Destined to Die: Apoptosis and Pediatric Cancers


{kind=link}
{kind=link}
Abstract
:1. Introduction
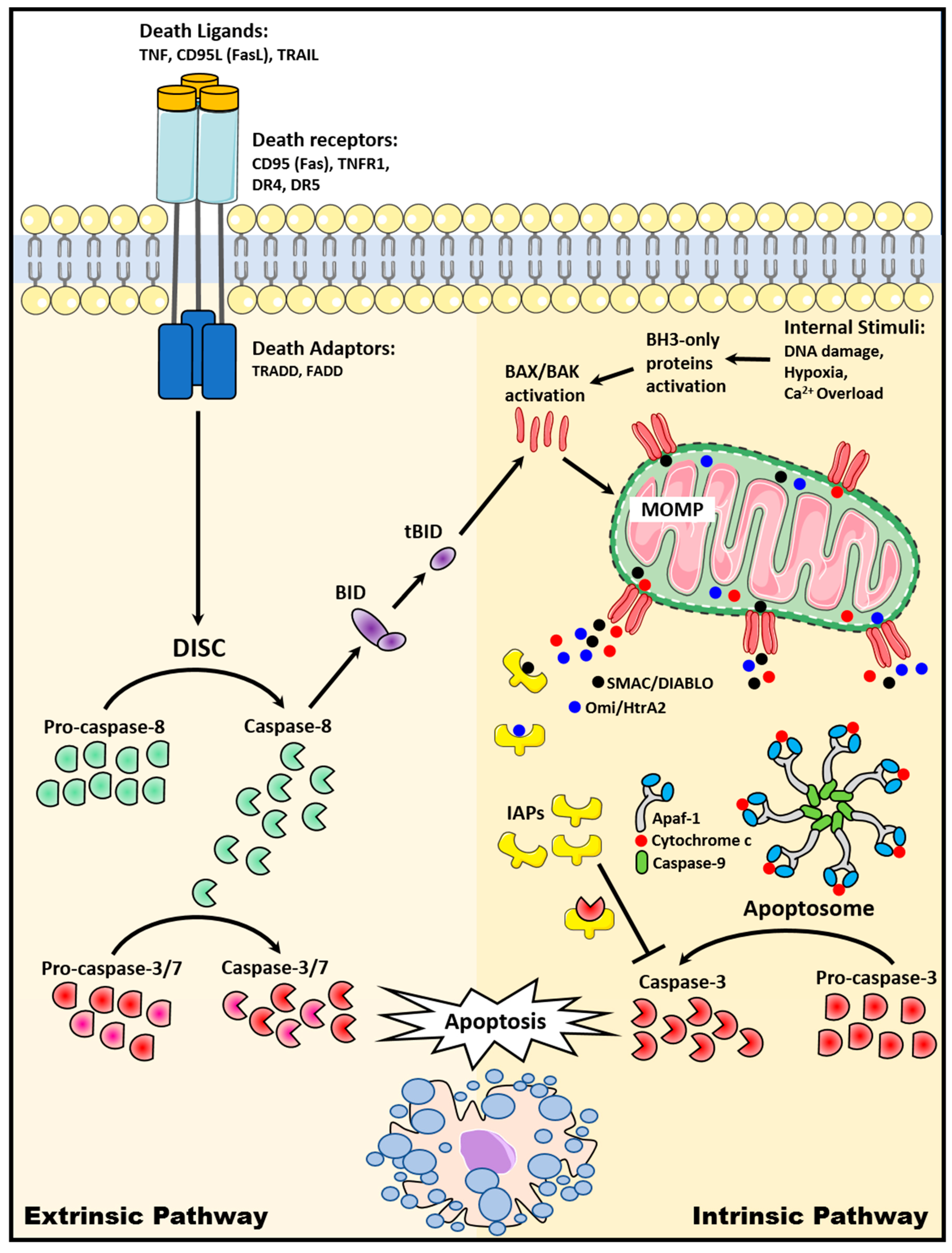
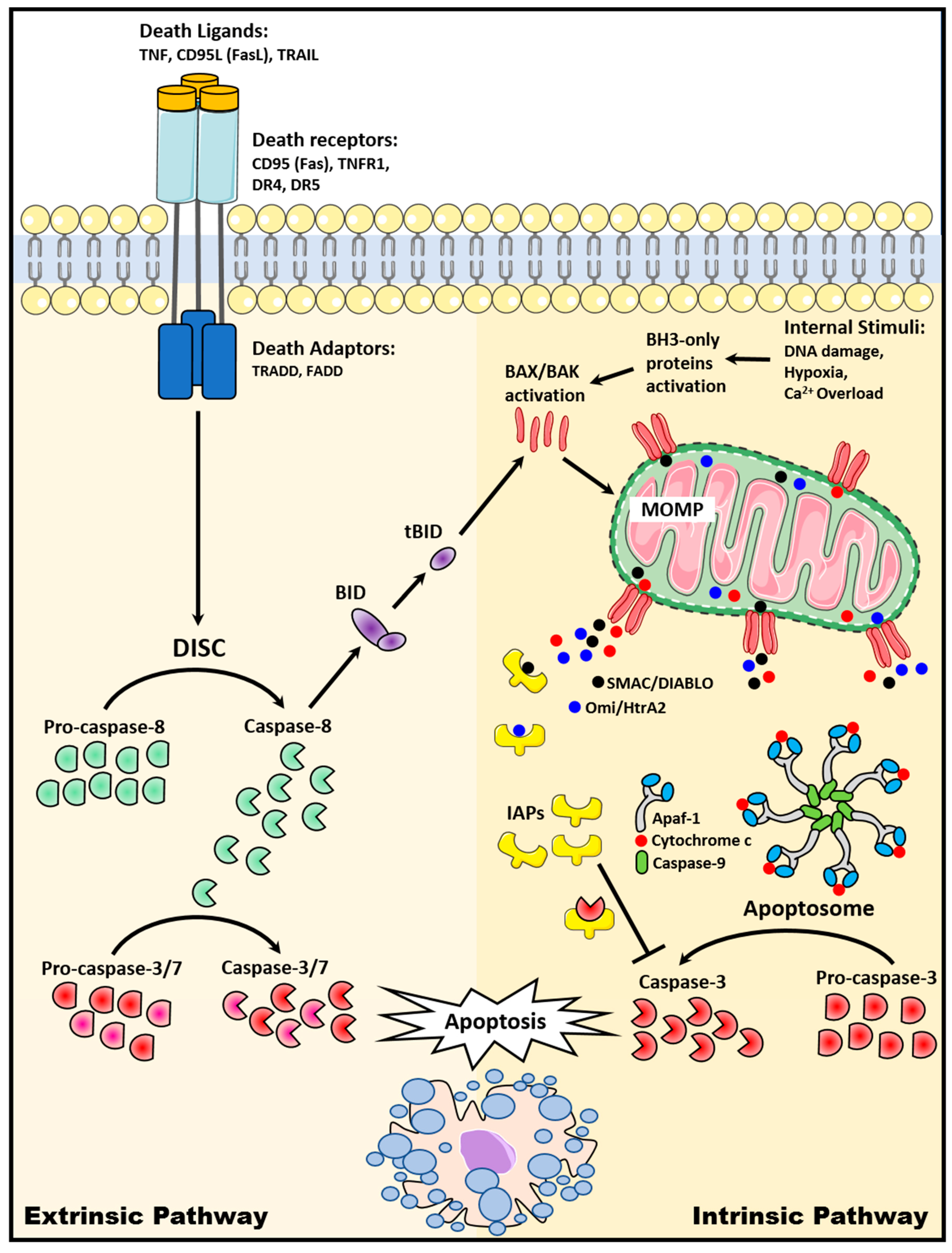
2. Apoptosis Signaling Pathways
2.1. The Extrinsic Death Receptor Pathway
2.2. The Intrinsic Mitochondrial Pathway
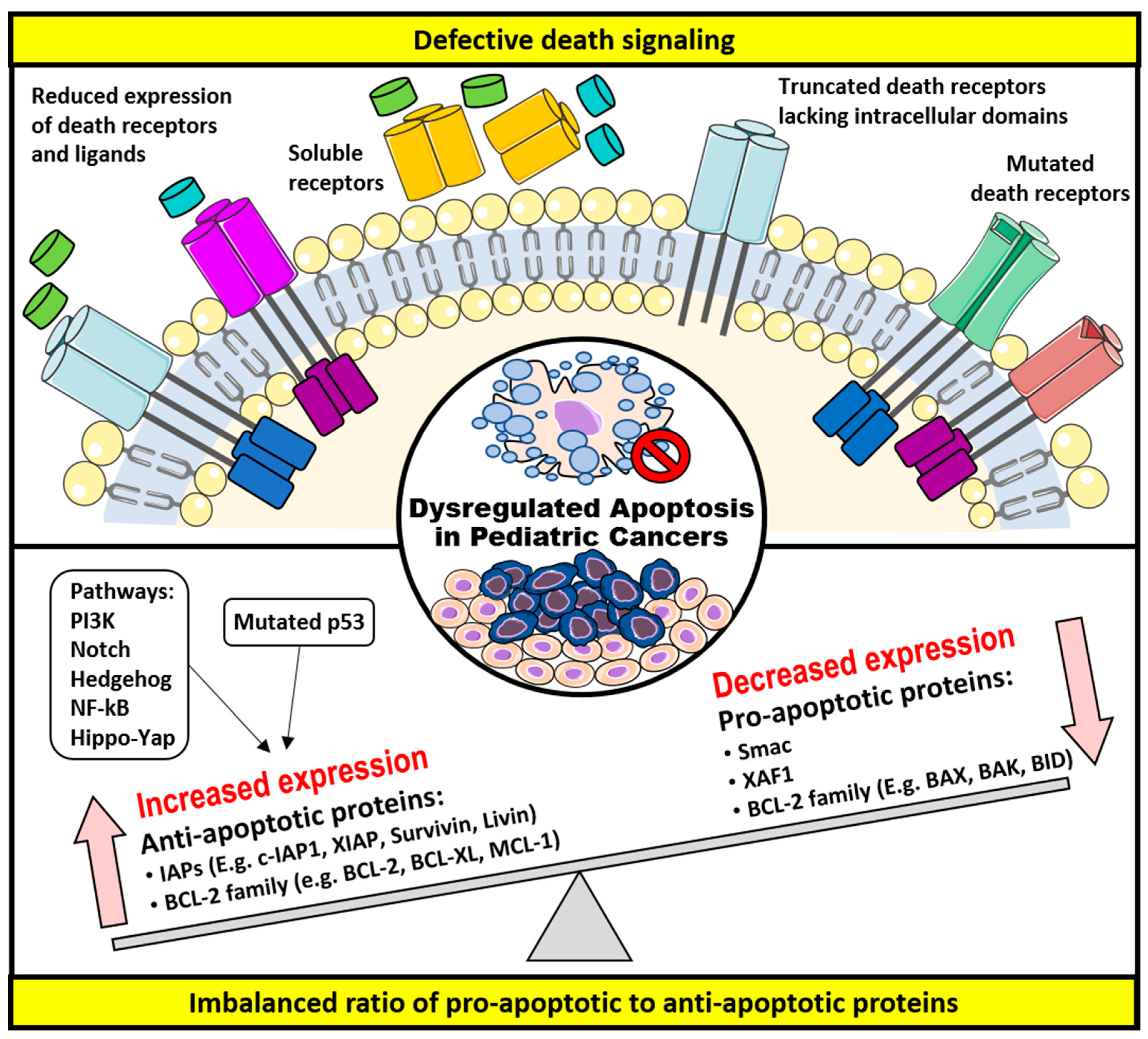
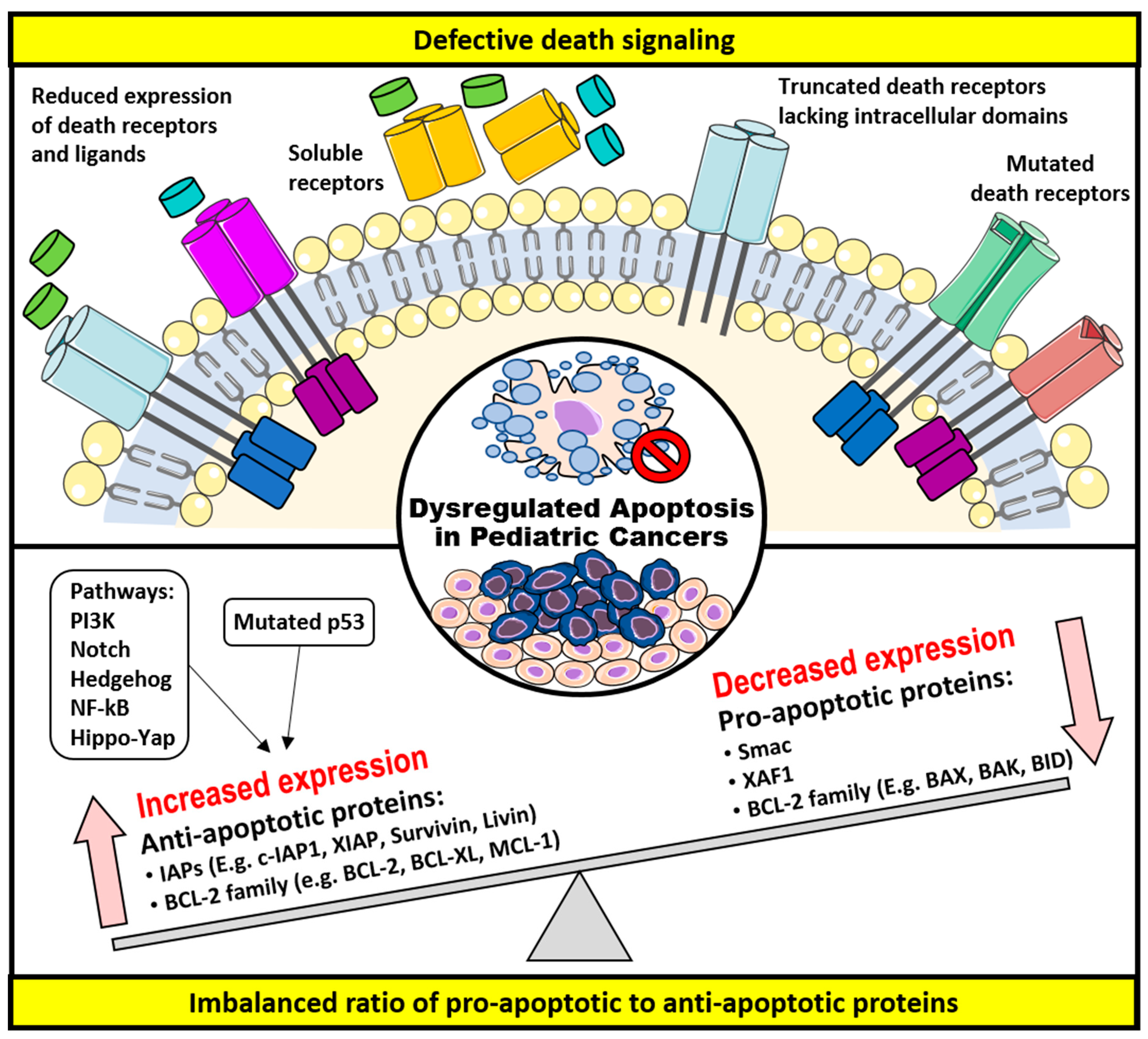
3. Dysregulation of Apoptosis and Apoptosis-Targeted Therapies in Childhood Cancers
3.1. Defective Death Receptor Signaling
3.2. Imbalanced Ratio of Pro-Apoptotic to Anti-Apoptotic Proteins
3.2.1. The BCL-2 Family of Proteins
3.2.2. The IAPs
4. Conclusions
Funding
Conflicts of Interest
References
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Norbury, C.J.; Hickson, I.D. Cellular responses to DNA damage. