PIK3CA Cooperates with KRAS to Promote MYC Activity and Tumorigenesis via the Bromodomain Protein BRD9


Abstract
1. Introduction
2. Results
2.1. Contribution of Oncogenic PIK3CA and KRAS to Anchorage-Independent Growth, Proliferation and Migration
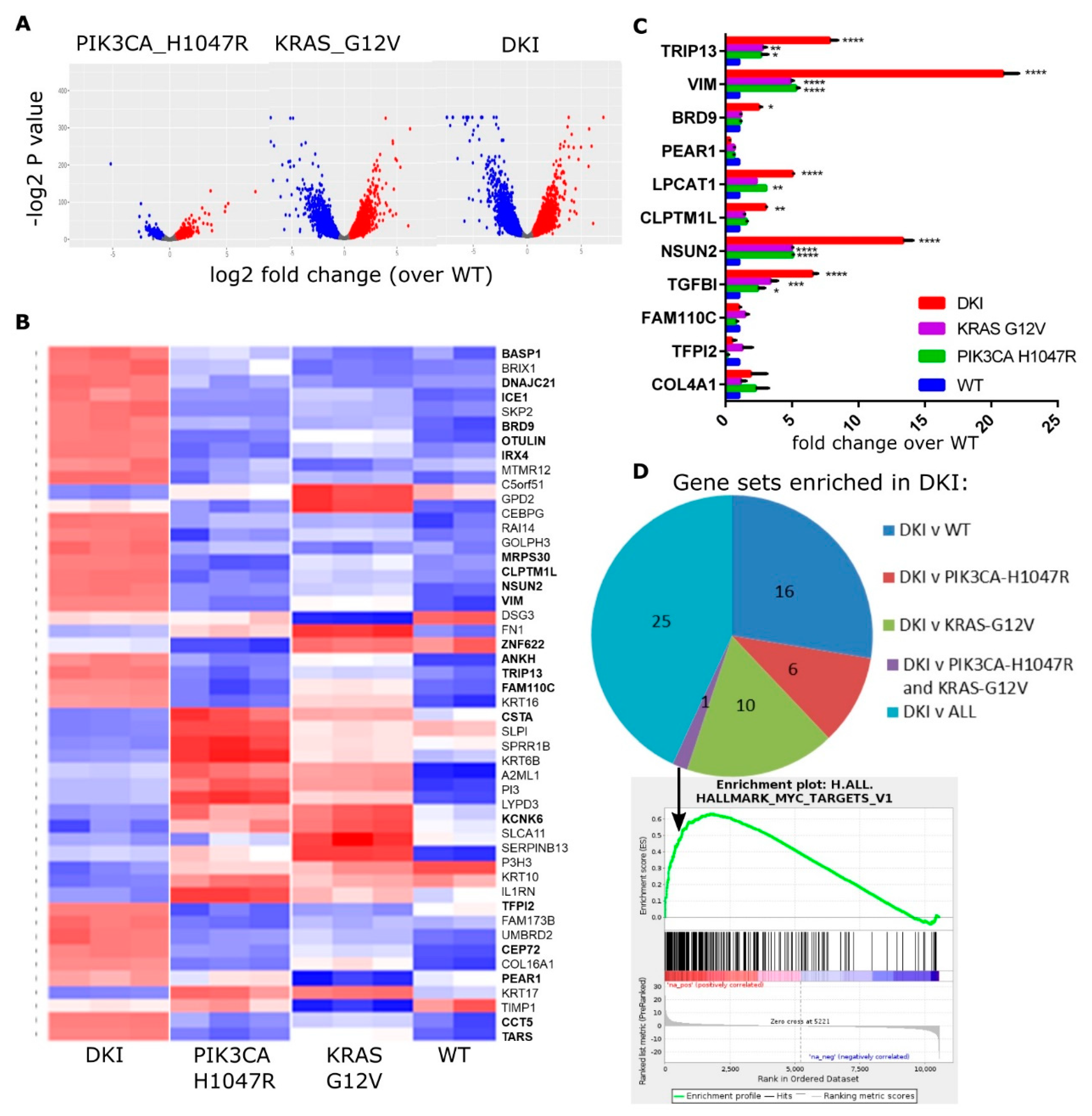
2.2. KRAS and PIK3CA Oncogenes Cooperate to Generate a Transcriptional Signature Enriched for EMT, Cell Cycle and MYC Target Genes
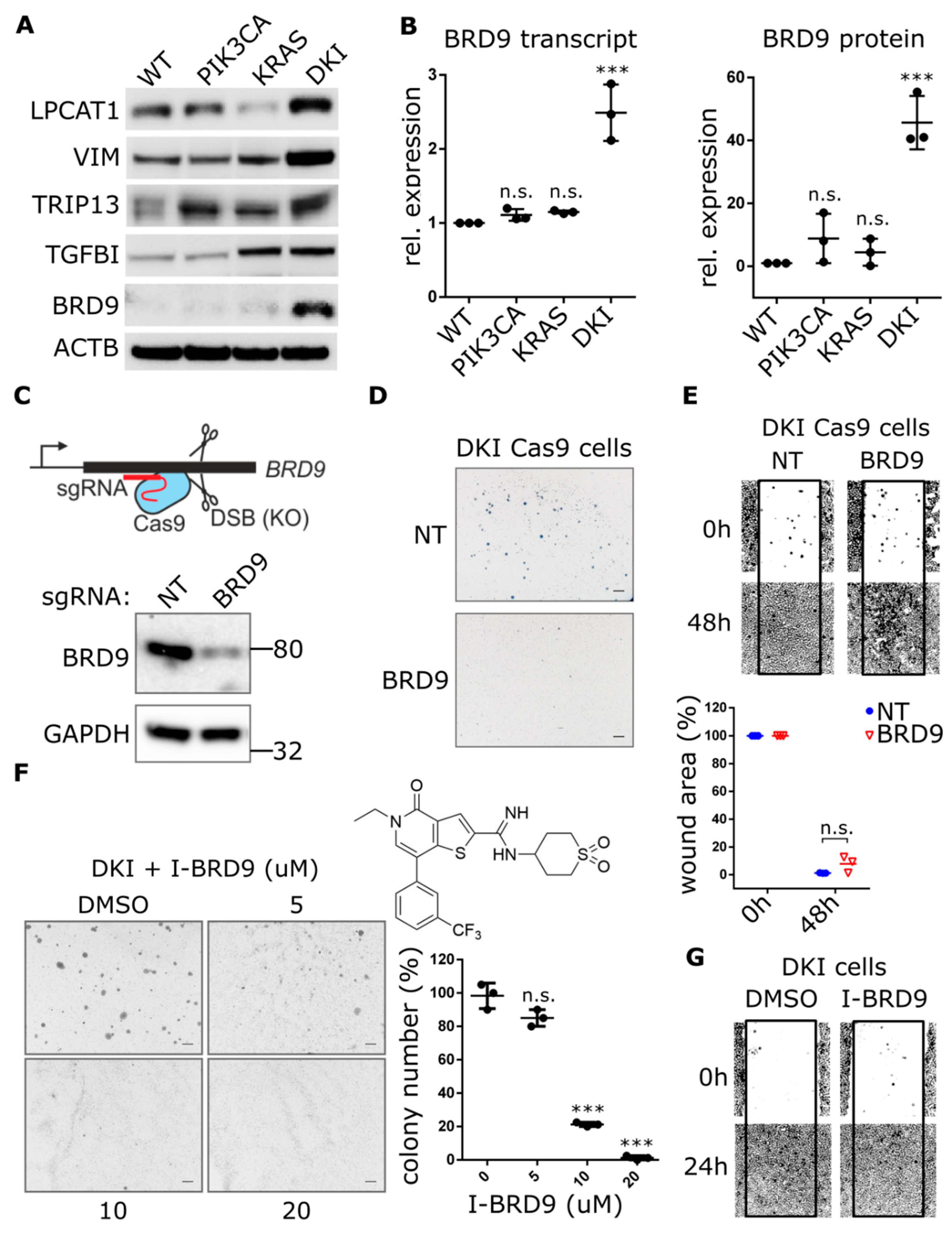
2.3. Oncogenic PIK3CA and KRAS Induce the Expression of the Bromodomain-Containing Protein BRD9
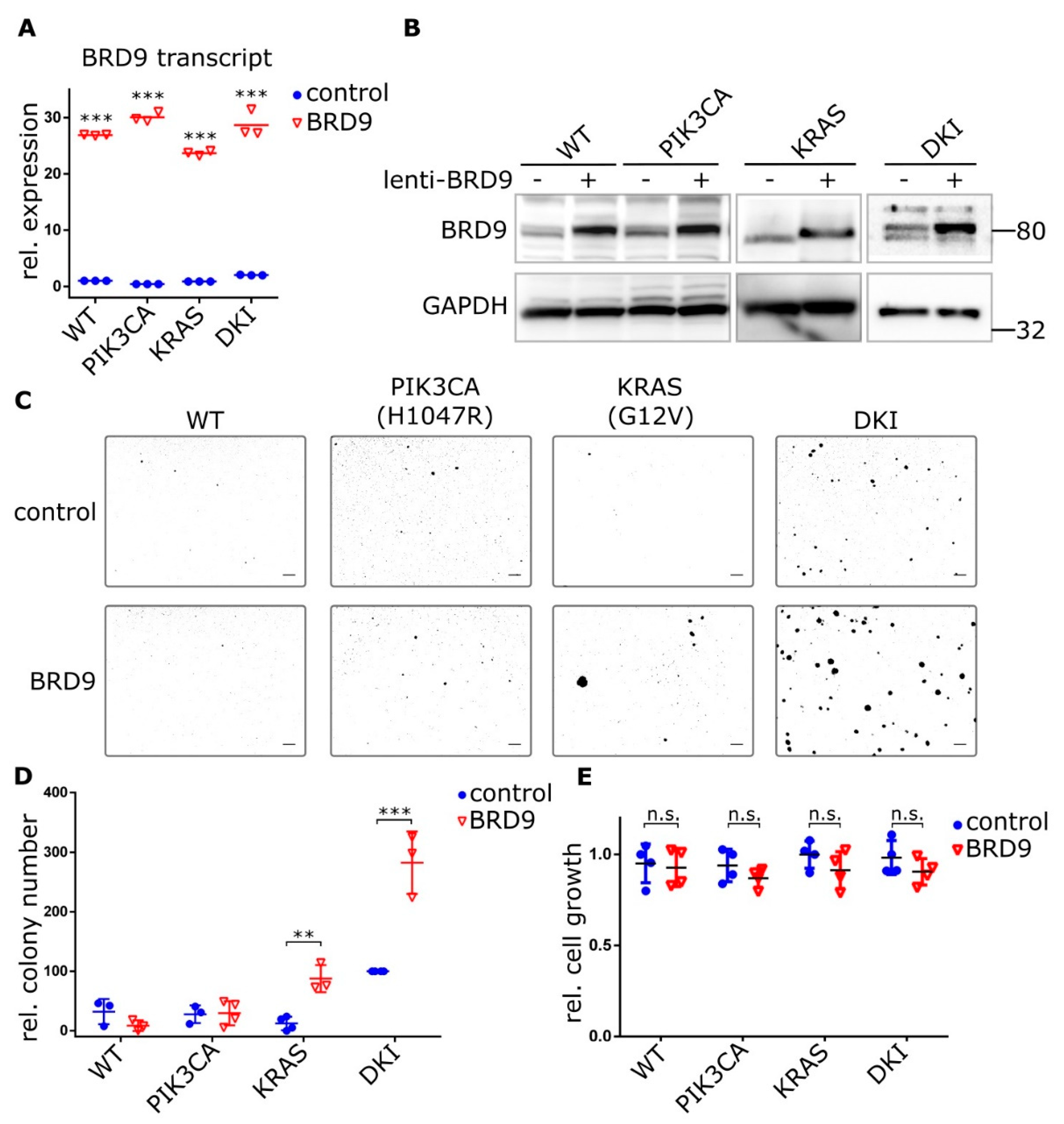
2.4. BRD9 is Part of the SWI-SNF Complex and Augments MYC Activity
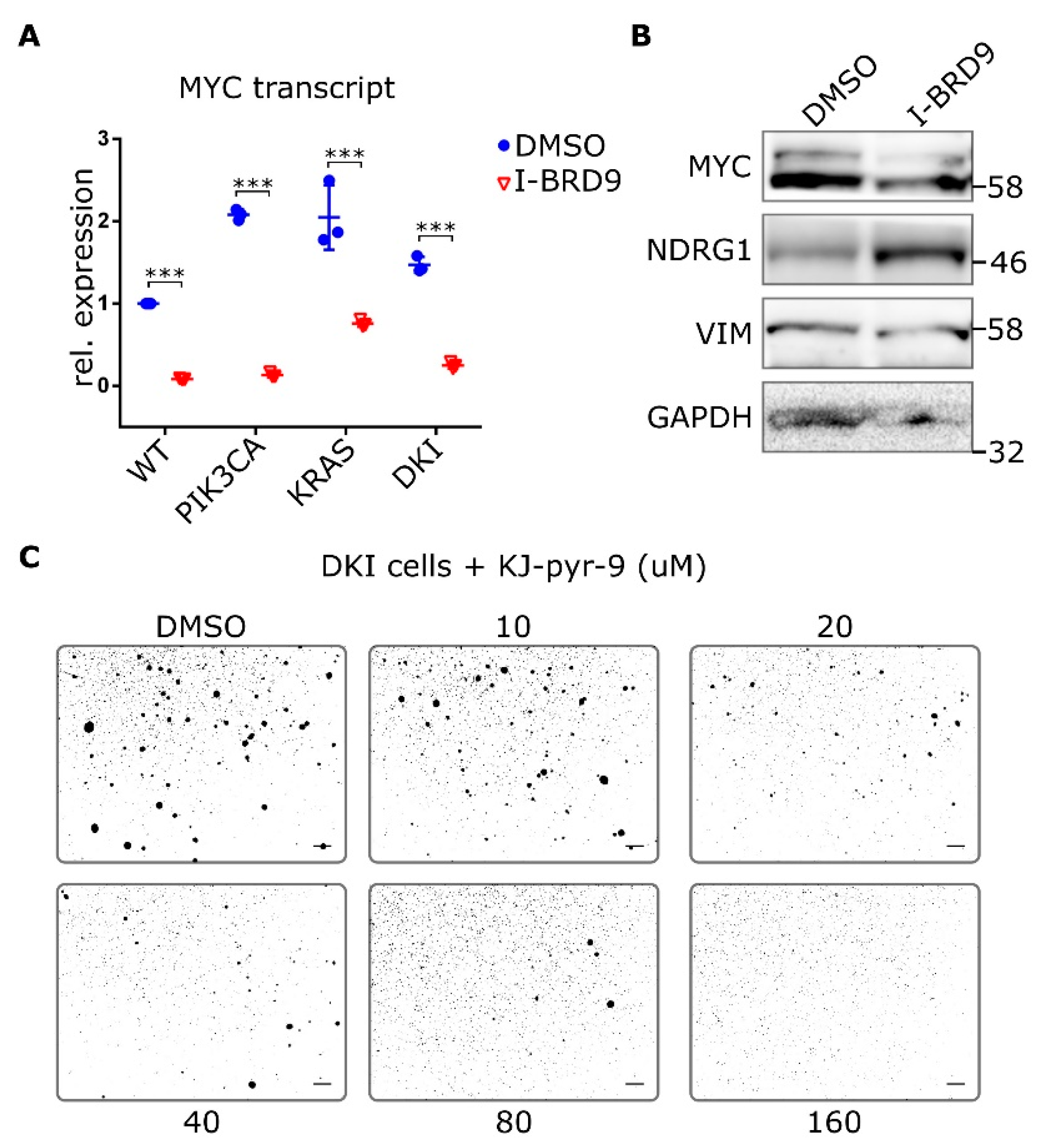
2.5. BRD9 Inhibition Blocks MYC Expression and MYC Inhibition Blocks Anchorage-Independent Growth
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Viability Assays
4.2. Next-Generation Sequencing
4.3. Gene Expression Analysis
4.4. Plasmids and CRISPR-Cas9 Gene Editing
4.5. RT-qPCR
4.6. Immunoblotting
4.7. Immunoprecipitation
4.8. Chromatin Immunoprecipitation
4.9. Colony Formation in Semisolid Medium
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Tomasetti, C.; Marchionni, L.; Nowak, M.A.; Parmigiani, G.; Vogelstein, B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Knapp, S.; Ahmed, A.A. The structural basis of PI3K cancer mutations: From mechanism to therapy. Cancer Res. 2014, 74, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed]
- Stachler, M.D.; Rinehart, E.M.; Garcia, E.; Lindeman, N.I. PIK3CA Mutations are Common in Many Tumor Types and are Often Associated with Other Driver Mutations. Appl. Immunohistochem. Mol. Morphol. 2016, 24, 313–319. [Google Scholar] [CrossRef]
