Intra-Patient Heterogeneity of Circulating Tumor Cells and Circulating Tumor DNA in Blood of Melanoma Patients

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
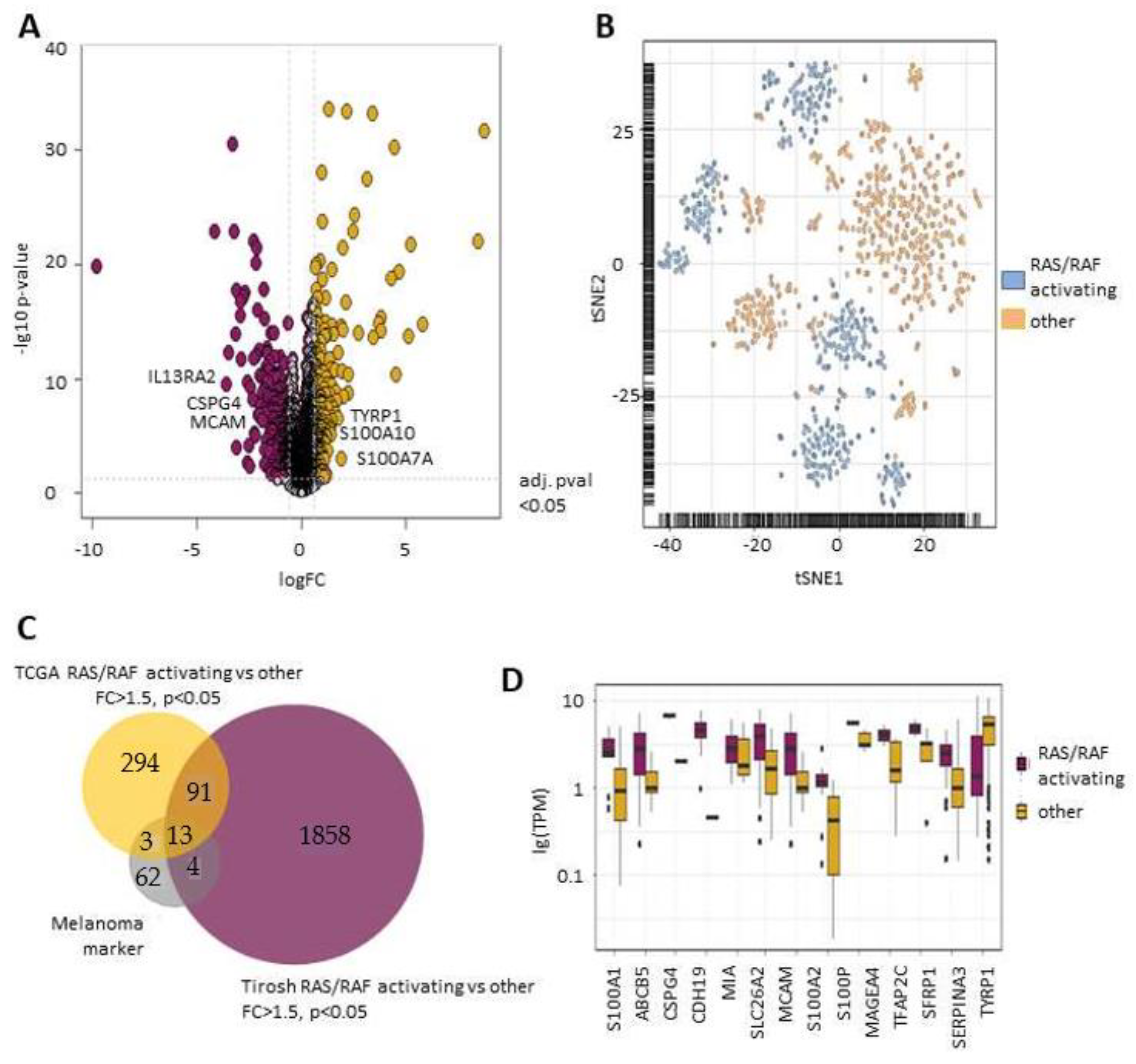
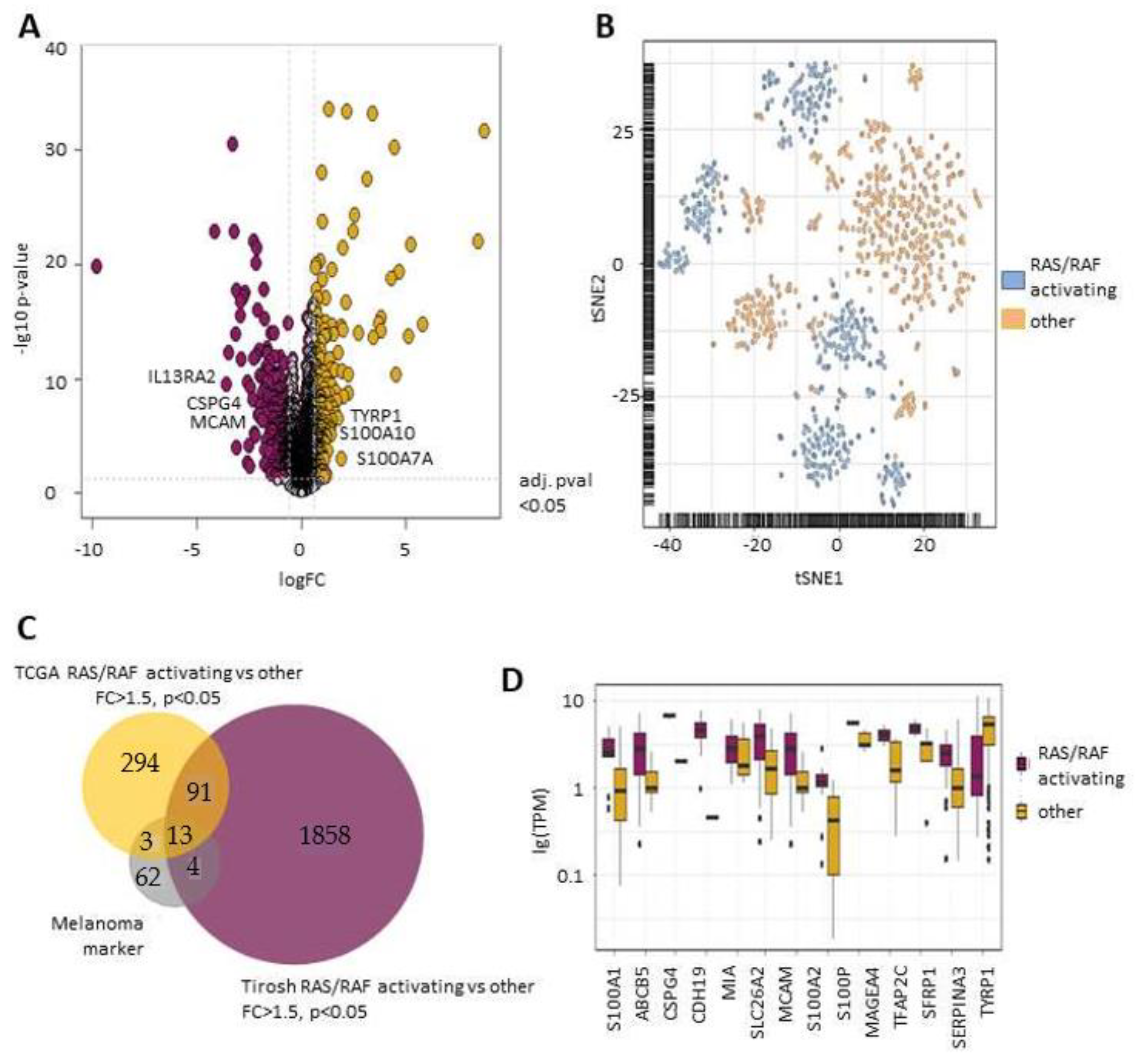
2.1. RAS/RAF Activating Mutations Lead to A Distinct Melanoma Marker Expression Pattern
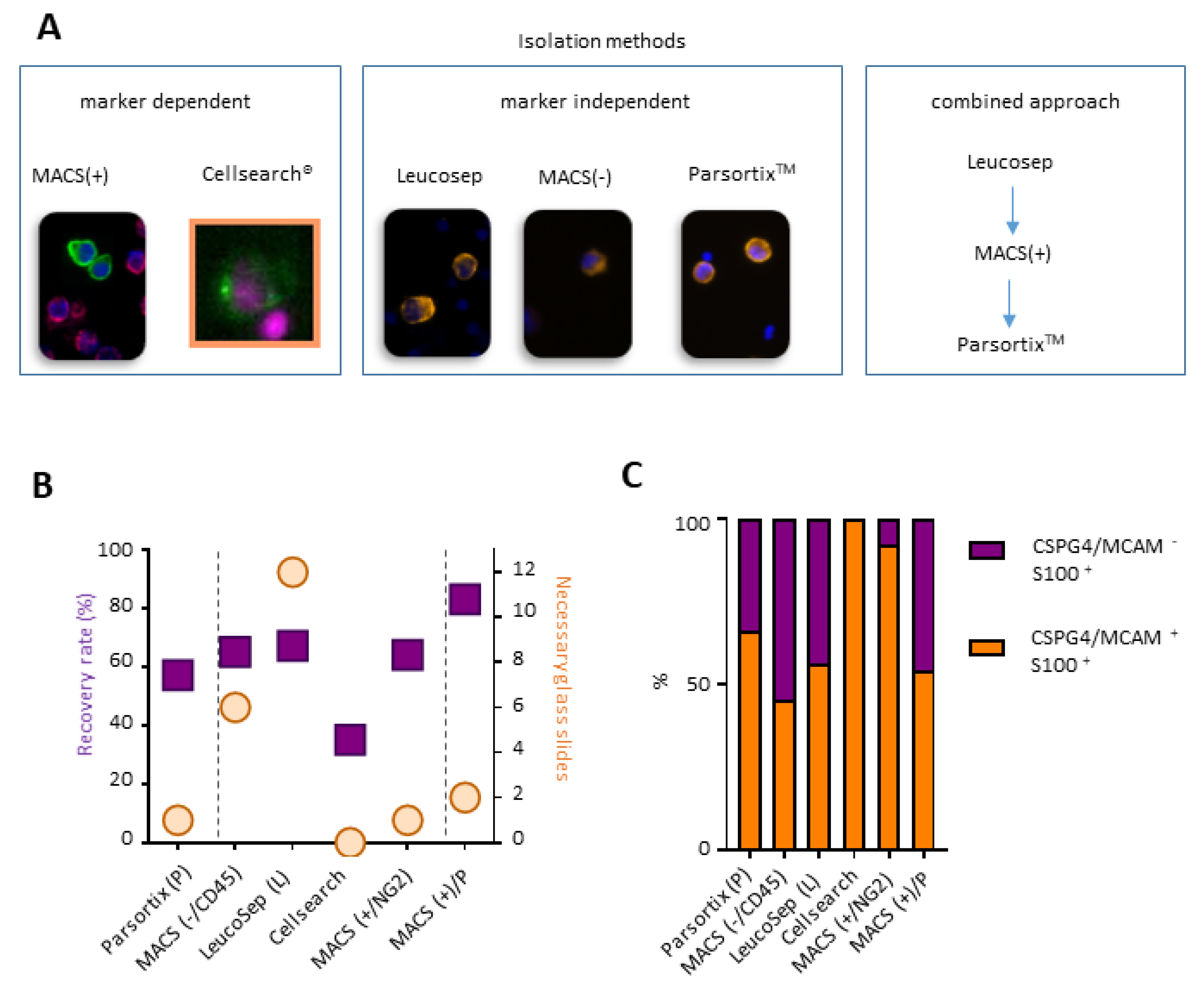
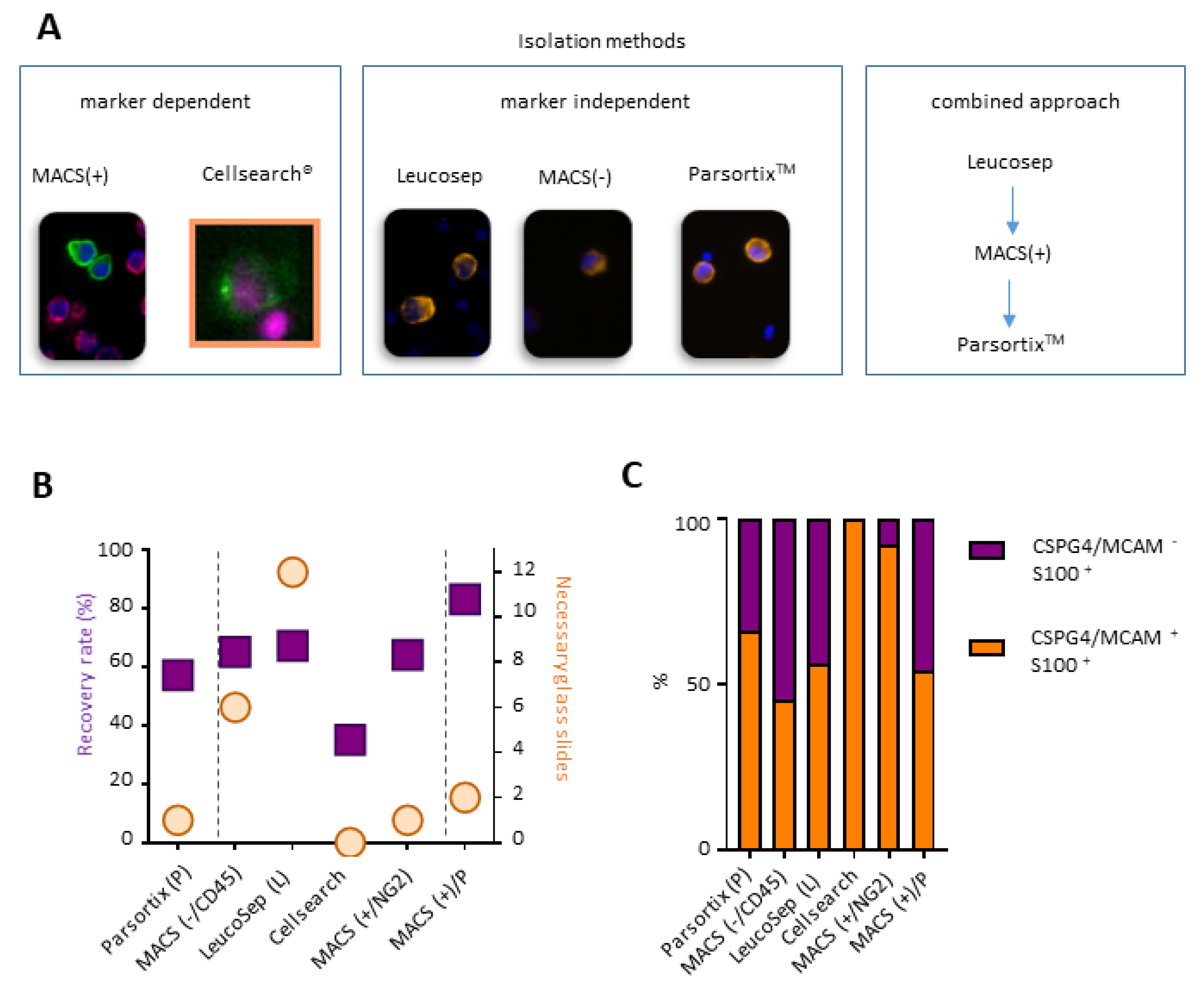
2.2. A Combined Enrichment Approach Allows the Detection of CTC Subpopulations
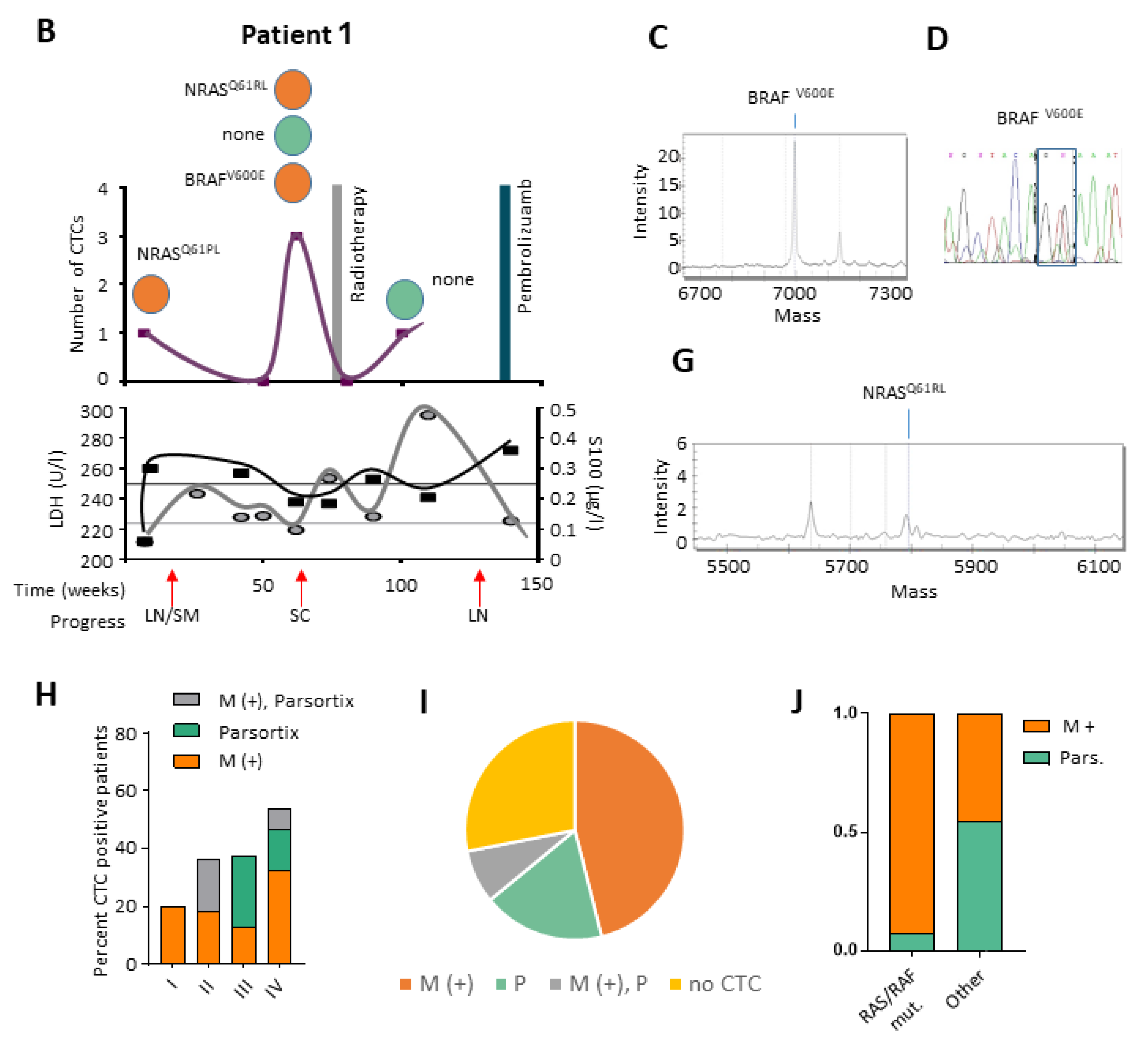
2.3. Targeted Sequencing Reveals Mutational CTC Subclones
2.4. RNA Expression Pattern on Selected CTCs
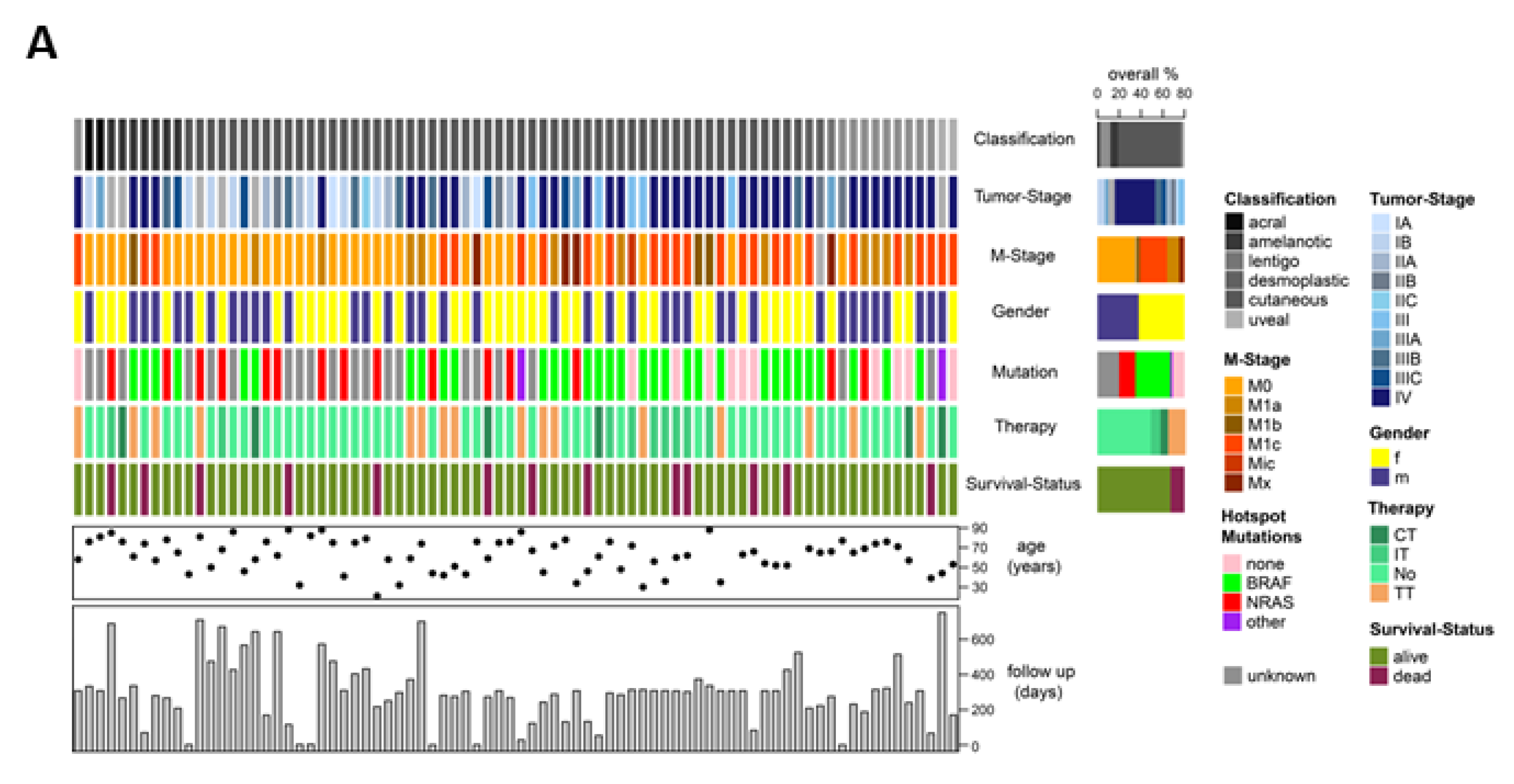
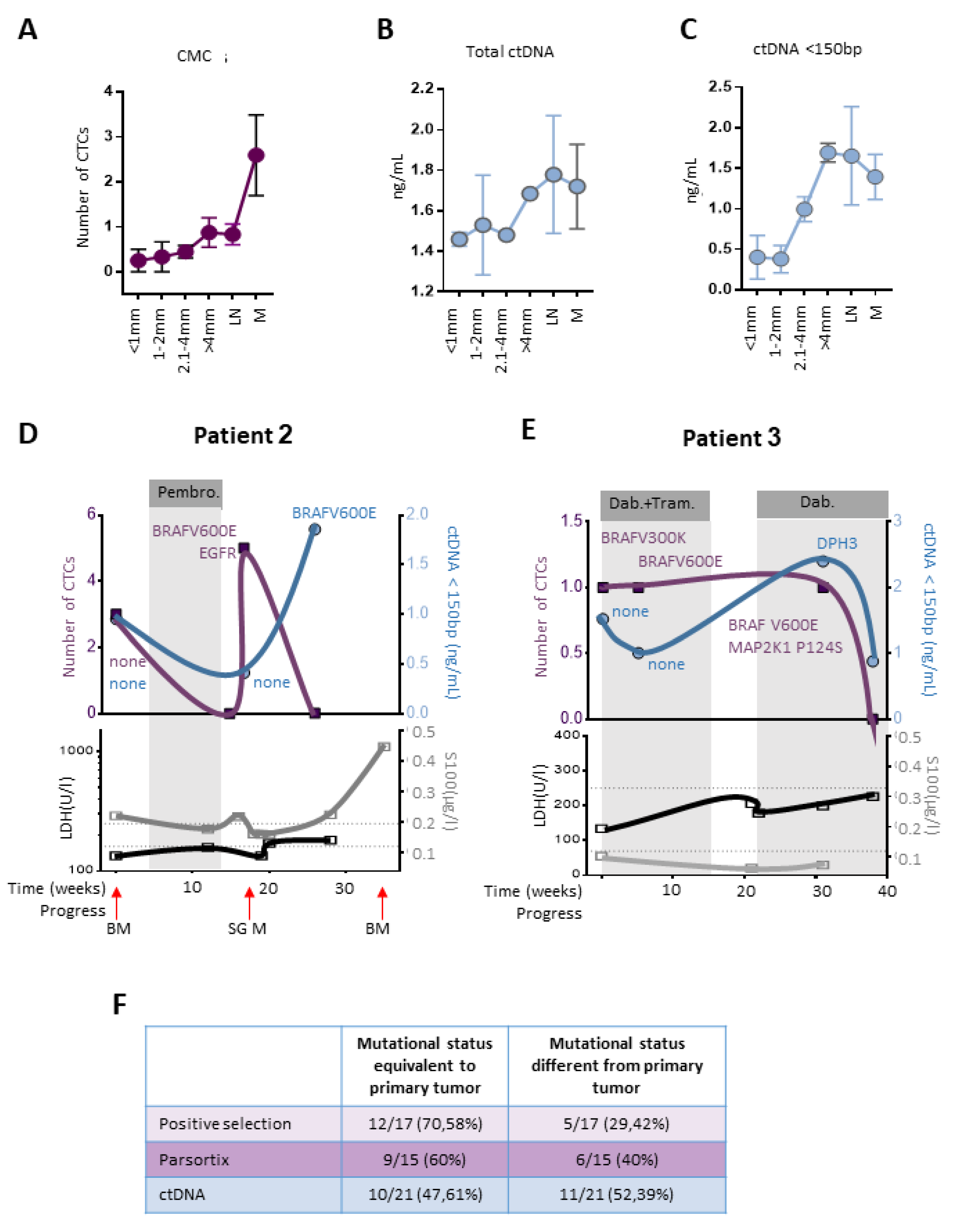
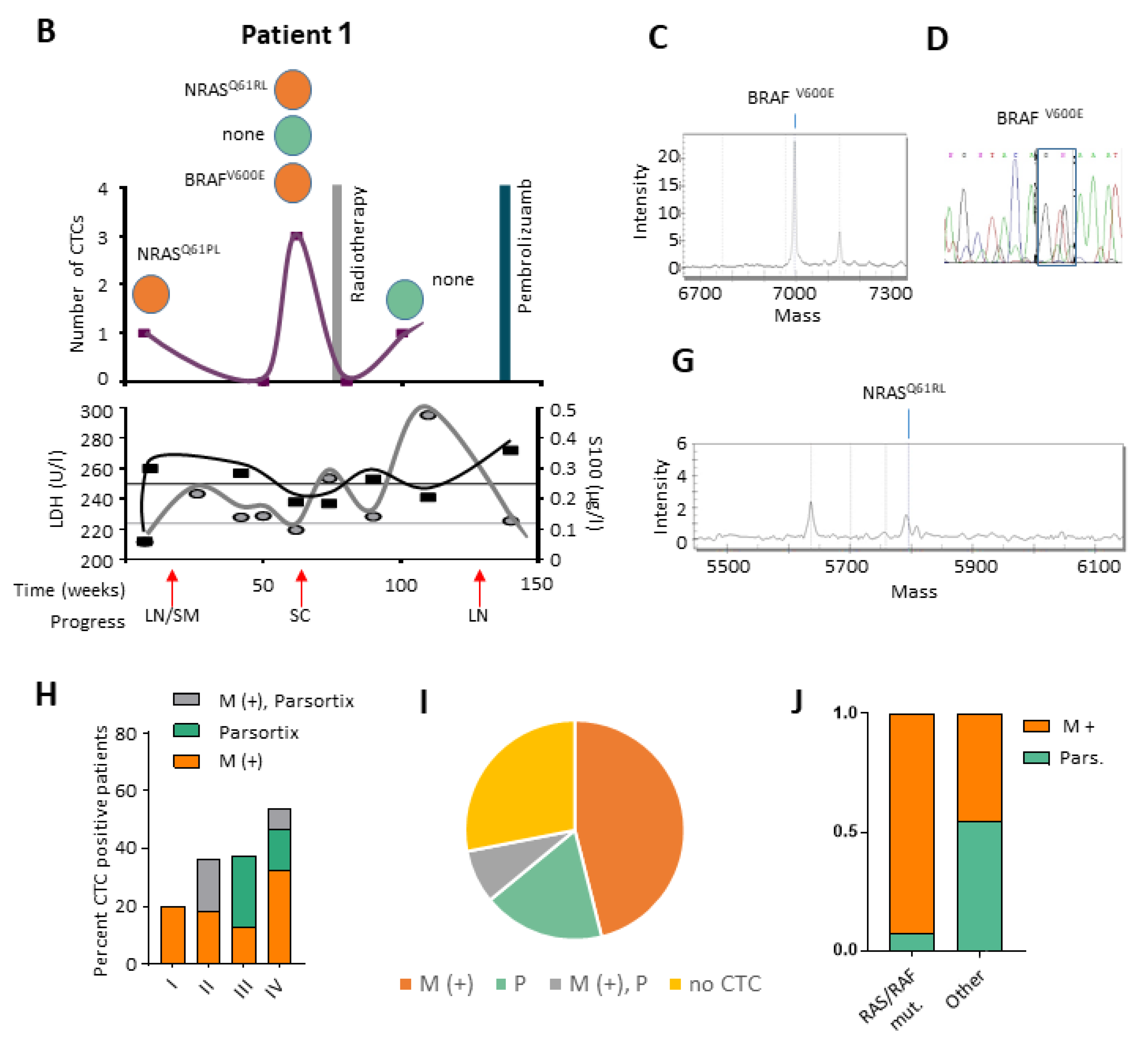
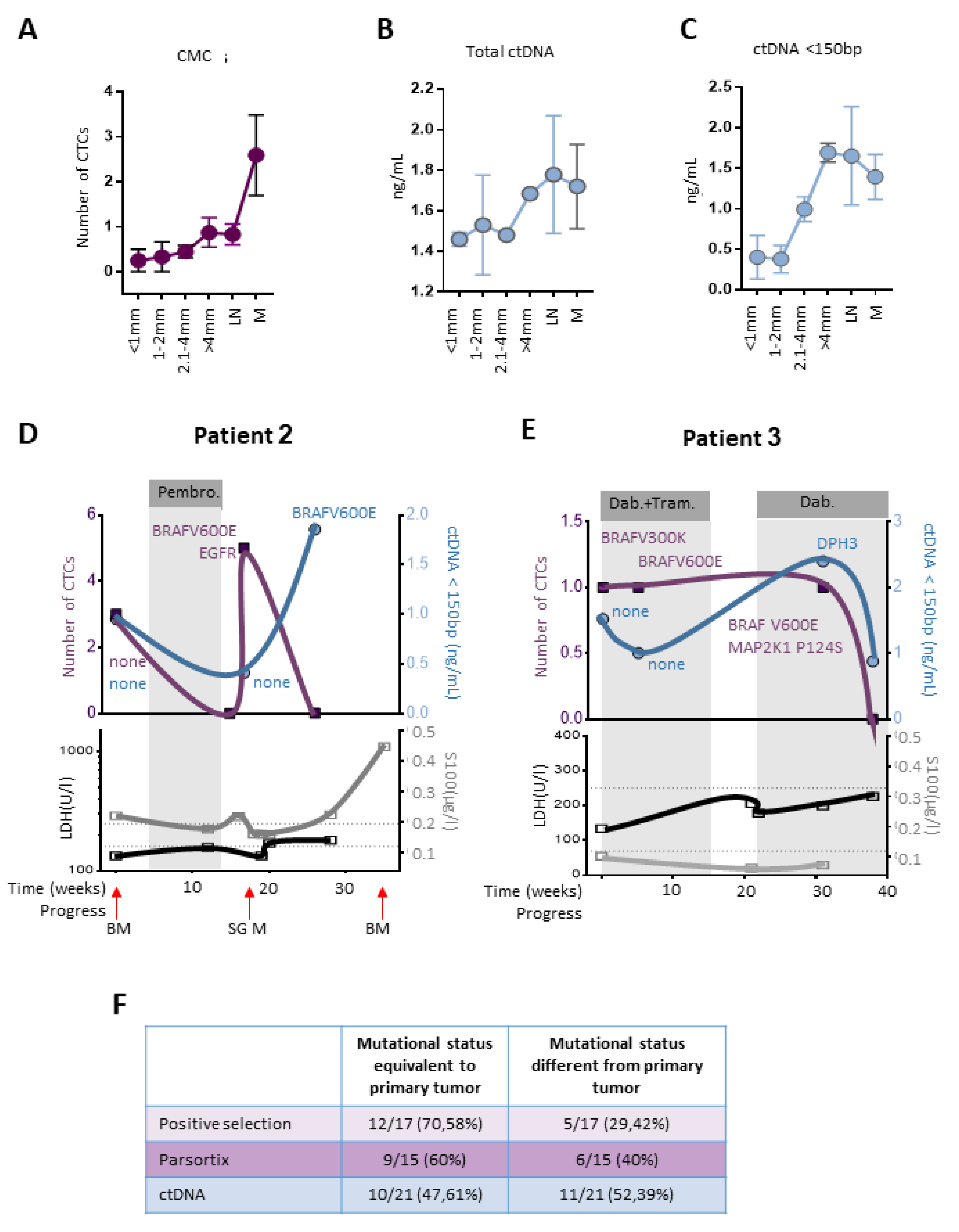
2.5. CTCs and ctDNA Provide Synergistic Clinical Information
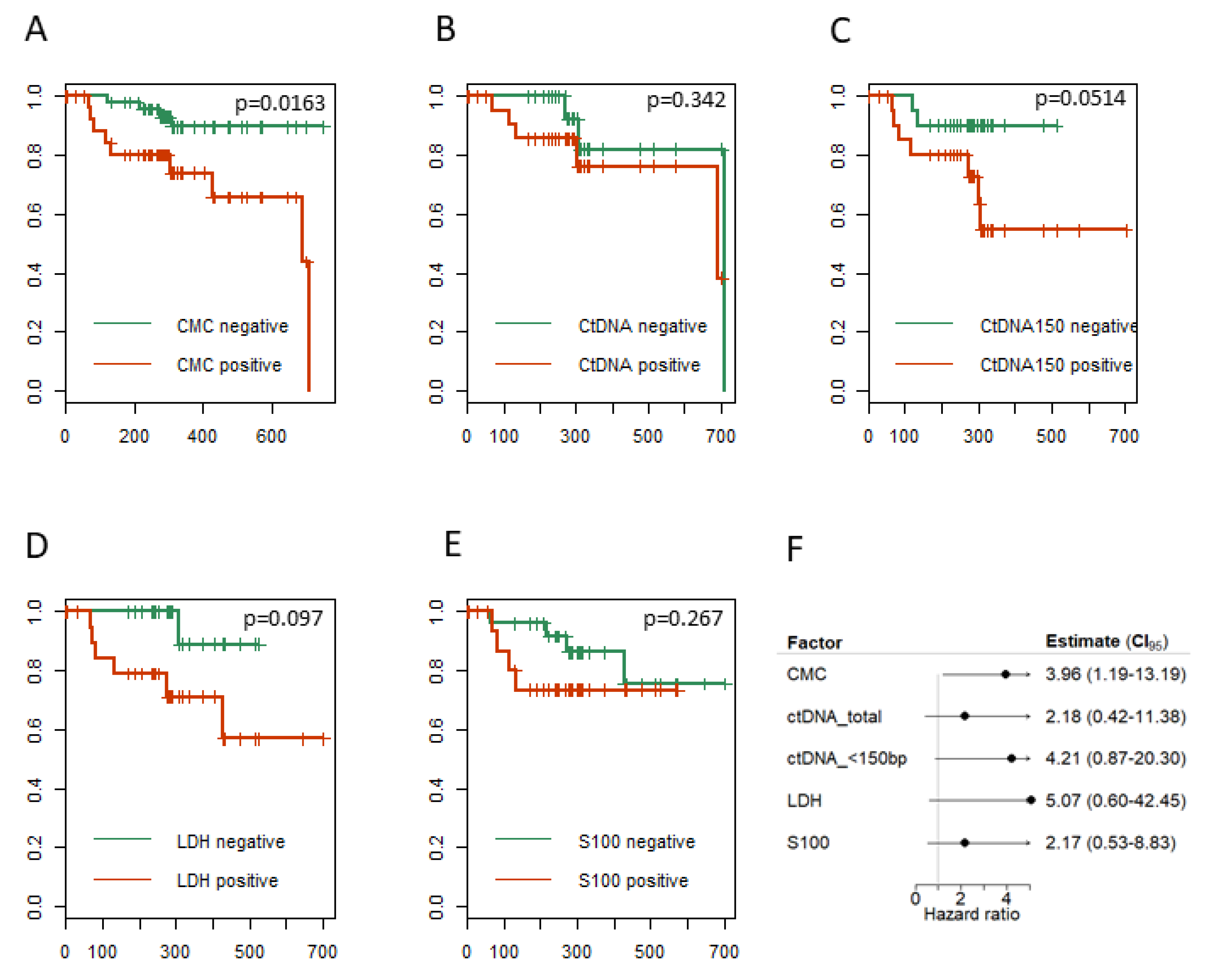
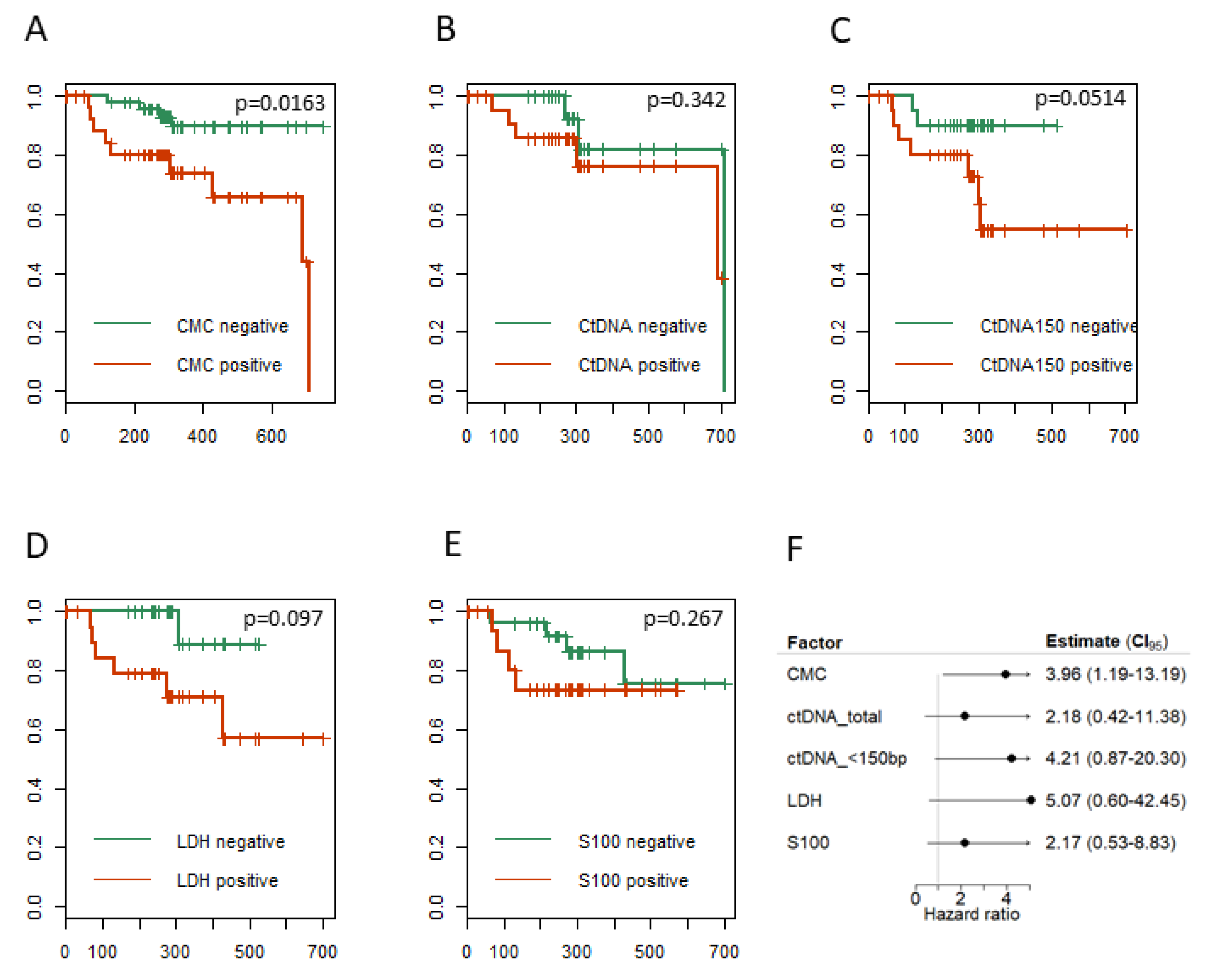
2.6. CTCs, ctDNA < 150 bp and LDH Predict Clinical Outcome
3. Discussion
4. Material and Methods
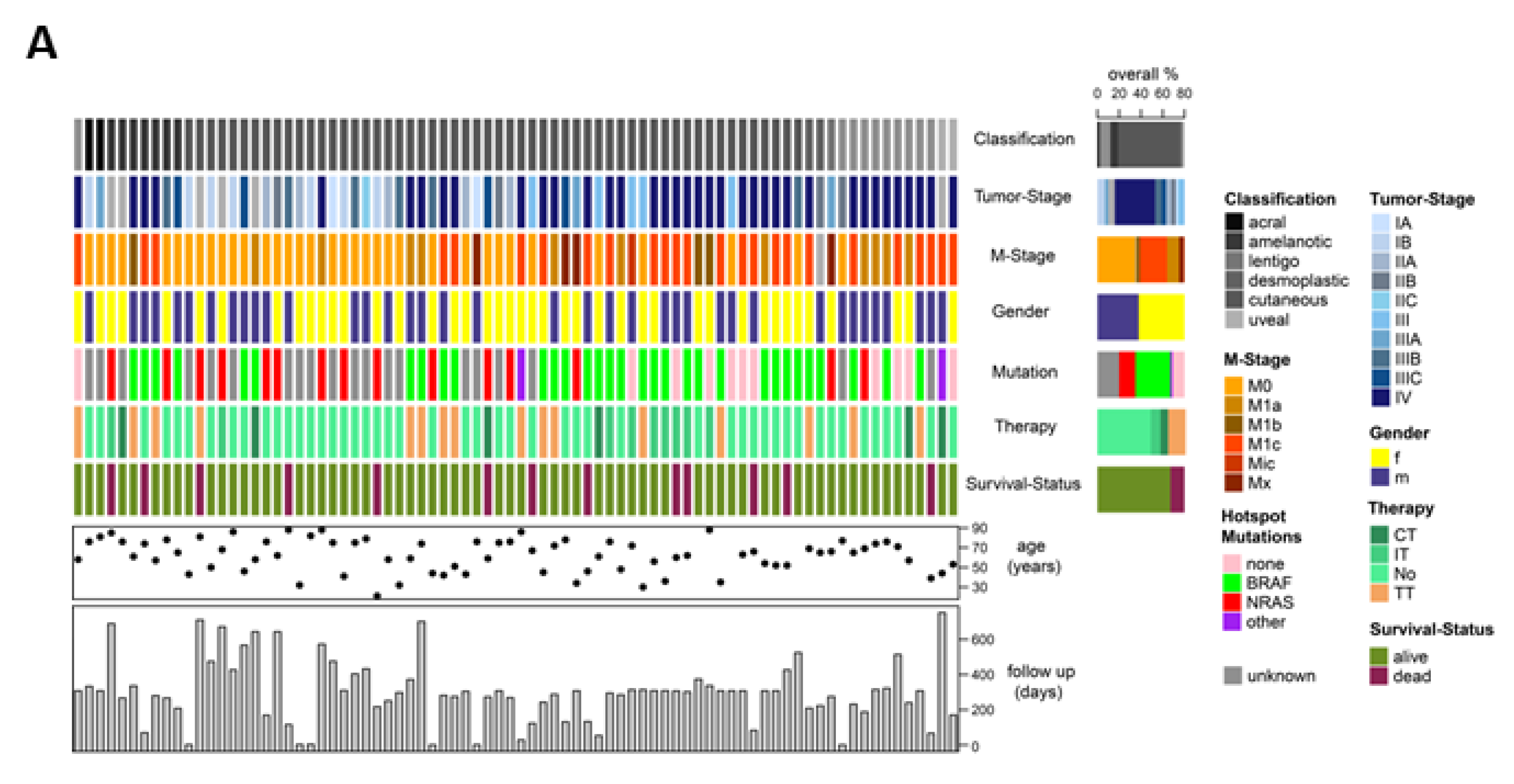
4.1. Patient Samples
4.2. Tumor Cell Enrichment
4.2.1. Marker-Dependent Approach
4.2.2. Marker-Independent Approach
4.2.3. Combined Approach (MACS and Partsortix®)
4.3. Immunofluorescence Staining
4.4. Whole Genome Amplification
4.5. cfDNA Extraction
4.6. Quantification and Size Fragment Distribution of cfDNA
4.7. Mutational Analysis
4.8. RNA Analysis
4.9. Bioinformatical and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Karimkhani, C.; Green, A.C.; Nijsten, T.; Weinstock, M.A.; Dellavalle, R.P.; Naghavi, M.; Fitzmaurice, C. The global burden of melanoma: Results from the Global Burden of Disease Study 2015. Br. J. Dermatol. 