SLUG Directs the Precursor State of Human Brain Tumor Stem Cells

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
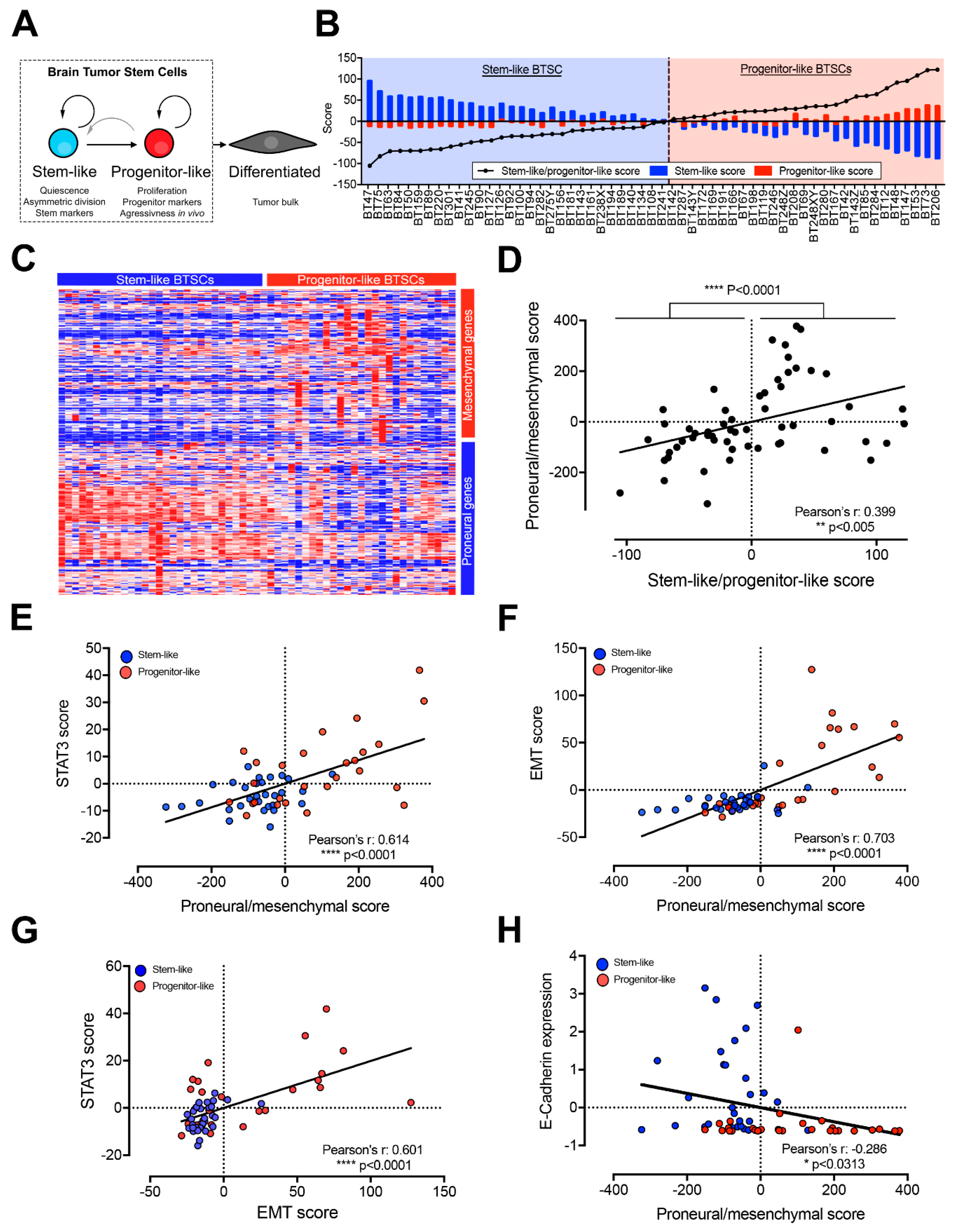
2.1. Progenitor-Like BTSCs Display a Mesenchymal Expression Profile Associated with Over-Activated STAT3 and EMT Pathways
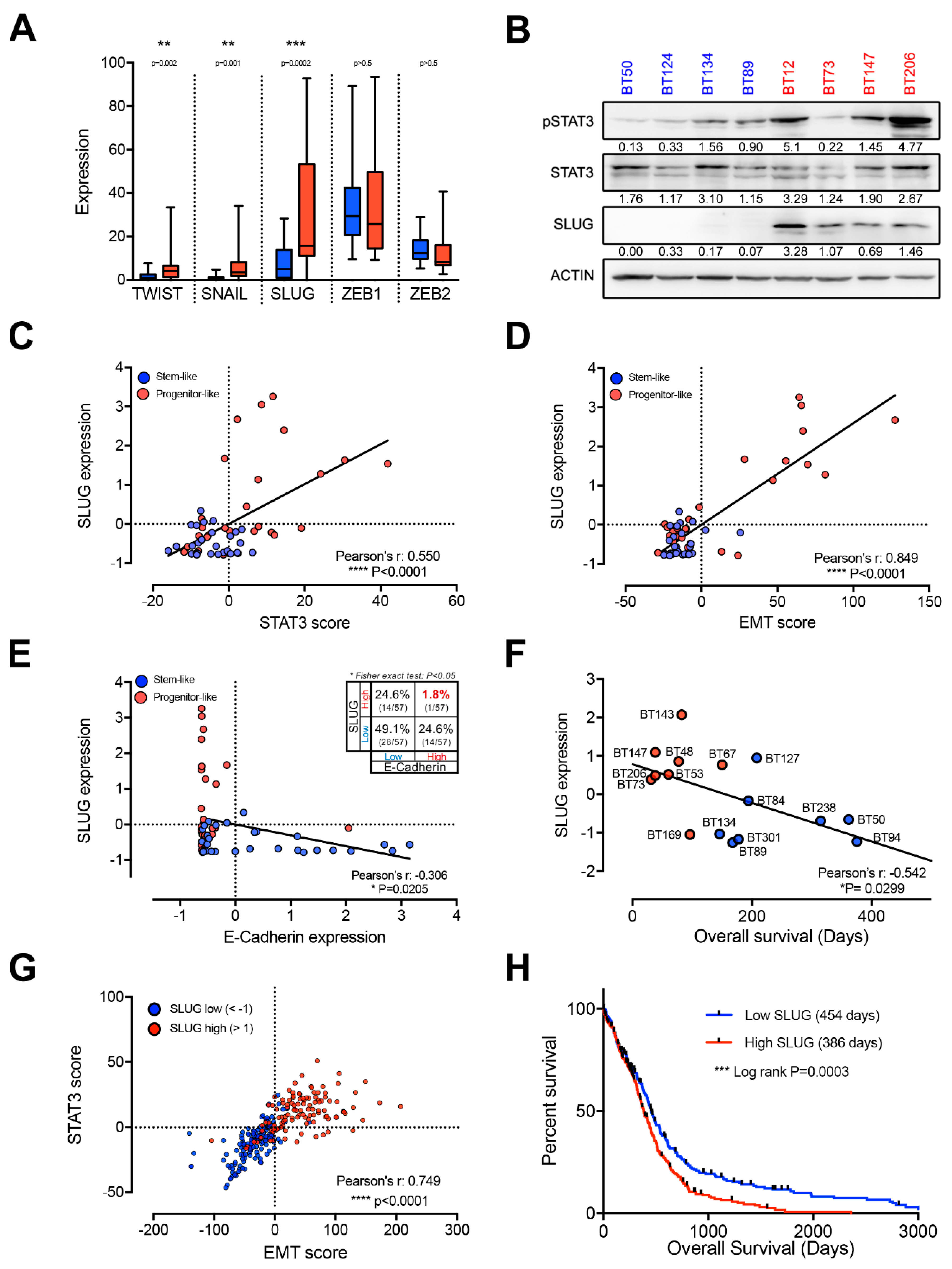
2.2. EMT Master Regulators Are Enriched in Progenitor-Like BTSCs
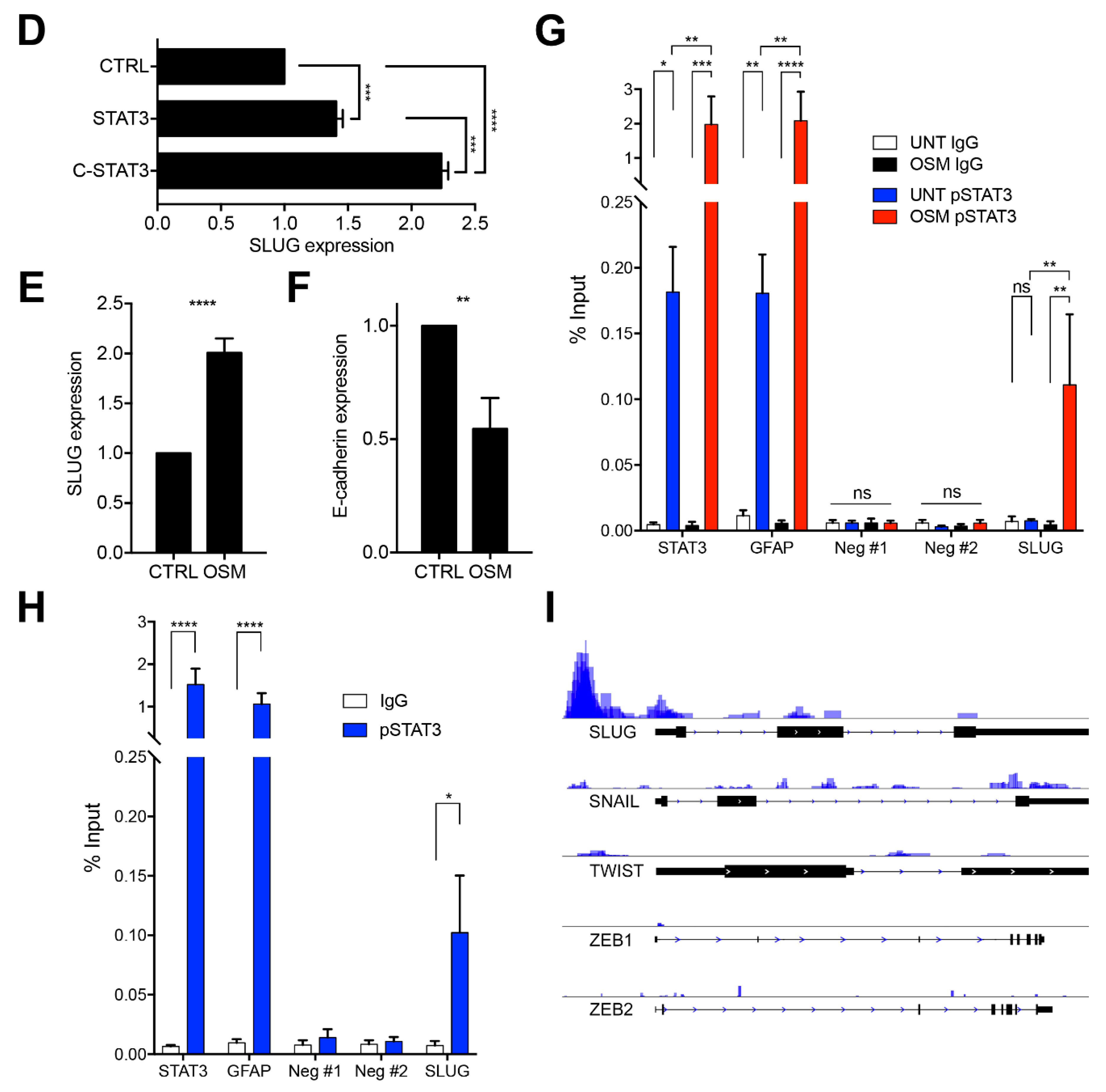
2.3. SLUG Is the Main EMT Master Regulator Directly Regulated by STAT3 in BTSCs
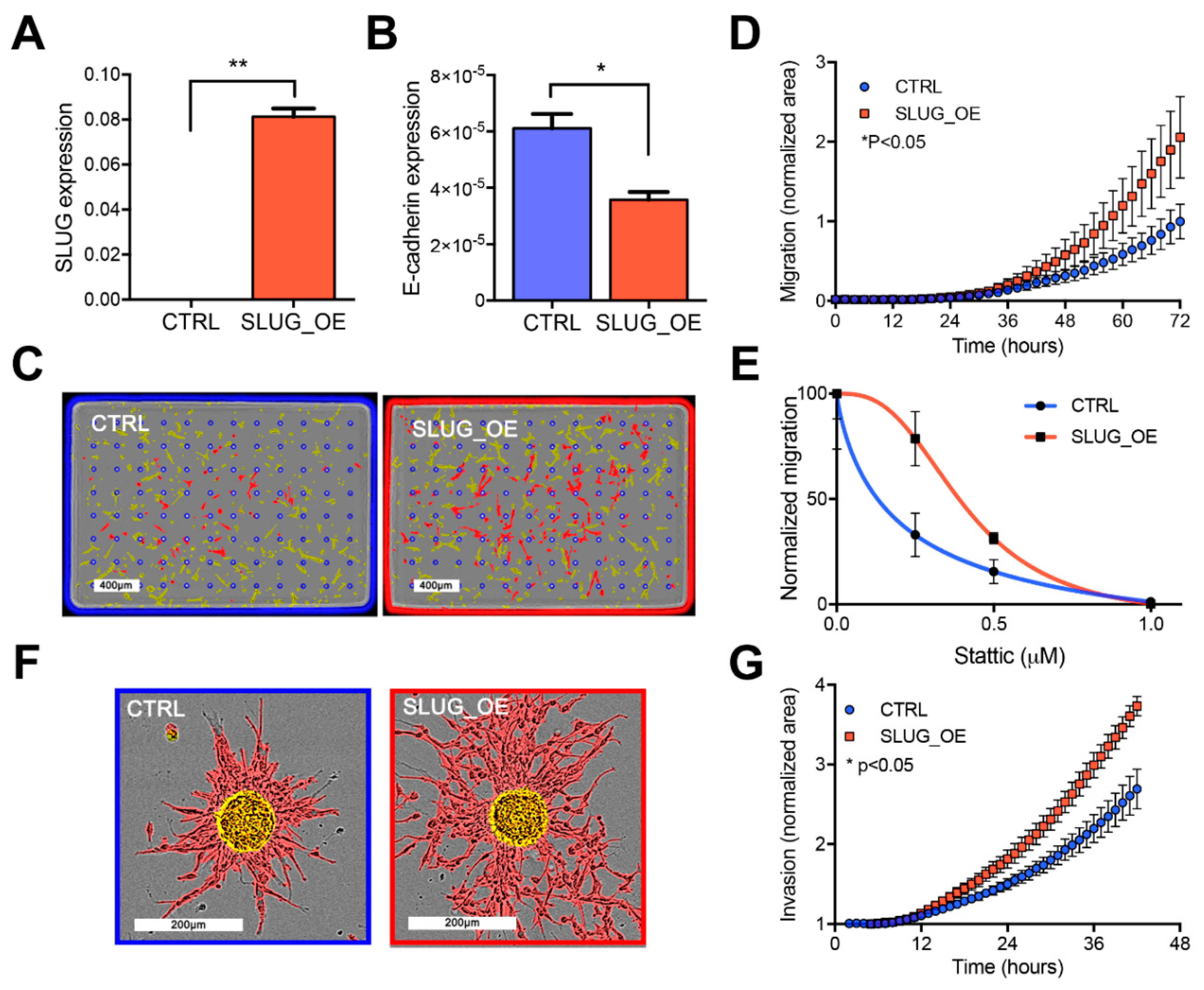
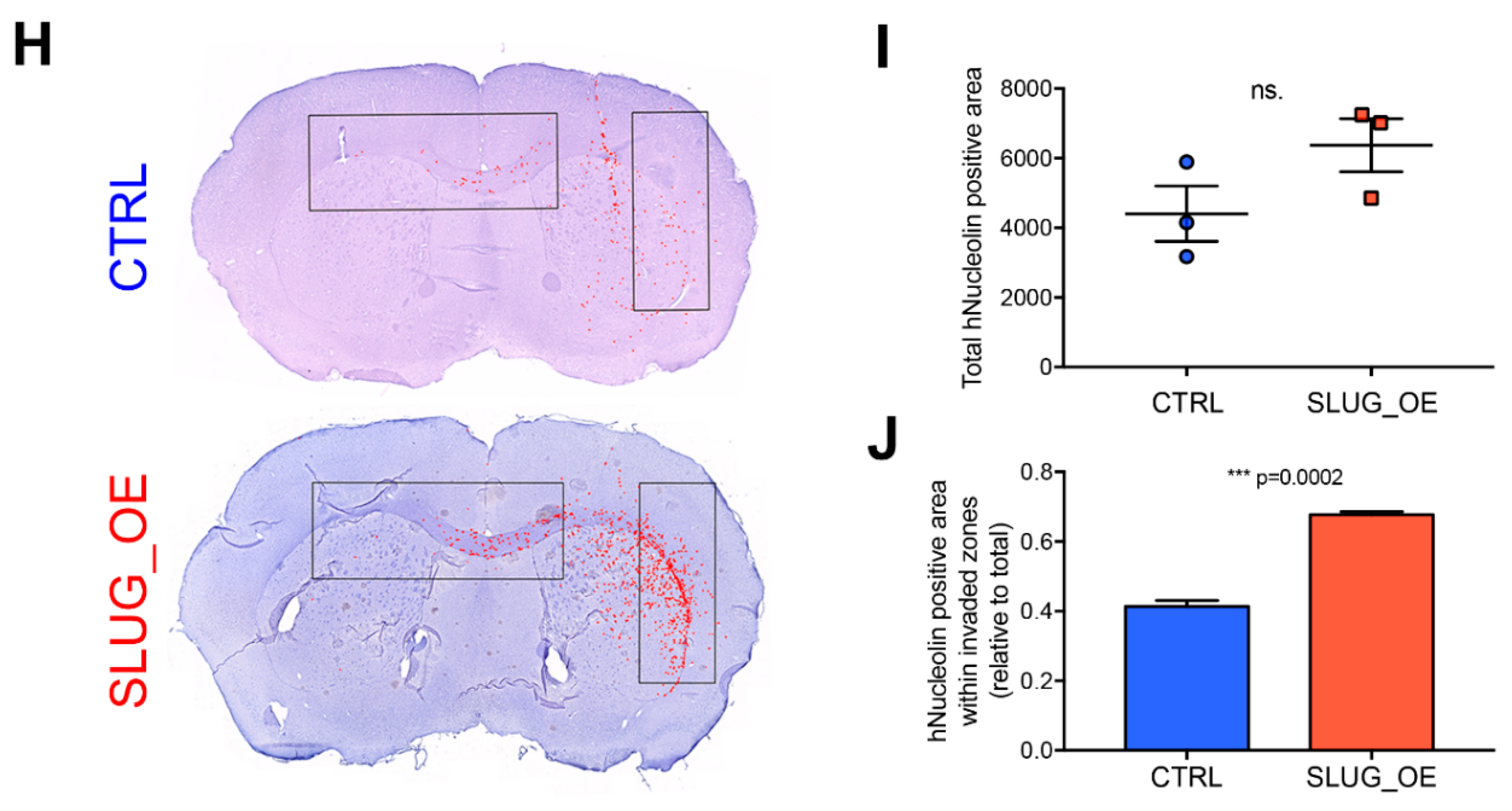
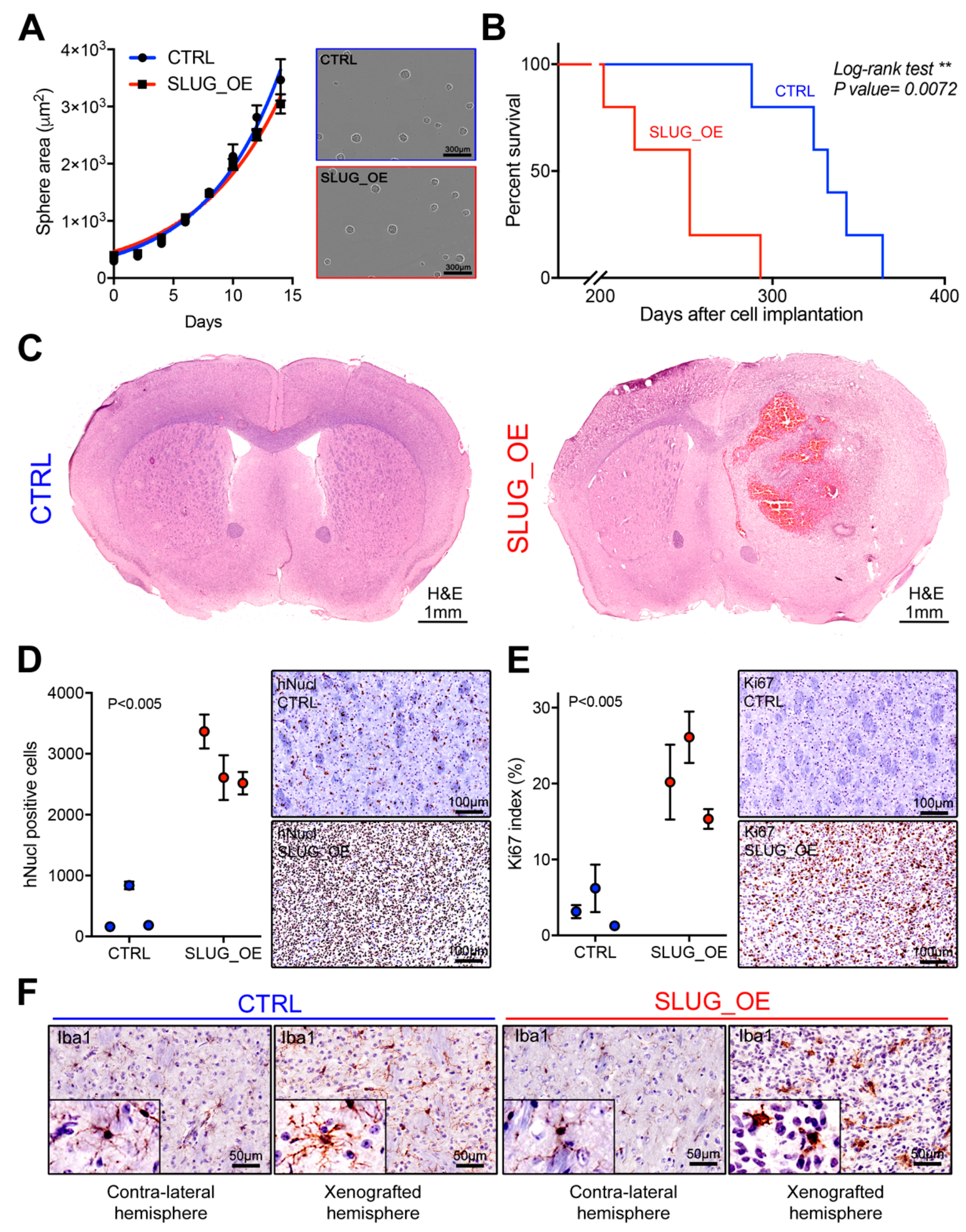
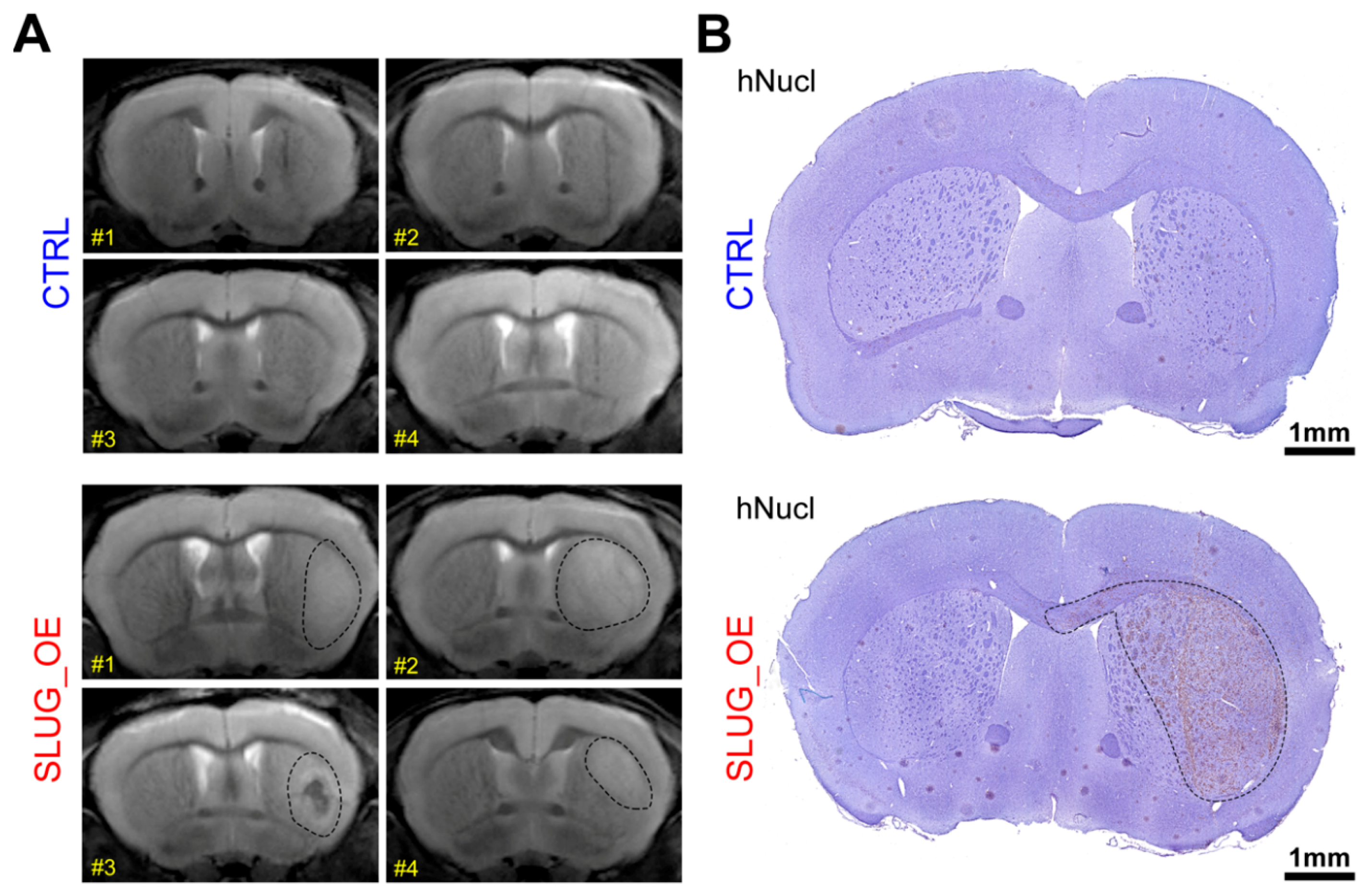
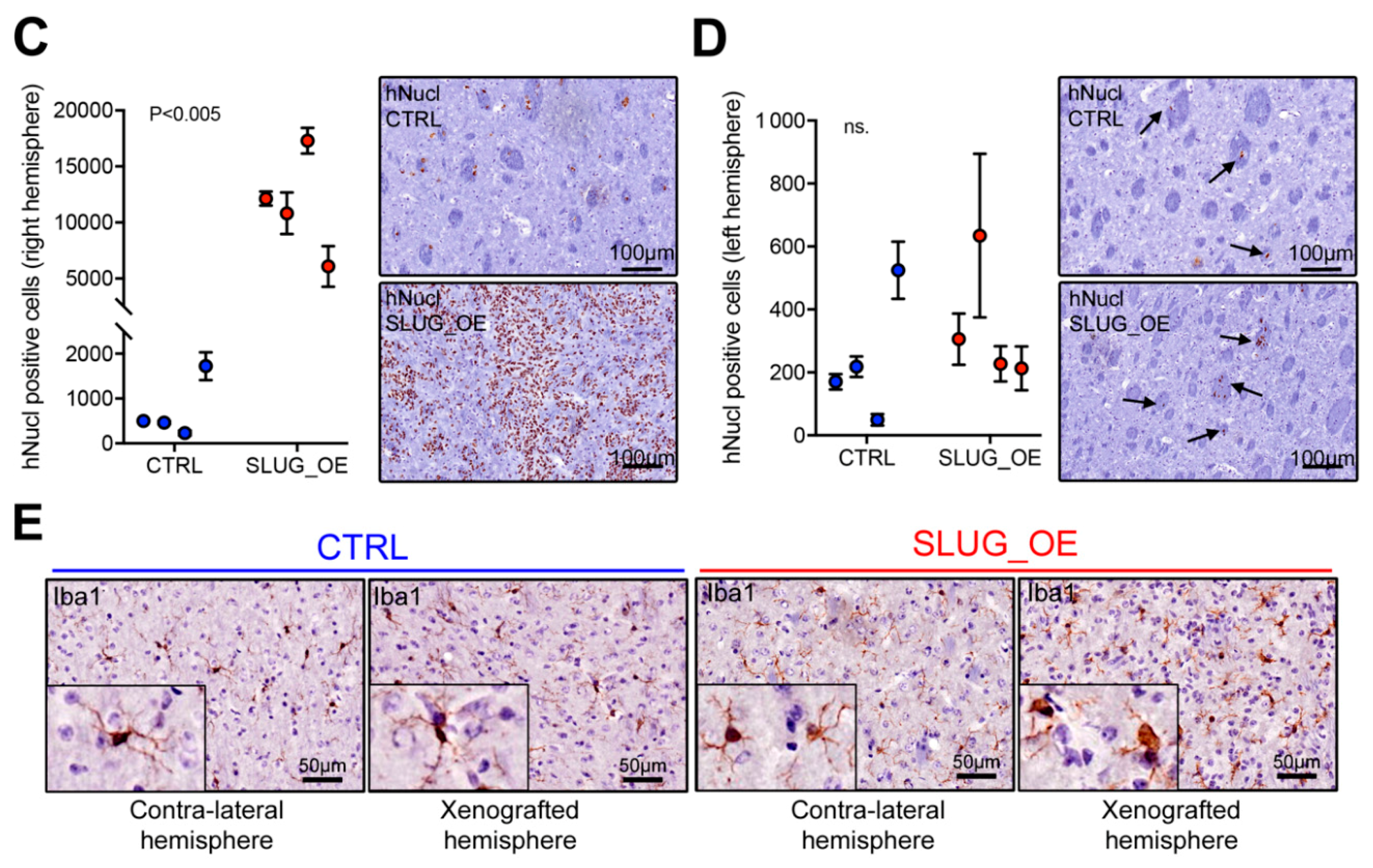
2.4. SLUG Overexpression Promotes BTSC Migration and Invasion Both In Vitro and In Vivo
2.5. SLUG Overexpression Leads to Shorter Survival In Vivo
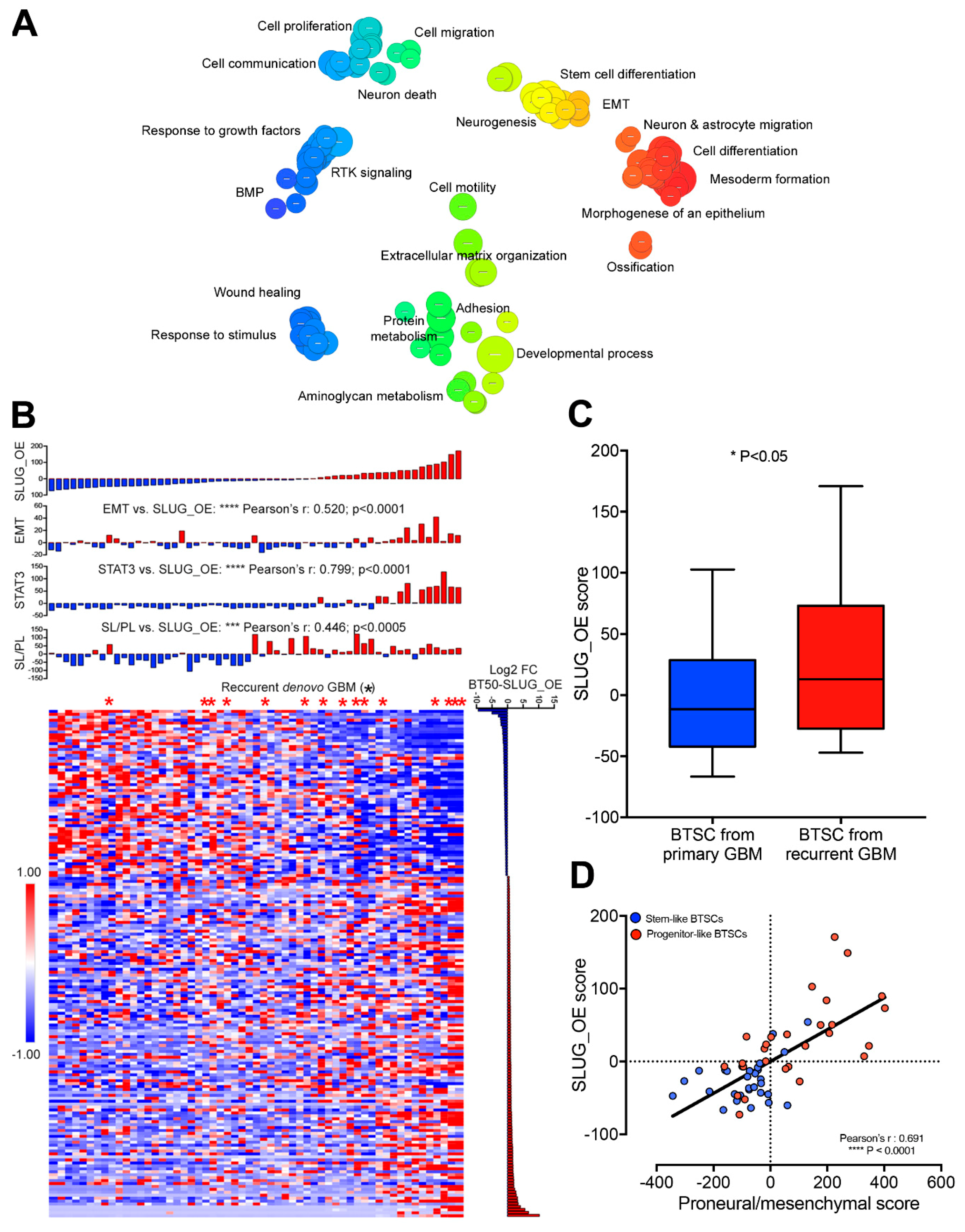
2.6. SLUG Drives a Shift toward an Aggressive Progenitor-Like Precursor State
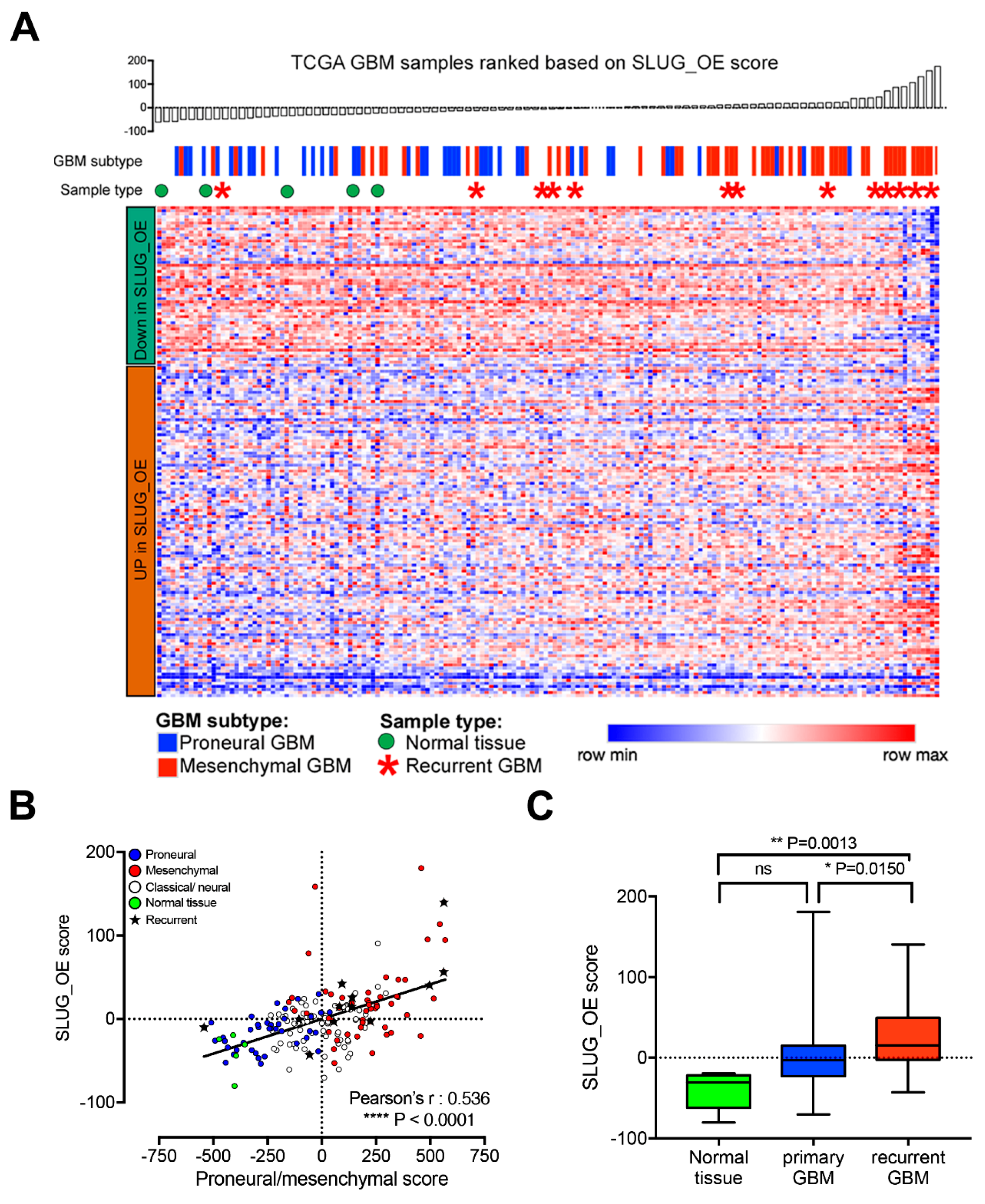
2.7. SLUG-Induced Transcriptional Changes Associate with Progenitor-Like BTSC Precursor State and Recurrence
3. Discussion
4. Materials and Methods
4.1. BTSC Lines
4.2. Cell Culture
4.3. Immunoblotting
4.4. Real-Time Quantitative PCR
- ACTIN: F: CATGTACGTTGCTATCCAGGC; R: CTCCTTAATGTCACGCACGAT
- SNAI2: F: TCGGACCCACACATTACCTTG; R: AAAAAGGCTTCTCCCCCGTGT
- CDH1: F: CCCAATACATCTCCCTTCACAG; R: CCACCTCTAAGGCCATCTTTG
4.5. Transcriptomic Data
4.6. Signature Scores
4.7. Differential Genes Expression Analysis and Gene Ontology Analysis
4.8. Overexpression of STAT3 and SLUG in BTSCs
4.9. Chromatin Immunoprecipitation (ChIP)
- STAT3: F: 5′CTCCCTGAGTTGGCTGTTCT3′; R: 5′GAGCCGTATCAGGGCATTTA3′
- GFAP: F: 5′TCCGAGAAGCCCATTGAG3′; R: 5′TGTGCTGCTTTTATCCCAAGA3′
