Genomic-Destabilization-Associated Mutagenesis and Clonal Evolution of Cells with Mutations in Tumor-Suppressor Genes

Abstract
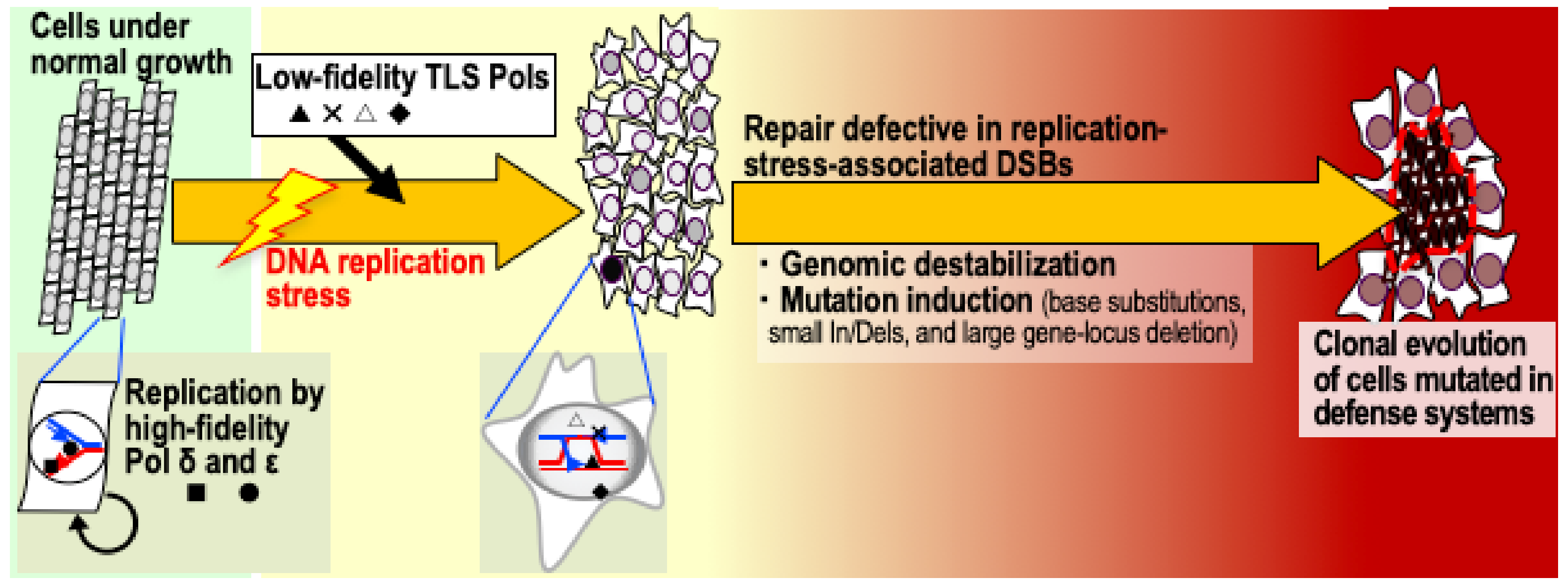
1. Introduction
2. Mutations Induced along with Genomic Destabilization
2.1. Mutation Rate and Its Association with Cancer Risk
2.2. Mutation Induction when Replication-Stress-Associated DSBs Accumulate
2.3. Senescence-Associated Increase in the Risk of Genomic Destabilization
2.4. Genomic-Destabilization-Associated Mutagenesis in Cancer Development
2.5. Epigenetic Alterations as a Cause of Genomic Destabilization
2.6. Avoidable or Unavoidable?
2.7. Mutations in Cancer-Suppressor Genes
3. Clonal Evolution of Cells with Abrogated Defense Systems
3.1. Genomic-Destabilization-Triggered Clonal Evolution
3.2. Transcription Variety and Epigenetic Alteration for Clonal Evolution
3.3. Association of Immune Response with Clonal Evolution
4. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B.; Dubal, P.M.; Svider, P.F.; Kanumuri, V.V.; Patel, A.A.; Baredes, S.; Eloy, J.A.; Danaei, G.; Hoorn, S.V.; et al. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Durrett, R.; Kimmel, M.; Lambert, A.; Parmigiani, G.; Zauber, A.; Vogelstein, B. Role of stem-cell divisions in cancer risk. Nature 2017, 548, E13–E14. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Fowler, J.C.; Wabik, A.; Lawson, A.R.J.; Abascal, F.; Hall, M.W.J.; Cagan, A.; Murai, K.; Mahbubani, K.; Stratton, M.R.; et al. Somatic mutant clones colonize the human esophagus with age. Science 2018, 362, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Kakiuchi, N.; Yoshizato, T.; Nannya, Y.; Suzuki, H.; Takeuchi, Y.; Shiozawa, Y.; Sato, Y.; Aoki, K.; Kim, S.K.; et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 2019, 565, 312–317. [Google Scholar] [CrossRef]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef]
- Matsuno, Y.; Atsumi, Y.; Shimizu, A.; Katayama, K.; Fujimori, H.; Hyodo, M.; Minakawa, Y.; Nakatsu, Y.; Kaneko, S.; Hamamoto, R.; et al. Replication stress triggers microsatellite destabilization and hypermutation leading to clonal expansion in vitro. Nat. Commun. 2019, 10, 3925. [Google Scholar] [CrossRef]
- Duijf, P.H.G.; Nanayakkara, D.; Nones, K.; Srihari, S.; Kalimutho, M.; Khanna, K.K. Mechanisms of Genomic Instability in Breast Cancer. Trends Mol. Med. 2019, 25, 595–611. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.; Vogelstein, B. Genetic instability in colorectal cancers. Nature 1997, 386, 623–627. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef]
- Lee, S.; Hurwitz, J. Mechanism of elongation of primed DNA by DNA polymerase 6, proliferating cell nuclear antigen, and activator 1. Proc. Natl. Acad. Sci. USA 1990, 87, 5672–5676. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Pan, Z.Q.; Kwongs, A.D.; Burgersqll, P.M.J.; Hurwitz, J. Synthesis of DNA by DNA Polymerase E in Vitro*. J. Biol. Chem. 1991, 266, 22707–22717. [Google Scholar] [PubMed]
- Burgers, P.M.J.; Kunkel, T.A. Eukaryotic DNA Replication Fork. Annu. Rev. Biochem. 2017, 86, 417–438. [Google Scholar] [CrossRef] [PubMed]
