Stem Cell Marker CD133 and Src Protein Promotes Focal Adhesion Kinase (FAK) Phosphorylation and Cell Migration. J. Biol. Chem. 2016, 291, 15540–15550. [Google Scholar] [CrossRef]
- Begum, A.; Ewachiw, T.; Jung, C.; Huang, A.; Norberg, K.J.; Marchionni, L.; McMillan, R.; Penchev, V.; Rajeshkumar, N.V.; Maitra, A.; et al. The extracellular matrix and focal adhesion kinase signaling regulate cancer stem cell function in pancreatic ductal adenocarcinoma. PLoS ONE 2017, 12, e0180181. [Google Scholar] [CrossRef]
- Ou, J.; Deng, J.; Wei, X.; Xie, G.; Zhou, R.; Yu, L.; Liang, H. Fibronectin extra domain A (EDA) sustains CD133(+)/CD44(+) subpopulation of colorectal cancer cells. Stem Cell Res. 2013, 11, 820–833. [Google Scholar] [CrossRef]
- Guan, J.L. Integrin signaling through FAK in the regulation of mammary stem cells and breast cancer. IUBMB Life 2010, 62, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.-J.; Bak, Y.; Pham, T.-H.; Kwon, S.-B.; Kim, B.-Y.; Hong, J.; Yoon, D.-Y. STK899704 inhibits stemness of cancer stem cells and migration via the FAK-MEK-ERK pathway in HT29 cells. BMB Rep. 2018, 51, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Liu, S.; Diebel, M.E.; Korkaya, H.; Luo, M.; Brown, M.; Wicinski, J.; Cabaud, O.; Charafe-Jauffret, E.; Birnbaum, D.; et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J. Clin. Investig. 2010, 120, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Luo, Q.; Liu, L.; Yang, X.; Zhu, S.; Song, G. Salinomycin attenuates liver cancer stem cell motility by enhancing cell stiffness and increasing F-actin formation via the FAK-ERK1/2 signalling pathway. Toxicology 2017, 384, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Thakur, R.; Trivedi, R.; Rastogi, N.; Singh, M.; Mishra, D.P. Inhibition of STAT3, FAK and Src mediated signaling reduces cancer stem cell load, tumorigenic potential and metastasis in breast cancer. Sci. Rep. 2015, 5, 10194. [Google Scholar] [CrossRef]
- Blum, W.; Pecze, L.; Felley-Bosco, E.; Wu, L.; de Perrot, M.; Schwaller, B. Stem Cell Factor-Based Identification and Functional Properties of In Vitro-Selected Subpopulations of Malignant Mesothelioma Cells. Stem Cell Rep. 2017, 8, 1005–1017. [Google Scholar] [CrossRef]
- Shapiro, I.M.; Kolev, V.N.; Vidal, C.M.; Kadariya, Y.; Ring, J.E.; Wright, Q.; Weaver, D.T.; Menges, C.; Padval, M.; McClatchey, A.I.; et al. Merlin deficiency predicts FAK inhibitor sensitivity: A synthetic lethal relationship. Sci. Transl. Med. 2014, 6, 237–268. [Google Scholar] [CrossRef]
- Luo, M.; Fan, H.; Nagy, T.; Wei, H.; Wang, C.; Liu, S.; Wicha, M.S.; Guan, J.L. Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Res. 2009, 69, 466–474. [Google Scholar] [CrossRef]
- Kolev, V.N.; Tam, W.F.; Wright, Q.G.; McDermott, S.P.; Vidal, C.M.; Shapiro, I.M.; Xu, Q.; Wicha, M.S.; Pachter, J.A.; Weaver, D.T. Inhibition of FAK kinase activity preferentially targets cancer stem cells. Oncotarget 2017, 8, 51733–51747. [Google Scholar] [CrossRef]
- Schober, M.; Fuchs, E. Tumor-initiating stem cells of squamous cell carcinomas and their control by TGF-beta and integrin/focal adhesion kinase (FAK) signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 10544–10549. [Google Scholar] [CrossRef]