- SLUG: F: 5′GCACCACATAAAAGCAGGGG3′; R: 5′GGGGGCAAGAGGTAACTGTC3′
4.10. Migration and Invasion
4.11. Intracranial BTSC Xenografts
4.12. MRI
4.13. Immunohistochemistry
4.14. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.E.; Patel, M.; Ruzevick, J.; Jackson, C.M.; Lim, M. STAT3 Activation in Glioblastoma: Biochemical and Therapeutic Implications. Cancers 2014, 6, 376–395. [Google Scholar] [CrossRef] [Green Version]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010, 463, 318–325. [Google Scholar] [CrossRef]
- Halliday, J.; Helmy, K.; Pattwell, S.S.; Pitter, K.L.; LaPlant, Q.; Ozawa, T.; Holland, E.C. In vivo radiation response of proneural glioma characterized by protective p53 transcriptional program and proneural-mesenchymal shift. Proc. Natl. Acad. Sci. USA 2014, 111, 5248–5253. [Google Scholar] [CrossRef] [Green Version]
- Lau, J.; Ilkhanizadeh, S.; Wang, S.; Miroshnikova, Y.A.; Salvatierra, N.A.; Wong, R.A.; Schmidt, C.; Weaver, V.M.; Weiss, W.A.; Persson, A.I. STAT3 blockade inhibits radiation-induced malignant progression in glioma. Cancer Res. 2015. [Google Scholar] [CrossRef]
- McKeever, P.E.; Davenport, R.D.; Shakui, P. Patterns of antigenic expression of human glioma cells. Crit. Rev. Neurobiol. 1991, 6, 119–147. [Google Scholar]
- Tso, C.L.; Freije, W.A.; Day, A.; Chen, Z.; Merriman, B.; Perlina, A.; Lee, Y.; Dia, E.Q.; Yoshimoto, K.; Mischel, P.S.; et al. Distinct transcription profiles of primary and secondary glioblastoma subgroups. Cancer Res. 2006, 66, 159–167. [Google Scholar] [CrossRef]
- Wood, M.D.; Reis, G.F.; Reuss, D.E.; Phillips, J.J. Protein Analysis of Glioblastoma Primary and Posttreatment Pairs Suggests a Mesenchymal Shift at Recurrence. J. Neuropathol. Exp. Neurol. 2016, 75, 925–935. [Google Scholar] [CrossRef]