- Modrich, P.; Lahue, R. Mismatch Repair in Replication Fidelity, Genetic Recombination, and Cancer Biology. Annu. Rev. Biochem. 1996, 65, 101–133. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T.A.; Erie, D.A. DNA Mismatch Repair. Annu. Rev. Biochem. 2005, 74, 681–710. [Google Scholar] [CrossRef] [PubMed]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Tissier, A.; McDonald, J.P.; Frank, E.G.; Woodgate, R. Poliota, a remarkably error-prone human DNA polymerase. Genes Dev. 2000, 14, 1642–1650. [Google Scholar]
- Sebesta, M.; Burkovics, P.; Juhasz, S.; Zhang, S.; Szabo, J.E.; Lee, M.Y.W.T.; Haracska, L.; Krejci, L. Role of PCNA and TLS polymerases in D-loop extension during homologous recombination in humans. DNA Repair 2013, 12, 691–698. [Google Scholar] [CrossRef]
- McVey, M.; Khodaverdian, V.Y.; Meyer, D.; Cerqueira, P.G.; Heyer, W.-D. Eukaryotic DNA Polymerases in Homologous Recombination. Annu. Rev. Genet. 2016, 50, 393–421. [Google Scholar] [CrossRef]
- Goodman, M.F.; Woodgate, R. Translesion DNA polymerases. Cold Spring Harb. Perspect. Biol. 2013, 5, a010363. [Google Scholar] [CrossRef]
- Vaisman, A.; Woodgate, R. Translesion DNA polymerases in eukaryotes: What makes them tick? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 274–303. [Google Scholar] [CrossRef] [PubMed]
- Ichijima, Y.; Yoshioka, K.; Yoshioka, Y.; Shinohe, K.; Fujimori, H.; Unno, J.; Takagi, M.; Goto, H.; Inagaki, M.; Mizutani, S.; et al. DNA lesions induced by replication stress trigger mitotic aberration and tetraploidy development. PLoS ONE 2010, 5, e8821. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.Q.; Bailey, D.; Bronson, D.; Goss, P.E.; Hogg, D. Loss of heterozygosity of tumor suppressor genes in testis cancer. Cancer Res. 1995, 55, 2871–2875. [Google Scholar] [PubMed]
- Deming, S.L.; Nass, S.J.; Dickson, R.B.; Trock, B.J. C-myc amplification in breast cancer: A meta-analysis of its occurrence and prognostic relevance. Br. J. Cancer 2000, 83, 1688–1695. [Google Scholar] [CrossRef]
- Berggren, P.; Kumar, R.; Sakano, S.; Hemminki, L.; Wada, T.; Steineck, G.; Adolfsson, J.; Larsson, P.; Norming, U.; Wijkström, H.; et al. Detecting homozygous deletions in the CDKN2A(p16(INK4a))/ARF(p14(ARF)) gene in urinary bladder cancer using real-time quantitative PCR. Clin. Cancer Res. 2003, 4, 441–444. [Google Scholar]
- The Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [CrossRef]
- Ahn, S.M.; Ansari, A.A.; Kim, J.; Kim, D.; Chun, S.M.; Kim, J.; Kim, T.W.; Park, I.; Yu, C.S.; Jang, S.J. The somatic POLE P286R mutation defines a unique subclass of colorectal cancer featuring hypermutation, representing a potential genomic biomarker for immunotherapy. Oncotarget 2016, 7, 68638–68649. [Google Scholar] [CrossRef]
- Peto, R.; Roe, F.J.; Lee, P.N.; Levy, L.; Clack, J. Cancer and ageing in mice and men. Br. J. Cancer 1975, 32, 411–426. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef]
- Makova, K.D.; Hardison, R.C. The effects of chromatin organization on variation in mutation rates in the genome. Nat. Rev. Genet. 2015, 16, 213. [Google Scholar] [CrossRef] [PubMed]