2017, 177, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Reid, A.L.; Freeman, J.B.; Millward, M.; Ziman, M.; Gray, E.S. Detection of BRAF-V600E and V600K in melanoma circulating tumour cells by droplet digital PCR. Clin. Biochem. 2015, 48, 999–1002. [Google Scholar] [CrossRef]
- Bardelli, A.; Pantel, K. Liquid Biopsies, What We Do Not Know (Yet). Cancer Cell 2017, 31, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Pantel, K.; Alix-Panabieres, C. Liquid biopsy and minimal residual disease—Latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef]
- Alix-Panabieres, C.; Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Quandt, D.; Dieter Zucht, H.; Amann, A.; Wulf-Goldenberg, A.; Borrebaeck, C.; Cannarile, M.; Lambrechts, D.; Oberacher, H.; Garrett, J.; Nayak, T.; et al. Implementing liquid biopsies into clinical decision making for cancer immunotherapy. Oncotarget 2017, 8, 48507–48520. [Google Scholar] [CrossRef] [Green Version]
- Pantel, K.; Alix-Panabieres, C. Real-time liquid biopsy in cancer patients: Fact or fiction? Cancer Res. 2013, 73, 6384–6388. [Google Scholar] [CrossRef]
- Koyanagi, K.; Kuo, C.; Nakagawa, T.; Mori, T.; Ueno, H.; Lorico, A.R., Jr.; Wang, H.J.; Hseuh, E.; O’Day, S.J.; Hoon, D.S. Multimarker quantitative real-time PCR detection of circulating melanoma cells in peripheral blood: Relation to disease stage in melanoma patients. Clin. Chem. 2005, 51, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Hoshimoto, S.; Shingai, T.; Morton, D.L.; Kuo, C.; Faries, M.B.; Chong, K.; Elashoff, D.; Wang, H.J.; Elashoff, R.M.; Hoon, D.S. Association between circulating tumor cells and prognosis in patients with stage III melanoma with sentinel lymph node metastasis in a phase III international multicenter trial. J. Clin. Oncol. 2012, 30, 3819–3826. [Google Scholar] [CrossRef] [PubMed]