- Cheng, W.Y.; Kandel, J.J.; Yamashiro, D.J.; Canoll, P.; Anastassiou, D. A multi-cancer mesenchymal transition gene expression signature is associated with prolonged time to recurrence in glioblastoma. PLoS ONE 2012, 7, e34705. [Google Scholar] [CrossRef] [PubMed]
- Han, S.P.; Kim, J.H.; Han, M.E.; Sim, H.E.; Kim, K.S.; Yoon, S.; Baek, S.Y.; Kim, B.S.; Oh, S.O. SNAI1 is involved in the proliferation and migration of glioblastoma cells. Cell. Mol. Neurobiol. 2011, 31, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Mikheeva, S.A.; Mikheev, A.M.; Petit, A.; Beyer, R.; Oxford, R.G.; Khorasani, L.; Maxwell, J.P.; Glackin, C.A.; Wakimoto, H.; Gonzalez-Herrero, I.; et al. TWIST1 promotes invasion through mesenchymal change in human glioblastoma. Mol. Cancer 2010, 9, 194. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.W.; Menon, L.G.; Black, P.M.; Carroll, R.S.; Johnson, M.D. SNAI2/Slug promotes growth and invasion in human gliomas. BMC Cancer 2010, 10, 301. [Google Scholar] [CrossRef]
- Reynolds, B.A.; Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 1992, 255, 1707–1710. [Google Scholar] [CrossRef]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef]
- Yuan, X.; Curtin, J.; Xiong, Y.; Liu, G.; Waschsmann-Hogiu, S.; Farkas, D.L.; Black, K.L.; Yu, J.S. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene 2004, 23, 9392–9400. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Kelly, J.J.; Stechishin, O.; Chojnacki, A.; Lun, X.; Sun, B.; Senger, D.L.; Forsyth, P.; Auer, R.N.; Dunn, J.F.; Cairncross, J.G.; et al. Proliferation of human glioblastoma stem cells occurs independently of exogenous mitogens. Stem Cells 2009, 27, 1722–1733. [Google Scholar] [CrossRef]