- Schuster-Böckler, B.; Lehner, B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 2012, 488, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Bert, S.A.; Armstrong, N.J.; Caldon, C.E.; Song, J.Z.; Nair, S.S.; Gould, C.M.; Luu, P.L.; Peters, T.; Khoury, A.; et al. Replication timing and epigenome remodelling are associated with the nature of chromosomal rearrangements in cancer. Nat. Commun. 2019, 10, 416. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.S.; Takata, K.I.; Wood, R.D. DNA polymerases and cancer. Nat. Rev. Cancer 2011, 11, 96–110. [Google Scholar] [CrossRef]
- Janssen, A.; van der Burg, M.; Szuhai, K.; Kops, G.J.P.L.; Medema, R.H. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011, 333, 1895–1898. [Google Scholar] [CrossRef]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.-C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.W.; Fugger, K.; West, S.C. Unresolved recombination intermediates lead to ultra-fine anaphase bridges, chromosome breaks and aberrations. Nat. Cell Biol. 2018, 20, 92–103. [Google Scholar] [CrossRef]
- Stephens, P.J.; McBride, D.J.; Lin, M.-L.; Varela, I.; Pleasance, E.D.; Simpson, J.T.; Stebbings, L.A.; Leroy, C.; Edkins, S.; Mudie, L.J.; et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 2009, 462, 1005–1010. [Google Scholar] [CrossRef]
- Pleasance, E.D.; Cheetham, R.K.; Stephens, P.J.; McBride, D.J.; Humphray, S.J.; Greenman, C.D.; Varela, I.; Lin, M.-L.; Ordóñez, G.R.; Bignell, G.R.; et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010, 463, 191–196. [Google Scholar] [CrossRef]
- Peto, J. Cancer epidemiology in the last century and the next decade. Nature 2001, 411, 390–395. [Google Scholar] [CrossRef]
- Edwards, B.K.; Howe, H.L.; Ries, L.A.G.; Thun, M.J.; Rosenberg, H.M.; Yancik, R.; Wingo, P.A.; Jemal, A.; Feigal, E.G. Annual Report to the Nation on the status of cancer, 1973-1999, featuring implications of age and aging on U.S. cancer burden. Cancer 2002, 94, 2766–2792. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 2004, 6, 168–170. [Google Scholar] [CrossRef] [PubMed]
- Di Fagagna, F.D.A.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Jeyapalan, J.C.; Ferreira, M.; Sedivy, J.M.; Herbig, U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech. Ageing Dev. 2007, 128, 36–44. [Google Scholar] [CrossRef]
- Nijnik, A.; Woodbine, L.; Marchetti, C.; Dawson, S.; Lambe, T.; Liu, C.; Rodrigues, N.P.; Crockford, T.L.; Cabuy, E.; Vindigni, A.; et al. DNA repair is limiting for haematopoietic stem cells during ageing. Nature 2007, 447, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Horikawa, I.; Redon, C.; Nakamura, A.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M. Delayed kinetics of DNA double-strand break processing in normal and pathological aging. Aging Cell 2008, 7, 89–100. [Google Scholar] [CrossRef]
- Rübe, C.E.; Fricke, A.; Widmann, T.A.; Fürst, T.; Madry, H.; Pfreundschuh, M.; Rübe, C. Accumulation of DNA Damage in Hematopoietic Stem and Progenitor Cells during Human Aging. PLoS ONE 2011, 6, e17487. [Google Scholar] [CrossRef]