- Koyanagi, K.; Mori, T.; O’Day, S.J.; Martinez, S.R.; Wang, H.J.; Hoon, D.S. Association of circulating tumor cells with serum tumor-related methylated DNA in peripheral blood of melanoma patients. Cancer Res. 2006, 66, 6111–6117. [Google Scholar] [CrossRef] [PubMed]
- Koyanagi, K.; O’Day, S.J.; Gonzalez, R.; Lewis, K.; Robinson, W.A.; Amatruda, T.T.; Wang, H.J.; Elashoff, R.M.; Takeuchi, H.; Umetani, N.; et al. Serial monitoring of circulating melanoma cells during neoadjuvant biochemotherapy for stage III melanoma: Outcome prediction in a multicenter trial. J. Clin. Oncol. 2005, 23, 8057–8064. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Fernandez-Landazuri, S.; Rodriguez, C.; Zarate, R.; Lozano, M.D.; Zubiri, L.; Perez-Gracia, J.L.; Martin-Algarra, S.; Gonzalez, A. Quantitative cell-free circulating BRAFV600E mutation analysis by use of droplet digital PCR in the follow-up of patients with melanoma being treated with BRAF inhibitors. Clin. Chem. 2015, 61, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.J.; Gremel, G.; Marshall, A.; Myers, K.A.; Fisher, N.; Dunn, J.A.; Dhomen, N.; Corrie, P.G.; Middleton, M.R.; Lorigan, P.; et al. Circulating tumor DNA predicts survival in patients with resected high-risk stage II/III melanoma. Ann. Oncol. 2018, 29, 490–496. [Google Scholar] [CrossRef]
- Valpione, S.; Gremel, G.; Mundra, P.; Middlehurst, P.; Galvani, E.; Girotti, M.R.; Lee, R.J.; Garner, G.; Dhomen, N.; Lorigan, P.C.; et al. Plasma total cell-free DNA (cfDNA) is a surrogate biomarker for tumour burden and a prognostic biomarker for survival in metastatic melanoma patients. Eur. J. Cancer 2018, 88, 1–9. [Google Scholar] [CrossRef]