- Stechishin, O.D.; Luchman, H.A.; Ruan, Y.; Blough, M.D.; Nguyen, S.A.; Kelly, J.J.; Cairncross, J.G.; Weiss, S. On-target JAK2/STAT3 inhibition slows disease progression in orthotopic xenografts of human glioblastoma brain tumor stem cells. Neuro Oncol. 2013, 15, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Luchman, H.A.; Stechishin, O.D.; Dang, N.H.; Blough, M.D.; Chesnelong, C.; Kelly, J.J.; Nguyen, S.A.; Chan, J.A.; Weljie, A.M.; Cairncross, J.G.; et al. An in vivo patient-derived model of endogenous IDH1-mutant glioma. Neuro Oncol. 2012, 14, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Luchman, H.A.; Stechishin, O.D.; Nguyen, S.A.; Lun, X.Q.; Cairncross, J.G.; Weiss, S. Dual mTORC1/2 blockade inhibits glioblastoma brain tumor initiating cells in vitro and in vivo and synergizes with temozolomide to increase orthotopic xenograft survival. Clin. Cancer Res. 2014, 20, 5756–5767. [Google Scholar] [CrossRef] [PubMed]
- Cusulin, C.; Chesnelong, C.; Bose, P.; Bilenky, M.; Kopciuk, K.; Chan, J.A.; Cairncross, J.G.; Jones, S.J.; Marra, M.A.; Luchman, H.A.; et al. Precursor States of Brain Tumor Initiating Cell Lines Are Predictive of Survival in Xenografts and Associated with Glioblastoma Subtypes. Stem Cell Rep. 2015, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal Differentiation Mediated by NF-κB Promotes Radiation Resistance in Glioblastoma. Cancer Cell 2013, 24. [Google Scholar] [CrossRef]
- Lottaz, C.; Beier, D.; Meyer, K.; Kumar, P.; Hermann, A.; Schwarz, J.; Junker, M.; Oefner, P.J.; Bogdahn, U.; Wischhusen, J.; et al. Transcriptional profiles of CD133+ and CD133− glioblastoma-derived cancer stem cell lines suggest different cells of origin. Cancer Res. 2010, 70, 2030–2040. [Google Scholar] [CrossRef]