- Dickey, J.S.; Solier, S.; Pommier, Y.; Nakamura, A.J.; Redon, C.E.; Sedelnikova, O.A.; Bonner, W.M. γH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar]
- Atsumi, Y.; Fujimori, H.; Fukuda, H.; Inase, A.; Shinohe, K.; Yoshioka, Y.; Shikanai, M.; Ichijima, Y.; Unno, J.; Mizutani, S.; et al. Onset of Quiescence Following p53 Mediated Down-Regulation of H2AX in Normal Cells. PLoS ONE 2011, 6, e23432. [Google Scholar] [CrossRef]
- Osawa, T.; Atsumi, Y.; Sugihara, E.; Saya, H.; Kanno, M.; Tashiro, F.; Masutani, M.; Yoshioka, K. Arf and p53 act as guardians of a quiescent cellular state by protecting against immortalization of cells with stable genomes. Biochem. Biophys. Res. Commun. 2013, 432, 34–39. [Google Scholar] [CrossRef]
- Atsumi, Y.; Inase, A.; Osawa, T.; Sugihara, E.; Sakasai, R.; Fujimori, H.; Teraoka, H.; Saya, H.; Kanno, M.; Tashiro, F.; et al. The Arf/p53 Protein Module, Which Induces Apoptosis, Down-regulates Histone H2AX to Allow Normal Cells to Survive in the Presence of Anti-cancer Drugs. J. Biol. Chem. 2013, 288, 13269–13277. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, Y.; Minakawa, Y.; Ono, M.; Dobashi, S.; Shinohe, K.; Shinohara, A.; Takeda, S.; Takagi, M.; Takamatsu, N.; Nakagama, H.; et al. ATM and SIRT6/SNF2H Mediate Transient H2AX Stabilization When DSBs Form by Blocking HUWE1 to Allow Efficient γH2AX Foci Formation. Cell Rep. 2015, 13, 2728–2740. [Google Scholar] [CrossRef] [PubMed]
- Minakawa, Y.; Atsumi, Y.; Shinohara, A.; Murakami, Y.; Yoshioka, K. Gamma-irradiated quiescent cells repair directly induced double-strand breaks but accumulate persistent double-strand breaks during subsequent DNA replication. Genes to Cells 2016, 21, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Wedge, D.C.; Alexandrov, L.B.; Petljak, M.; Butler, A.P.; Bolli, N.; Davies, H.R.; Knappskog, S.; Martin, S.; Papaemmanuil, E.; et al. Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nat. Genet. 2014, 46, 487–491. [Google Scholar] [CrossRef]
- Nikkilä, J.; Kumar, R.; Campbell, J.; Brandsma, I.; Pemberton, H.N.; Wallberg, F.; Nagy, K.; Scheer, I.; Vertessy, B.G.; Serebrenik, A.A.; et al. Elevated APOBEC3B expression drives a kataegic-like mutation signature and replication stress-related therapeutic vulnerabilities in p53-defective cells. Br. J. Cancer 2017, 117, 113–123. [Google Scholar] [CrossRef]
- Shinohara, M.; Io, K.; Shindo, K.; Matsui, M.; Sakamoto, T.; Tada, K.; Kobayashi, M.; Kadowaki, N.; Takaori-Kondo, A. APOBEC3B can impair genomic stability by inducing base substitutions in genomic DNA in human cells. Sci. Rep. 2012, 2, 806. [Google Scholar] [CrossRef]
- Hoopes, J.I.I.; Cortez, L.M.M.; Mertz, T.M.M.; Malc, E.P.P.; Mieczkowski, P.A.A.; Roberts, S.A.A. APOBEC3A and APOBEC3B Preferentially Deaminate the Lagging Strand Template during DNA Replication. Cell Rep. 2016, 14, 1273–1282. [Google Scholar] [CrossRef]