- Vishnubalaji, R.; Manikandan, M.; Fahad, M.; Hamam, R.; Alfayez, M.; Kassem, M.; Aldahmash, A.; Alajez, N.M. Molecular profiling of ALDH1(+) colorectal cancer stem cells reveals preferential activation of MAPK, FAK, and oxidative stress pro-survival signalling pathways. Oncotarget 2018, 9, 13551–13564. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Zhao, X.; Chen, S.; Liu, S.; Wicha, M.S.; Guan, J.L. Distinct FAK activities determine progenitor and mammary stem cell characteristics. Cancer Res. 2013, 73, 5591–5602. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Mok, T.S.K.; Nishio, M.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): Key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir. Med. 2019, 7, 387–401. [Google Scholar] [CrossRef]
- Gandini, S.; Massi, D.; Mandala, M. PD-L1 expression in cancer patients receiving anti PD-1/PD-L1 antibodies: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2016, 100, 88–98. [Google Scholar] [CrossRef]
- Ott, P.A.; Elez, E.; Hiret, S.; Kim, D.-W.; Morosky, A.; Saraf, S.; Piperdi, B.; Mehnert, J.M. Pembrolizumab in Patients With Extensive-Stage Small-Cell Lung Cancer: Results From the Phase Ib KEYNOTE-028 Study. J. Clin. Oncol. 2017, 35, 3823–3829. [Google Scholar] [CrossRef]
- Wang, W.; Hodkinson, P.; McLaren, F.; MacKinnon, A.; Wallace, W.; Howie, S.; Sethi, T. Small cell lung cancer tumour cells induce regulatory T lymphocytes, and patient survival correlates negatively with FOXP3+ cells in tumour infiltrate. Int. J. Cancer 2012, 131, 928–937. [Google Scholar] [CrossRef]
- Serrels, A.; Lund, T.; Serrels, B.; Byron, A.; McPherson, R.C.; von Kriegsheim, A.; Gomez-Cuadrado, L.; Canel, M.; Muir, M.; Ring, J.E.; et al. Nuclear FAK controls chemokine transcription, Tregs, and evasion of anti-tumor immunity. Cell 2015, 163, 160–173. [Google Scholar] [CrossRef]
- Ring, J.; Li, Y.; Shapiro, I.; Wang, Y.; Weaver, D.; Pachter, J. FAK/PYK2 inhibitors defactinib and VS-4718 enhance immune checkpoint inhibitor efficacy. J. Immunother. Cancer 2015, 3. [Google Scholar] [CrossRef]
- Hoelzinger, D.B.; Smith, S.E.; Mirza, N.; Dominguez, A.L.; Manrique, S.Z.; Lustgarten, J. Blockade of CCL1 inhibits T regulatory cell suppressive function enhancing tumor immunity without affecting T effector responses. J. Immunol. 2010, 184, 6833–6842. [Google Scholar] [CrossRef]
- Kuehnemuth, B.; Piseddu, I.; Wiedemann, G.M.; Lauseker, M.; Kuhn, C.; Hofmann, S.; Schmoeckel, E.; Endres, S.; Mayr, D.; Jeschke, U.; et al. CCL1 is a major regulatory T cell attracting factor in human breast cancer. BMC Cancer 2018, 18, 1278. [Google Scholar] [CrossRef]
- Wang, X.; Lang, M.; Zhao, T.; Feng, X.; Zheng, C.; Huang, C.; Hao, J.; Dong, J.; Luo, L.; Li, X.; et al. Cancer-FOXP3 directly activated CCL5 to recruit FOXP3(+)Treg cells in pancreatic ductal adenocarcinoma. Oncogene 2017, 36, 3048–3058. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.C.; Goedegebuure, P.S.; Belt, B.A.; Flaherty, B.; Sankpal, N.; Gillanders, W.E.; Eberlein, T.J.; Hsieh, C.S.; Linehan, D.C. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J. Immunol. 2009, 182, 1746–1755. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Sharma, S.C.; Das, S. Dynamics of regulatory T cells (Tregs ) in patients with oral squamous cell carcinoma. J. Surg. Oncol. 2017, 116, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Puig, P.E.; Roux, S.; Parcellier, A.; Schmitt, E.; Solary, E.; Kroemer, G.; Martin, F.; Chauffert, B.; Zitvogel, L. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J. Exp. Med. 2005, 202, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Tian, T.; Gu, X.; Zhang, B.; Liu, Y.; Yuan, C.; Shao, L.; Guo, Y.; Fan, K. Increased circulating CD14(+)HLA-DR-/low myeloid-derived suppressor cells are associated with poor prognosis in patients with small-cell lung cancer. Cancer Biomark. 2015, 15, 425–432. [Google Scholar] [CrossRef]