- Mao, P.; Joshi, K.; Li, J.; Kim, S.H.; Li, P.; Santana-Santos, L.; Luthra, S.; Chandran, U.R.; Benos, P.V.; Smith, L.; et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. USA 2013, 110, 8644–8649. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, T.; Riester, M.; Cheng, Y.K.; Huse, J.T.; Squatrito, M.; Helmy, K.; Charles, N.; Michor, F.; Holland, E.C. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell 2014, 26, 288–300. [Google Scholar] [CrossRef]
- Deuse, T.; Velotta, J.B.; Hoyt, G.; Govaert, J.A.; Taylor, V.; Masuda, E.; Herlaar, E.; Park, G.; Carroll, D.; Pelletier, M.P.; et al. Novel immunosuppression: R348, a JAK3- and Syk-inhibitor attenuates acute cardiac allograft rejection. Transplantation 2008, 85, 885–892. [Google Scholar] [CrossRef]
- Jensen, K.V.; Cseh, O.; Aman, A.; Weiss, S.; Luchman, H.A. The JAK2/STAT3 inhibitor pacritinib effectively inhibits patient-derived GBM brain tumor initiating cells in vitro and when used in combination with temozolomide increases survival in an orthotopic xenograft model. PLoS ONE 2017, 12, e0189670. [Google Scholar] [CrossRef]
- Haftchenary, S.; Luchman, H.A.; Jouk, A.O.; Veloso, A.J.; Page, B.D.; Cheng, X.R.; Dawson, S.S.; Grinshtein, N.; Shahani, V.M.; Kerman, K.; et al. Potent Targeting of the STAT3 Protein in Brain Cancer Stem Cells: A Promising Route for Treating Glioblastoma. ACS Med. Chem. Lett. 2013, 4, 1102–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef]
- Esmaeili, M.; Stensjøen, A.L.; Berntsen, E.M.; Solheim, O.; Reinertsen, I. The Direction of Tumour Growth in Glioblastoma Patients. Sci. Rep. 2018, 8, 1199. [Google Scholar] [CrossRef]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain J. Neurol. 2019, 142, 847–866. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Lo, H.W.; Hsu, S.C.; Xia, W.; Cao, X.; Shih, J.Y.; Wei, Y.; Abbruzzese, J.L.; Hortobagyi, G.N.; Hung, M.C. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007, 67, 9066–9076. [Google Scholar] [CrossRef]