- Ramjaun, A.R.; Downward, J. Ras and phosphoinositide 3-kinase: Partners in development and tumorigenesis. Cell Cycle 2007, 6, 2902–2905. [Google Scholar] [CrossRef][Green Version]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136. [Google Scholar] [CrossRef]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Luby-Phelps, K.; Das, B.; Shu, X.; Xia, Y.; Mosteller, R.D.; Krishna, U.M.; Falck, J.R.; White, M.A.; Broek, D. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science 1998, 279, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Ramjaun, A.R.; Haiko, P.; Wang, Y.; Warne, P.H.; Nicke, B.; Nye, E.; Stamp, G.; Alitalo, K.; Downward, J. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell 2007, 129, 957–968. [Google Scholar] [CrossRef]
- Mandelker, D.; Gabelli, S.B.; Schmidt-Kittler, O.; Zhu, J.; Cheong, I.; Huang, C.H.; Kinzler, K.W.; Vogelstein, B.; Amzel, L.M. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc. Natl. Acad. Sci. USA 2009, 106, 16996–17001. [Google Scholar] [CrossRef]
- Wang, G.M.; Wong, H.Y.; Konishi, H.; Blair, B.G.; Abukhdeir, A.M.; Gustin, J.P.; Rosen, D.M.; Denmeade, S.R.; Rasheed, Z.; Matsui, W.; et al. Single copies of mutant KRAS and mutant PIK3CA cooperate in immortalized human epithelial cells to induce tumor formation. Cancer Res. 2013, 73, 3248–3261. [Google Scholar] [CrossRef]
- Ignatoski, K.M.; Lapointe, A.J.; Radany, E.H.; Ethier, S.P. ERBB-2 overexpression in human mammary epithelial cells confers growth factor independence. Endocrinology 1999, 140, 3615–3622. [Google Scholar] [CrossRef][Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EDGER: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R.; et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 2004, 91, 355–358. [Google Scholar] [CrossRef]