- Ampofo, E.; Schmitt, B.M.; Menger, M.D.; Laschke, M.W. The regulatory mechanisms of NG2/CSPG4 expression. Cell Mol. Biol. Lett. 2017, 22, 4. [Google Scholar] [CrossRef]
- Kennedy, R.A.; Kemp, T.J.; Sugden, P.H.; Clerk, A. Using U0126 to dissect the role of the extracellular signal-regulated kinase 1/2 (ERK1/2) cascade in the regulation of gene expression by endothelin-1 in cardiac myocytes. J. Mol. Cell. Cardiol. 2006, 41, 236–247. [Google Scholar] [CrossRef]
- Theodosakis, N.; Held, M.A.; Marzuka-Alcala, A.; Meeth, K.M.; Micevic, G.; Long, G.V.; Scolyer, R.A.; Stern, D.F.; Bosenberg, M.W. BRAF Inhibition Decreases Cellular Glucose Uptake in Melanoma in Association with Reduction in Cell Volume. Mol. Cancer Ther. 2015, 14, 1680–1692. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, X.; Sullivan, R.J.; Kalinich, M.; Kwan, T.T.; Giobbie-Hurder, A.; Pan, S.; LiCausi, J.A.; Milner, J.D.; Nieman, L.T.; Wittner, B.S.; et al. Molecular signatures of circulating melanoma cells for monitoring early response to immune checkpoint therapy. Proc. Natl. Acad. Sci. USA 2018, 115, 2467–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Underhill, H.R.; Kitzman, J.O.; Hellwig, S.; Welker, N.C.; Daza, R.; Baker, D.N.; Gligorich, K.M.; Rostomily, R.C.; Bronner, M.P.; Shendure, J. Fragment Length of Circulating Tumor DNA. PLoS Genet. 2016, 12, e1006162. [Google Scholar] [CrossRef] [PubMed]
- Girotti, M.R.; Gremel, G.; Lee, R.; Galvani, E.; Rothwell, D.; Viros, A.; Mandal, A.K.; Lim, K.H.; Saturno, G.; Furney, S.J.; et al. Application of Sequencing, Liquid Biopsies, and Patient-Derived Xenografts for Personalized Medicine in Melanoma. Cancer Discov. 2016, 6, 286–299. [Google Scholar] [CrossRef]