- Herman, J.G.; Umar, A.; Polyak, K.; Graff, J.R.; Ahuja, N.; Issa, J.P.J.; Markowitz, S.; Willson, J.K.V.; Hamilton, S.R.; Kinzler, K.W.; et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc. Natl. Acad. Sci. USA 1998, 95, 6870–6875. [Google Scholar] [CrossRef]
- Pal, T.; Permuth-Wey, J.; Kumar, A.; Sellers, T.A. Systematic review and meta-analysis of ovarian cancers: Estimation of microsatellite-high frequency and characterization of mismatch repair deficient tumor histology. Clin. Cancer Res. 2008, 14, 6847–6854. [Google Scholar] [CrossRef]
- Koopman, M.; Kortman, G.A.M.; Mekenkamp, L.; Ligtenberg, M.J.L.; Hoogerbrugge, N.; Antonini, N.F.; Punt, C.J.A.; Van Krieken, J.H.J.M. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br. J. Cancer 2009, 100, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Marchionni, L.; Nowak, M.A.; Parmigiani, G.; Vogelstein, B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Nobori, T.; Miura, K.; Wu, D.J.; Lois, A.; Takabayashi, K.; Carson, D.A. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature 1994, 368, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, A.; Demetrickt, D.J.; Spillare, E.A.; Hagiwara, K.; Perwez Hussain, S.; Bennett, W.P.; Forrester, K.; Gerwin, B.; Serranot, M.; Beacht, D.H.; et al. Mutations and altered expression of p16INK4 in human cancer. Proc. Natl. Acad. Sci. USA 1994, 91, 11045–11049. [Google Scholar] [CrossRef] [PubMed]
- Fargnoli, M.C.; Chimenti, S.; Peris, K.; Keller, G.; Soyer, H.P.; Pozzo, V.D.; Höfler, H. CDKN2a/p16INK4a Mutations and Lack of p19ARF Involvement in Familial Melanoma Kindreds. J. Invest. Dermatol. 1998, 111, 1202–1206. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Mutation and Cancer: Statistical Study of Retinoblastoma. Sciences 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Mutation and Human Cancer. Adv. Cancer Res. 1973, 17, 317–352. [Google Scholar]
- Comings, D.E. A General Theory of Carcinogenesis (genes/viruses). Proc. Natl. Acad. Sci. USA 1973, 70, 3324–3328. [Google Scholar] [CrossRef]
- Vitale, I.; Galluzzi, L.; Senovilla, L.; Criollo, A.; Jemaà, M.; Castedo, M.; Kroemer, G. Illicit survival of cancer cells during polyploidization and depolyploidization. Cell Death Differ. 2011, 18, 1403–1413. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Jasin, M. Loss of heterozygosity induced by a chromosomal double-strand break. Proc. Natl. Acad. Sci. USA 1997, 94, 8988–8993. [Google Scholar] [CrossRef]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, K.-I.; Atsumi, Y.; Fukuda, H.; Masutani, M.; Teraoka, H. The quiescent cellular state is Arf/p53-dependent and associated with H2AX downregulation and genome stability. Int. J. Mol. Sci. 2012, 13, 6492–6506. [Google Scholar] [CrossRef] [PubMed]
- Minakawa, Y.; Shimizu, A.; Matsuno, Y.; Yoshioka, K. Genomic Destabilization Triggered by Replication Stress during Senescence. Cancers 2017, 9, 159. [Google Scholar] [CrossRef]
- Rosenberg, S.; Longerich, S.; Gee, P.; Harris, R. Adaptive mutation by deletions in small mononucleotide repeats. Science 1994, 265, 405–407. [Google Scholar] [CrossRef] [PubMed]