- Hart, J.R.; Zhang, Y.; Liao, L.; Ueno, L.; Du, L.; Jonkers, M.; Yates, J.R., 3rd; Vogt, P.K. The butterfly effect in cancer: A single base mutation can remodel the cell. Proc. Natl. Acad. Sci. USA 2015, 112, 1131–1136. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.; Measures, A.R.; Wilson, B.G.; Cortopassi, W.A.; Alexander, R.; Hoss, M.; Hewings, D.S.; Rooney, T.P.; Paton, R.S.; Conway, S.J. Small molecule inhibitors of bromodomain-acetyl-lysine interactions. ACS Chem. Biol. 2015, 10, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, A.F.; Martin, L.J.; Minder, J.L.; Roe, J.S.; Shi, J.; Steurer, S.; Bader, G.; McConnell, D.; Pearson, M.; Gerstberger, T.; et al. Sensitivity and engineered resistance of myeloid leukemia cells to BRD9 inhibition. Nat. Chem. Biol. 2016, 12, 672–679. [Google Scholar] [CrossRef]
- Poon, H.; Quirk, C.; DeZiel, C.; Heckerman, D. Literome: PubMed-scale genomic knowledge base in the cloud. Bioinformatics 2014, 30, 2840–2842. [Google Scholar] [CrossRef]
- Hart, J.R.; Garner, A.L.; Yu, J.; Ito, Y.; Sun, M.; Ueno, L.; Rhee, J.K.; Baksh, M.M.; Stefan, E.; Hartl, M.; et al. Inhibitor of MYC identified in a Krohnke pyridine library. Proc. Natl. Acad. Sci. USA 2014, 111, 12556–12561. [Google Scholar] [CrossRef]
- Jonsson, G.; Staaf, J.; Olsson, E.; Heidenblad, M.; Vallon-Christersson, J.; Osoegawa, K.; De Jong, P.; Oredsson, S.; Ringner, M.; Hoglund, M.; et al. High-resolution genomic profiles of breast cancer cell lines assessed by tiling BAC array comparative genomic hybridization. Genes Chromosomes Cancer 2007, 46, 543–558. [Google Scholar] [CrossRef]
- Worsham, M.J.; Pals, G.; Schouten, J.P.; Miller, F.; Tiwari, N.; Van Spaendonk, R.; Wolman, S.R. High-resolution mapping of molecular events associated with immortalization, transformation, and progression to breast cancer in the MCF10 model. Breast Cancer Res. Treat. 2006, 96, 177–186. [Google Scholar] [CrossRef]