- Saitoh, M.; Endo, K.; Furuya, S.; Minami, M.; Fukasawa, A.; Imamura, T.; Miyazawa, K. STAT3 integrates cooperative Ras and TGF-beta signals that induce Snail expression. Oncogene 2016, 35, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Chen, C.; Shi, M.; Wang, F.; Chen, X.; Diao, D.; Hu, M.; Yu, M.; Qian, L.; Guo, N. Stat3-coordinated Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain oncostatin M-driven epithelial-mesenchymal transition. Oncogene 2013, 32, 5272–5282. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C.; Tsai, J.T.; Chao, T.Y.; Ma, H.I.; Liu, W.H. The STAT3/Slug Axis Enhances Radiation-Induced Tumor Invasion and Cancer Stem-like Properties in Radioresistant Glioblastoma. Cancers 2018, 10, 512. [Google Scholar] [CrossRef] [PubMed]
- Ring, A.; Kim, Y.M.; Kahn, M. Wnt/catenin signaling in adult stem cell physiology and disease. Stem Cell Rev. 2014, 10, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Heuberger, J.; Birchmeier, W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002915. [Google Scholar] [CrossRef] [PubMed]
- Fragoso, M.A.; Patel, A.K.; Nakamura, R.E.; Yi, H.; Surapaneni, K.; Hackam, A.S. The Wnt/beta-catenin pathway cross-talks with STAT3 signaling to regulate survival of retinal pigment epithelium cells. PLoS ONE 2012, 7, e46892. [Google Scholar] [CrossRef] [PubMed]
- Howe, L.R.; Watanabe, O.; Leonard, J.; Brown, A.M. Twist is up-regulated in response to Wnt1 and inhibits mouse mammary cell differentiation. Cancer Res. 2003, 63, 1906–1913. [Google Scholar]