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 367–401. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.B. Apoptosis in the pathogenesis and treatment of disease. Science 1995, 267, 1456–1462. [Google Scholar] [CrossRef]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Evan, G.I.; Vousden, K.H. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef]
- Makin, G. Targeting apoptosis in cancer chemotherapy. Expert Opin. Ther. Targets 2002, 6, 73–84. [Google Scholar] [CrossRef]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Nagata, S. Apoptosis by death factor. Cell 1997, 88, 355–365. [Google Scholar] [CrossRef]
- Falschlehner, C.; Schaefer, U.; Walczak, H. Following TRAIL’s path in the immune system. Immunology 2009, 127, 145–154. [Google Scholar] [CrossRef]
- Sedger, L.M.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants—Past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef]
- Wallach, D.; Varfolomeev, E.E.; Malinin, N.L.; Goltsev, Y.V.; Kovalenko, A.V.; Boldin, M.P. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu. Rev. Immunol. 1999, 17, 331–367. [Google Scholar] [CrossRef]
- Walczak, H.; Krammer, P.H. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp. Cell Res. 2000, 256, 58–66. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Saelens, X.; Festjens, N.; Vande Walle, L.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar] [CrossRef] [Green Version]
- Shamas-Din, A.; Kale, J.; Leber, B.; Andrews, D.W. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008714. [Google Scholar] [CrossRef]
- Riedl, S.J.; Salvesen, G.S. The apoptosome: Signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 2007, 8, 405–413. [Google Scholar] [CrossRef]