- Tsao, S.C.; Weiss, J.; Hudson, C.; Christophi, C.; Cebon, J.; Behren, A.; Dobrovic, A. Monitoring response to therapy in melanoma by quantifying circulating tumour DNA with droplet digital PCR for BRAF and NRAS mutations. Sci. Rep. 2015, 5, 11198. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Hoon, D.S.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- Shinozaki, M.; O’Day, S.J.; Kitago, M.; Amersi, F.; Kuo, C.; Kim, J.; Wang, H.J.; Hoon, D.S. Utility of circulating B-RAF DNA mutation in serum for monitoring melanoma patients receiving biochemotherapy. Clin. Cancer Res. 2007, 13, 2068–2074. [Google Scholar] [CrossRef]
- Auer, M.; Heitzer, E.; Ulz, P.; Geigl, J.B.; Speicher, M.R. Single circulating tumor cell sequencing for monitoring. Oncotarget 2013, 4, 812–813. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.S.; Witkowski, T.; Pereira, M.; Calapre, L.; Herron, K.; Irwin, D.; Chapman, B.; Khattak, M.A.; Raleigh, J.; Hatzimihalis, A.; et al. Genomic Analysis of Circulating Tumor DNA Using a Melanoma-Specific UltraSEEK Oncogene Panel. J. Mol. Diagn. 2019, 21, 418–426. [Google Scholar] [CrossRef]
- Balch, C.M.; Gershenwald, J.E.; Soong, S.J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final version of 2009 AJCC melanoma staging and classification. J. Clin. Oncol. 2009, 27, 6199–6206. [Google Scholar] [CrossRef]
- Wroblewski, F.; Gregory, K.F. Lactic dehydrogenase isozymes and their distribution in normal tissues and plasma and in disease states. Ann. NY Acad. Sci. 1961, 94, 912–932. [Google Scholar] [CrossRef] [PubMed]