- Conacci-Sorrell, M.; Simcha, I.; Ben-Yedidia, T.; Blechman, J.; Savagner, P.; Ben-Ze’ev, A. Autoregulation of E-cadherin expression by cadherin-cadherin interactions: The roles of beta-catenin signaling, Slug, and MAPK. J. Cell Biol. 2003, 163, 847–857. [Google Scholar] [CrossRef]
- Wang, H.; Xiao, Z.; Zheng, J.; Wu, J.; Hu, X.L.; Yang, X.; Shen, Q. ZEB1 Represses Neural Differentiation and Cooperates with CTBP2 to Dynamically Regulate Cell Migration during Neocortex Development. Cell Rep. 2019, 27, 2335–2353.e2336. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Suwala, A.K.; Raabe, E.H.; Siebzehnrubl, F.A.; Suarez, M.J.; Orr, B.A.; Bar, E.E.; Maciaczyk, J.; Eberhart, C.G. ZEB1 Promotes Invasion in Human Fetal Neural Stem Cells and Hypoxic Glioma Neurospheres. Brain Pathol. 2015, 25, 724–732. [Google Scholar] [CrossRef]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Shen, Y.; Grisdale, C.J.; Islam, S.A.; Bose, P.; Lever, J.; Zhao, E.Y.; Grinshtein, N.; Ma, Y.; Mungall, A.J.; Moore, R.A.; et al. Comprehensive genomic profiling of glioblastoma tumors, BTICs, and xenografts reveals stability and adaptation to growth environments. Proc. Natl. Acad. Sci. USA 2019, 116, 19098–19108. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Smyth, G.K. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 2009, 347, 70–78. [Google Scholar] [CrossRef]
- Hart, S.; Goh, K.C.; Novotny-Diermayr, V.; Hu, C.Y.; Hentze, H.; Tan, Y.C.; Madan, B.; Amalini, C.; Loh, Y.K.; Ong, L.C.; et al. SB1518, a novel macrocyclic pyrimidine-based JAK2 inhibitor for the treatment of myeloid and lymphoid malignancies. Leukemia 2011, 25, 1751–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, R.; Bainbridge, M.; Fejes, A.; Hirst, M.; Krzywinski, M.; Pugh, T.; McDonald, H.; Varhol, R.; Jones, S.; Marra, M. Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing. BioTechniques 2008, 45, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, Y.S.; Kreitzman, M.; Thiessen, N.; Corbett, R.D.; Li, Y.; Pang, J.; Ma, Y.P.; Jones, S.J.; Birol, I. JAGuaR: Junction alignments to genome for RNA-seq reads. PLoS ONE 2014, 9, e102398. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Lo, H.W. STAT3 Target Genes Relevant to Human Cancers. Cancers 2014, 6, 897–925. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Watkinson, J.; Varadan, V.; Anastassiou, D. Multi-cancer computational analysis reveals invasion-associated variant of desmoplastic reaction involving INHBA, THBS2 and COL11A1. BMC Med. Genomics 2010, 3, 51. [Google Scholar] [CrossRef]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chesnelong, C.; Hao, X.; Cseh, O.; Wang, A.Y.; Luchman, H.A.; Weiss, S. SLUG Directs the Precursor State of Human Brain Tumor Stem Cells. Cancers 2019, 11, 1635. https://doi.org/10.3390/cancers11111635
Chesnelong C, Hao X, Cseh O, Wang AY, Luchman HA, Weiss S. SLUG Directs the Precursor State of Human Brain Tumor Stem Cells. Cancers. 2019; 11(11):1635. https://doi.org/10.3390/cancers11111635
Chicago/Turabian StyleChesnelong, Charles, Xiaoguang Hao, Orsolya Cseh, Alice Yijun Wang, H. Artee Luchman, and Samuel Weiss. 2019. "SLUG Directs the Precursor State of Human Brain Tumor Stem Cells" Cancers 11, no. 11: 1635. https://doi.org/10.3390/cancers11111635
APA StyleChesnelong, C., Hao, X., Cseh, O., Wang, A. Y., Luchman, H. A., & Weiss, S. (2019). SLUG Directs the Precursor State of Human Brain Tumor Stem Cells. Cancers, 11(11), 1635. https://doi.org/10.3390/cancers11111635