- Bjedov, I.; Tenaillon, O.; Gérard, B.; Souza, V.; Denamur, E.; Radman, M.; Taddei, F.; Matic, I. Stress-induced mutagenesis in bacteria. Science 2003, 300, 1404–1409. [Google Scholar] [CrossRef]
- Gonzalez, C.; Hadany, L.; Ponder, R.G.; Price, M.; Hastings, P.J.; Rosenberg, S.M. Mutability and importance of a hypermutable cell subpopulation that produces stress-induced mutants in Escherichia coli. PLoS Genet. 2008, 4, e1000208. [Google Scholar] [CrossRef]
- Bos, J.; Zhang, Q.; Vyawahare, S.; Rogers, E.; Rosenberg, S.M.; Austin, R.H. Emergence of antibiotic resistance from multinucleated bacterial filaments. Proc. Natl. Acad. Sci. USA 2015, 112, 178. [Google Scholar] [CrossRef]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef]
- Nyce, J.; Leonard, S.; Canupp, D.; Schulz, S.; Wong, S.O. Epigenetic mechanisms of drug resistance: Drug-induced DNA hypermethylation and drug resistance. Proc. Natl. Acad. Sci. USA 1993, 90, 2960–2964. [Google Scholar] [CrossRef]
- Ohata, Y.; Shimada, S.; Akiyama, Y.; Mogushi, K.; Nakao, K.; Matsumura, S.; Aihara, A.; Mitsunori, Y.; Ban, D.; Ochiai, T.; et al. Acquired Resistance with Epigenetic Alterations Under Long-Term Antiangiogenic Therapy for Hepatocellular Carcinoma. Mol. Cancer Ther. 2017, 16, 1155–1165. [Google Scholar] [CrossRef]
- Bartkova, J.; Hořejší, Z.; Koed, K.; Krämer, A.; Tort, F.; Zleger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Vassiliou, L.-V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; DiTullio Jr, R.A.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Downing, J.R.; Head, D.R.; Parham, D.M.; Douglass, E.C.; Hulshof, M.G.; Link, M.P.; Motroni, T.A.; Grier, H.E.; Curcio-Brint, A.M.; Shapiro, D.N. Detection of the (11;22)(q24;q12) translocation of Ewing’s sarcoma and peripheral neuroectodermal tumor by reverse transcription polymerase chain reaction. Am. J. Pathol. 1993, 143, 1294–1300. [Google Scholar] [PubMed]
- Chaplin, B.T.; Beverloo, H.; Saha, V.; Hagemeijer, A.; Berger, R.; Young, B. The t(10; 11) Translocation in Acute Myeloid Leukemia (M5) Consistently Fuses the Leucine Zipper Motif of AFlO Onto the HRX Gene. Blood 1995, 86, 2073–2076. [Google Scholar] [CrossRef]
- Cerrone, M.; Cantile, M.; Collina, F.; Marra, L.; Liguori, G.; Franco, R.; De Chiara, A.; Botti, G. Molecular strategies for detecting chromosomal translocations in soft tissue tumors (Review). Int. J. Mol. Med. 2014, 33, 1379. [Google Scholar] [CrossRef]
- Patel, N.; Black, J.; Chen, X.; Marcondes, A.M.; Grady, W.M.; Lawlor, E.R.; Borinstein, S.C. DNA Methylation and Gene Expression Profiling of Ewing Sarcoma Primary Tumors Reveal Genes That Are Potential Targets of Epigenetic Inactivation. Sarcoma 2012, 2012, 1–11. [Google Scholar] [CrossRef]
- Huether, R.; Dong, L.; Chen, X.; Wu, G.; Parker, M.; Wei, L.; Ma, J.; Edmonson, M.N.; Hedlund, E.K.; Rusch, M.C.; et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat. Commun. 2014, 5, 3630. [Google Scholar] [CrossRef]