- Kadota, M.; Yang, H.H.; Gomez, B.; Sato, M.; Clifford, R.J.; Meerzaman, D.; Dunn, B.K.; Wakefield, L.M.; Lee, M.P. Delineating genetic alterations for tumor progression in the MCF10A series of breast cancer cell lines. PLoS ONE 2010, 5, e9201. [Google Scholar] [CrossRef]
- Johnson, L.; Mercer, K.; Greenbaum, D.; Bronson, R.T.; Crowley, D.; Tuveson, D.A.; Jacks, T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 2001, 410, 1111–1116. [Google Scholar] [CrossRef]
- Konishi, H.; Karakas, B.; Abukhdeir, A.M.; Lauring, J.; Gustin, J.P.; Garay, J.P.; Konishi, Y.; Gallmeier, E.; Bachman, K.E.; Park, B.H. Knock-in of mutant K-ras in nontumorigenic human epithelial cells as a new model for studying K-ras mediated transformation. Cancer Res. 2007, 67, 8460–8467. [Google Scholar] [CrossRef] [PubMed]
- Arena, S.; Isella, C.; Martini, M.; De Marco, A.; Medico, E.; Bardelli, A. Knock-in of oncogenic Kras does not transform mouse somatic cells but triggers a transcriptional response that classifies human cancers. Cancer Res. 2007, 67, 8468–8476. [Google Scholar] [CrossRef] [PubMed]
- Albers, J.; Danzer, C.; Rechsteiner, M.; Lehmann, H.; Brandt, L.P.; Hejhal, T.; Catalano, A.; Busenhart, P.; Gonçalves, A.F.; Brandt, S.; et al. A versatile modular vector system for rapid combinatorial mammalian genetics. J. Clin. Investig. 2015, 125, 1603–1619. [Google Scholar] [CrossRef] [PubMed]
- Compere, S.J.; Baldacci, P.; Sharpe, A.H.; Thompson, T.; Land, H.; Jaenisch, R. The ras and myc oncogenes cooperate in tumor induction in many tissues when introduced into midgestation mouse embryos by retroviral vectors. Proc. Natl. Acad. Sci. USA 1989, 86, 2224–2228. [Google Scholar] [CrossRef]