- Gogas, H.; Eggermont, A.M.; Hauschild, A.; Hersey, P.; Mohr, P.; Schadendorf, D.; Spatz, A.; Dummer, R. Biomarkers in melanoma. Ann. Oncol. 2009, 20. [Google Scholar] [CrossRef] [PubMed]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denisova, E.; Heidenreich, B.; Nagore, E.; Rachakonda, P.S.; Hosen, I.; Akrap, I.; Traves, V.; Garcia-Casado, Z.; Lopez-Guerrero, J.A.; Requena, C.; et al. Frequent DPH3 promoter mutations in skin cancers. Oncotarget 2015, 6, 35922–35930. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, N.J.; Elliott, K.; Filges, S.; Van den Eynden, J.; Stahlberg, A.; Larsson, E. Recurrent promoter mutations in melanoma are defined by an extended context-specific mutational signature. PLoS Genet. 2017, 13, e1006773. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shi, Y.; Ju, P.; Liu, R.; Yeo, S.P.; Xia, Y.; Owlanj, H.; Feng, Z. Silencing of diphthamide synthesis 3 (Dph3) reduces metastasis of murine melanoma. PLoS ONE 2012, 7, e49988. [Google Scholar] [CrossRef]
- Lee, J.H.; Long, G.V.; Boyd, S.; Lo, S.; Menzies, A.M.; Tembe, V.; Guminski, A.; Jakrot, V.; Scolyer, R.A.; Mann, G.J.; et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann. Oncol. 2017, 28, 1130–1136. [Google Scholar] [CrossRef]
- Rao, C.; Bui, T.; Connelly, M.; Doyle, G.; Karydis, I.; Middleton, M.R.; Clack, G.; Malone, M.; Coumans, F.A.; Terstappen, L.W. Circulating melanoma cells and survival in metastatic melanoma. Int. J. Oncol. 2011, 38, 755–760. [Google Scholar] [CrossRef] [Green Version]
- Karakousis, G.; Yang, R.; Xu, X. Circulating melanoma cells as a predictive biomarker. J. Investig. Dermatol. 2013, 133, 1460–1462. [Google Scholar] [CrossRef]