- Hao, J.J.; Lin, D.C.; Dinh, H.Q.; Mayakonda, A.; Jiang, Y.Y.; Chang, C.; Jiang, Y.; Lu, C.C.; Shi, Z.Z.; Xu, X.; et al. Spatial intratumoral heterogeneity and temporal clonal evolution in esophageal squamous cell carcinoma. Nat. Genet. 2016, 48, 1500–1507. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949. [Google Scholar] [CrossRef]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [PubMed]
- Bald, T.; Quast, T.; Landsberg, J.; Rogava, M.; Glodde, N.; Lopez-Ramos, D.; Kohlmeyer, J.; Riesenberg, S.; van den Boorn-Konijnenberg, D.; Hömig-Hölzel, C.; et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature 2014, 507, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.N.; Nakamura, A.; Haeno, H. The evolution of tumor metastasis during clonal expansion with alterations in metastasis driver genes. Sci. Rep. 2015, 5, 15886. [Google Scholar] [CrossRef] [PubMed]
- McCreery, M.Q.; Halliwill, K.D.; Chin, D.; Delrosario, R.; Hirst, G.; Vuong, P.; Jen, K.Y.; Hewinson, J.; Adams, D.J.; Balmain, A. Evolution of metastasis revealed by mutational landscapes of chemically induced skin cancers. Nat. Med. 2015, 21, 1514–1520. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, H.; Wu, X.; Ma, D.; Wu, J.; Wang, L.; Jiang, Y.; Fei, Y.; Zhu, C.; Tan, R.; et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 2018, 563, 131–136. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| During Normal Replication | Under Replication Stress | |
|---|---|---|
| MMR-proficient cells | ||
| Operating DNA polymerases | Polα, Polδ, and Polε | TLS pols (*1) |
| Mutation rate | Low | High |
| Genomic destabilization risk and genomic instability type caused | No | CIN |
| Genomic-instability-associated alterations risking cancer-driver mutations | No | Gene amplification, LOH, and deletion (*2) |
| MMR-deficient cells | ||
| Operating DNA polymerases | Polδ and Polε | TLS pols (*1) |
| Mutation rate | High | Very high |
| Genomic destabilization risk and genomic instability type caused | No | MSI |
| Genomic-instability-associated alterations risking cancer-driver mutations | No | LOH and deletion (*3) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshioka, K.-i.; Matsuno, Y.; Hyodo, M.; Fujimori, H. Genomic-Destabilization-Associated Mutagenesis and Clonal Evolution of Cells with Mutations in Tumor-Suppressor Genes. Cancers 2019, 11, 1643. https://doi.org/10.3390/cancers11111643
Yoshioka K-i, Matsuno Y, Hyodo M, Fujimori H. Genomic-Destabilization-Associated Mutagenesis and Clonal Evolution of Cells with Mutations in Tumor-Suppressor Genes. Cancers. 2019; 11(11):1643. https://doi.org/10.3390/cancers11111643
Chicago/Turabian StyleYoshioka, Ken-ichi, Yusuke Matsuno, Mai Hyodo, and Haruka Fujimori. 2019. "Genomic-Destabilization-Associated Mutagenesis and Clonal Evolution of Cells with Mutations in Tumor-Suppressor Genes" Cancers 11, no. 11: 1643. https://doi.org/10.3390/cancers11111643
APA StyleYoshioka, K.-i., Matsuno, Y., Hyodo, M., & Fujimori, H. (2019). Genomic-Destabilization-Associated Mutagenesis and Clonal Evolution of Cells with Mutations in Tumor-Suppressor Genes. Cancers, 11(11), 1643. https://doi.org/10.3390/cancers11111643