- Podsypanina, K.; Politi, K.; Beverly, L.J.; Varmus, H.E. Oncogene cooperation in tumor maintenance and tumor recurrence in mouse mammary tumors induced by Myc and mutant Kras. Proc. Natl. Acad. Sci. USA 2008, 105, 5242–5247. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Santiago, S.; Raeder, M.; Zhang, F.; Isabella, A.; Yang, J.; Semaan, D.J.; Chen, C.; Fox, E.A.; et al. Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway-dependent and PI3K pathway-independent mechanisms. Nat. Med. 2011, 17, 1116–1120. [Google Scholar] [CrossRef]
- Poli, V.; Fagnocchi, L.; Fasciani, A.; Cherubini, A.; Mazzoleni, S.; Ferrillo, S.; Miluzio, A.; Gaudioso, G.; Vaira, V.; Turdo, A.; et al. MYC-driven epigenetic reprogramming favors the onset of tumorigenesis by inducing a stem cell-like state. Nat. Commun. 2018, 9, 1024. [Google Scholar] [CrossRef]
- Gustin, J.P.; Karakas, B.; Weiss, M.B.; Abukhdeir, A.M.; Lauring, J.; Garay, J.P.; Cosgrove, D.; Tamaki, A.; Konishi, H.; Konishi, Y.; et al. Knockin of mutant PIK3CA activates multiple oncogenic pathways. Proc. Natl. Acad. Sci. USA 2009, 106, 2835–2840. [Google Scholar] [CrossRef]
- Grand, F.; Kulkarni, S.; Chase, A.; Goldman, J.M.; Gordon, M.; Cross, N.C. Frequent deletion of hSNF5/INI1, a component of the SWI/SNF complex, in chronic myeloid leukemia. Cancer Res. 1999, 59, 3870–3874. [Google Scholar]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef]
- Shi, J.; Whyte, W.A.; Zepeda-Mendoza, C.J.; Milazzo, J.P.; Shen, C.; Roe, J.S.; Minder, J.L.; Mercan, F.; Wang, E.; Eckersley-Maslin, M.A.; et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013, 27, 2648–2662. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Haynes, S.R.; Dollard, C.; Winston, F.; Beck, S.; Trowsdale, J.; Dawid, I.B. The bromodomain: A conserved sequence found in human, Drosophila and yeast proteins. Nucleic Acids Res. 1992, 20, 2603. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Puissant, A.; Frumm, S.M.; Alexe, G.; Bassil, C.F.; Qi, J.; Chanthery, Y.H.; Nekritz, E.A.; Zeid, R.; Gustafson, W.C.; Greninger, P.; et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013, 3, 308–323. [Google Scholar] [CrossRef] [PubMed]
- Bihani, T.; Ezell, S.A.; Ladd, B.; Grosskurth, S.E.; Mazzola, A.M.; Pietras, M.; Reimer, C.; Zinda, M.; Fawell, S.; D’Cruz, C.M. Resistance to everolimus driven by epigenetic regulation of MYC in ER+ breast cancers. Oncotarget 2015, 6, 2407–2420. [Google Scholar] [CrossRef]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef]
- Marquez-Vilendrer, S.B.; Rai, S.K.; Gramling, S.J.; Lu, L.; Reisman, D.N. Loss of the SWI/SNF ATPase subunits BRM and BRG1 drives lung cancer development. Oncoscience 2016, 3, 322–336. [Google Scholar] [CrossRef]
- Prochownik, E.V.; Vogt, P.K. Therapeutic Targeting of Myc. Genes Cancer 2010, 1, 650–659. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Shern, J.F.; Chen, L.; Chmielecki, J.; Wei, J.S.; Patidar, R.; Rosenberg, M.; Ambrogio, L.; Auclair, D.; Wang, J.; Song, Y.K.; et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014, 4, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Engstrom, P.G.; Steijger, T.; Sipos, B.; Grant, G.R.; Kahles, A.; Ratsch, G.; Goldman, N.; Hubbard, T.J.; Harrow, J.; Guigo, R.; et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat. Methods 2013, 10, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Steijger, T.; Abril, J.F.; Engstrom, P.G.; Kokocinski, F.; RGASP Consortium; Hubbard, T.J.; Guigo, R.; Harrow, J.; Bertone, P. Assessment of transcript reconstruction methods for RNA-seq. Nat. Methods 2013, 10, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Plagnol, V.; Curtis, J.; Epstein, M.; Mok, K.Y.; Stebbings, E.; Grigoriadou, S.; Wood, N.W.; Hambleton, S.; Burns, S.O.; Thrasher, A.J.; et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 2012, 28, 2747–2754. [Google Scholar] [CrossRef]