- Iriki, T.; Ohnishi, K.; Fujiwara, Y.; Horlad, H.; Saito, Y.; Pan, C.; Ikeda, K.; Mori, T.; Suzuki, M.; Ichiyasu, H.; et al. The cell-cell interaction between tumor-associated macrophages and small cell lung cancer cells is involved in tumor progression via STAT3 activation. Lung Cancer 2017, 106, 22–32. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Type | Specificity | Cancers Targeted | Study Phase | References |
|---|---|---|---|---|---|
| TAE-226 Novartis | Kinase inhibitor ATP competitive | FAK, IGF-IR, c-Met, Pyk2 | Glioma, ovarian | Preclinical | [47,62] |
| PF-573,228 Pfizer | Kinase inhibitor ATP competitive | FAK | Prostate, breast | Preclinical | [48] |
| GSK2256098 GlaxoSmithKline | Kinase inhibitor ATP competitive Reversible | FAK, UGT1A1 | Solid tumors (ovarian, pancreatic, meningioma, glioblastoma, malignant pleural mesothelioma) | Clinical: phase I & II | [34,35,36,44,49] NCT00996671, NCT02523014 |
| NVP-TAC544 | Kinase inhibitor ATP competitive | FAK | N/A | Preclinical | [50] |
| VS-4718 (PND-1186) Verastem | Kinase inhibitor ATP competitive Reversible | FAK, Pyk2 | Solid tumors (pancreas, breast, ovarian), acute myeloid leukemia, B-cell acute lymphoblastic leukemia | Clinical: phase I | [51] |
| VS-6062 (PF-562271 and PF271) Verastem | Kinase inhibitor ATP competitive Reversible | FAK, CDK2/CyclinE, CDK3/CyclinE, CDK1/CyclinB, Pyk2 | Prostate, pancreatic, head and neck | Clinical: phase I | [37,52] |
| VS-6063 (Defactinib) Verastem | Kinase inhibitor ATP competitive | FAK, Pyk2 | NSCLC, pancreatic cancer, ovarian, malignant pleural mesothelioma, hematologic | Clinical: phase I/Ib & II | [38,39,40,45,53] NCT02758587 NCT02004028 NCT03875820 NCT03727880, NCT02943317, NCT02913716, NCT02465060, NCT02546531 |
| 1H-Pyrrolo(2,3-b) Merk Serono | Kinase inhibitor non-ATP competitive | Hinge region of FAK | N/A | Preclinical | [54] |
| C4 CureFAKtor Pharmaceuticals | Scaffold inhibitor | FAK /VEGFR-3 | Neuroblastoma, pancreatic, breast | Preclinical | [55,56,57] |
| Compound R2 (Roslins) CureFAKtor Pharmaceuticals | Scaffold inhibitor | FAK, p53 | Colon, reast | Preclinical | [58] |
| Y11 CureFAKtor Pharmaceuticals | Scaffold inhibitor | FAK Y397 site | Colon, breast | Preclinical | [59] |
| BI853520 | ATP competitive inhibitor | FAK | Malignant pleural mesothelioma, non-hematologic malignancies | Preclinical, clinical | [42,43,60] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aboubakar Nana, F.; Vanderputten, M.; Ocak, S. Role of Focal Adhesion Kinase in Small-Cell Lung Cancer and Its Potential as a Therapeutic Target. Cancers 2019, 11, 1683. https://doi.org/10.3390/cancers11111683
Aboubakar Nana F, Vanderputten M, Ocak S. Role of Focal Adhesion Kinase in Small-Cell Lung Cancer and Its Potential as a Therapeutic Target. Cancers. 2019; 11(11):1683. https://doi.org/10.3390/cancers11111683
Chicago/Turabian StyleAboubakar Nana, Frank, Marie Vanderputten, and Sebahat Ocak. 2019. "Role of Focal Adhesion Kinase in Small-Cell Lung Cancer and Its Potential as a Therapeutic Target" Cancers 11, no. 11: 1683. https://doi.org/10.3390/cancers11111683
APA StyleAboubakar Nana, F., Vanderputten, M., & Ocak, S. (2019). Role of Focal Adhesion Kinase in Small-Cell Lung Cancer and Its Potential as a Therapeutic Target. Cancers, 11(11), 1683. https://doi.org/10.3390/cancers11111683