- Cayrefourcq, L.; De Roeck, A.; Garcia, C.; Stoebner, P.E.; Fichel, F.; Garima, F.; Perriard, F.; Daures, J.P.; Meunier, L.; Alix-Panabieres, C. S100-EPISPOT: A New Tool to Detect Viable Circulating Melanoma Cells. Cells 2019, 8, 755. [Google Scholar] [CrossRef]
- Schreuer, M.; Meersseman, G.; Van Den Herrewegen, S.; Jansen, Y.; Chevolet, I.; Bott, A.; Wilgenhof, S.; Seremet, T.; Jacobs, B.; Buyl, R.; et al. Quantitative assessment of BRAF V600 mutant circulating cell-free tumor DNA as a tool for therapeutic monitoring in metastatic melanoma patients treated with BRAF/MEK inhibitors. J. Transl. Med. 2016, 14, 95. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Walker, A.; Gagnon, R.; Mazumdar, J.; Casey, M.; Long, G.V.; Schadendorf, D.; Flaherty, K.; Kefford, R.; Hauschild, A.; Hwu, P.; et al. Correlation of BRAF Mutation Status in Circulating-Free DNA and Tumor and Association with Clinical Outcome across Four BRAFi and MEKi Clinical Trials. Clin. Cancer Res. 2016, 22, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Altman, D.G.; McShane, L.M.; Sauerbrei, W.; Taube, S.E. Reporting recommendations for tumor marker prognostic studies (REMARK): Explanation and elaboration. BMC Med. 2012, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabieres, C.; Pantel, K. Challenges in circulating tumour cell research. Nat. Rev. Cancer 2014, 14, 623–631. [Google Scholar] [CrossRef]
- Hvichia, G.E.; Parveen, Z.; Wagner, C.; Janning, M.; Quidde, J.; Stein, A.; Muller, V.; Loges, S.; Neves, R.P.; Stoecklein, N.H.; et al. A novel microfluidic platform for size and deformability based separation and the subsequent molecular characterization of viable circulating tumor cells. Int. J. Cancer 2016, 138, 2894–2904. [Google Scholar] [CrossRef]