- Conrad, D.F.; Pinto, D.; Redon, R.; Feuk, L.; Gokcumen, O.; Zhang, Y.; Aerts, J.; Andrews, T.D.; Barnes, C.; Campbell, P.; et al. Origins and functional impact of copy number variation in the human genome. Nature 2010, 464, 704–712. [Google Scholar] [CrossRef]
- Wang, K.; Bucan, M. Copy Number Variation Detection via High-Density SNP Genotyping. CSH Protoc. 2008, 2008, pdb top46. [Google Scholar] [CrossRef]
- Watson, J.V.; Chambers, S.H.; Smith, P.J. A pragmatic approach to the analysis of DNA histograms with a definable G1 peak. Cytometry 1987, 8, 1–8. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | DKI vs. WT | log2FC | DKI vs. PIK3CAH1047R | log2FC | DKI vs. KRASG12V | log2FC |
|---|---|---|---|---|---|---|
| 1 | COL4A1 | 4.19 | DHRS3 | 5.40 | CLPTM1L | 1.08 |
| 2 | TFPI2 | 4.09 | TFPI2 | 4.29 | CCT5 | 1.14 |
| 3 | FAM110C | 4.47 | TGFBI | 4.14 | MRPS30 | 1.06 |
| 4 | ARL4C | 3.84 | TINAGL1 | 4.04 | NSUN2 | 1.01 |
| 5 | TCF19 | 3.73 | COL4A1 | 3.97 | LPCAT1 | 1.06 |
| 6 | DHRS3 | 5.33 | FAM110C | 3.78 | BASP1 | 0.98 |
| 7 | TINAGL1 | 4.18 | TCF19 | 3.73 | PEAR1 | 2.18 |
| 8 | LMNB2 | 3.43 | LMNB2 | 3.67 | BRD9 | 1.07 |
| 9 | PROSER2 | 3.42 | MYBL2 | 3.59 | VIM | 1.22 |
| 10 | ADAM19 | 4.66 | CCT2 | 3.54 | ANKH | 1.16 |
| 11 | PHLDA1 | 3.40 | ADAM19 | 3.49 | TRIP13 | 1.05 |
| 12 | TGFBI | 4.23 | UBA2 | 3.44 | IRX4 | 1.17 |
| 13 | CCT2 | 2.80 | SRP14 | 3.38 | OTULIN | 0.83 |
| 14 | TXNRD1 | 2.91 | EIF1 | 3.18 | ICE1 | 0.94 |
| 15 | UBA2 | 3.25 | MCM2 | 3.17 | TARS | 0.93 |
| 16 | TRIP13 | 3.44 | RPL7 | 3.10 | DNAJC21 | 1.03 |
| 17 | LTBP1 | 3.06 | NSUN2 | 3.09 | LMBRD2 | 1.06 |