- Mosko, M.J.; Nakorchevsky, A.A.; Flores, E.; Metzler, H.; Ehrich, M.; van den Boom, D.J.; Sherwood, J.L.; Nygren, A.O. Ultrasensitive Detection of Multiplexed Somatic Mutations Using MALDI-TOF Mass Spectrometry. J. Mol. Diagn. 2016, 18, 23–31. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorges, K.; Wiltfang, L.; Gorges, T.M.; Sartori, A.; Hildebrandt, L.; Keller, L.; Volkmer, B.; Peine, S.; Babayan, A.; Moll, I.; et al. Intra-Patient Heterogeneity of Circulating Tumor Cells and Circulating Tumor DNA in Blood of Melanoma Patients. Cancers 2019, 11, 1685. https://doi.org/10.3390/cancers11111685
Gorges K, Wiltfang L, Gorges TM, Sartori A, Hildebrandt L, Keller L, Volkmer B, Peine S, Babayan A, Moll I, et al. Intra-Patient Heterogeneity of Circulating Tumor Cells and Circulating Tumor DNA in Blood of Melanoma Patients. Cancers. 2019; 11(11):1685. https://doi.org/10.3390/cancers11111685
Chicago/Turabian StyleGorges, Katharina, Lisa Wiltfang, Tobias M. Gorges, Alexander Sartori, Lina Hildebrandt, Laura Keller, Beate Volkmer, Sven Peine, Anna Babayan, Ingrid Moll, and et al. 2019. "Intra-Patient Heterogeneity of Circulating Tumor Cells and Circulating Tumor DNA in Blood of Melanoma Patients" Cancers 11, no. 11: 1685. https://doi.org/10.3390/cancers11111685
APA StyleGorges, K., Wiltfang, L., Gorges, T. M., Sartori, A., Hildebrandt, L., Keller, L., Volkmer, B., Peine, S., Babayan, A., Moll, I., Schneider, S. W., Twarock, S., Mohr, P., Fischer, J. W., & Pantel, K. (2019). Intra-Patient Heterogeneity of Circulating Tumor Cells and Circulating Tumor DNA in Blood of Melanoma Patients. Cancers, 11(11), 1685. https://doi.org/10.3390/cancers11111685