| 18 | NSUN2 | 3.04 | CSTA | 2.99 | ZNF622 | 0.93 |
| 19 | MCM2 | 3.61 | GLRX | 2.93 | CEP72 | 1.26 |
| 20 | UHRF1 | 3.52 | RPS7 | 2.86 | KCNK6 | 1.03 |
| DKI vs. WT | DKI vs. PIK3CA_H1047R | DKI vs. KRAS_G12V |
|---|---|---|
| EPITHELIAL_MESENCHYMAL_TRANSITION | E2F_TARGETS | MYC_TARGETS_V1 |
| E2F_TARGETS | G2M_CHECKPOINT | E2F_TARGETS |
| G2M_CHECKPOINT | EPITHELIAL_MESENCHYMAL_TRANSITION | OXIDATIVE_PHOSPHORYLATION |
| KRAS_SIGNALING_UP | KRAS_SIGNALING_UP | G2M_CHECKPOINT |
| APICAL_JUNCTION | MITOTIC_SPINDLE | MYC_TARGETS_V2 |
| TNFA_SIGNALING_VIA_NFKB | ESTROGEN_RESPONSE | MTORC1_SIGNALING |
| MTORC1_SIGNALING | APICAL_JUNCTION | APOPTOSIS |
| TGF_BETA_SIGNALING | MTORC1_SIGNALING | UNFOLDED_PROTEIN_RESPONSE |
| WNT_BETA_CATENIN_SIGNALING | MYC_TARGETS_V1 | |
| IL2_STAT5_SIGNALING | TNFA_SIGNALING_VIA_NFKB |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bell, C.M.; Raffeiner, P.; Hart, J.R.; Vogt, P.K. PIK3CA Cooperates with KRAS to Promote MYC Activity and Tumorigenesis via the Bromodomain Protein BRD9. Cancers 2019, 11, 1634. https://doi.org/10.3390/cancers11111634
Bell CM, Raffeiner P, Hart JR, Vogt PK. PIK3CA Cooperates with KRAS to Promote MYC Activity and Tumorigenesis via the Bromodomain Protein BRD9. Cancers. 2019; 11(11):1634. https://doi.org/10.3390/cancers11111634
Chicago/Turabian StyleBell, Catherine M., Philipp Raffeiner, Jonathan R. Hart, and Peter K. Vogt. 2019. "PIK3CA Cooperates with KRAS to Promote MYC Activity and Tumorigenesis via the Bromodomain Protein BRD9" Cancers 11, no. 11: 1634. https://doi.org/10.3390/cancers11111634
APA StyleBell, C. M., Raffeiner, P., Hart, J. R., & Vogt, P. K. (2019). PIK3CA Cooperates with KRAS to Promote MYC Activity and Tumorigenesis via the Bromodomain Protein BRD9. Cancers, 11(11), 1634. https://doi.org/10.3390/cancers11111634