- Vande Walle, L.; Lamkanfi, M.; Vandenabeele, P. The mitochondrial serine protease HtrA2/Omi: An overview. Cell Death Differ. 2008, 15, 453–460. [Google Scholar] [CrossRef]
- Murphy, S.L.; Xu, J.; Kochanek, K.D. Deaths: Final data for 2010. Natl. Vital Stat. Rep. 2013, 61, 1–117. [Google Scholar] [PubMed]
- Howlader, N.N.A.; Krapcho, M.; Garshell, J.; Neyman, N.; Altekruse, S.F.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Cho, H.; et al. (Eds.) SEER Cancer Statistics Review, 1975–2010. Available online: https://seer.cancer.gov/archive/csr/1975_2010/ (accessed on 13 August 2019).
- Ward, E.; De Santis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and adolescent cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Koesters, R.; Von Knebel Doeberitz, M. The Wnt signaling pathway in solid childhood tumors. Cancer Lett. 2003, 198, 123–138. [Google Scholar] [CrossRef]
- Oue, T.; Yoneda, A.; Uehara, S.; Yamanaka, H.; Fukuzawa, M. Increased expression of the hedgehog signaling pathway in pediatric solid malignancies. J. Pediatr. Surg. 2010, 45, 387–392. [Google Scholar] [CrossRef]
- Zweidler-McKay, P.A. Notch signaling in pediatric malignancies. Curr. Oncol. Rep. 2008, 10, 459–468. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Mohamed, A.D.; Gener, M.; Li, W.; Taboada, E. YAP and the Hippo pathway in pediatric cancer. Mol. Cell. Oncol. 2017, 4, e1295127. [Google Scholar] [CrossRef] [Green Version]
- Strasser, A.; Jost, P.J.; Nagata, S. The many roles of FAS receptor signaling in the immune system. Immunity 2009, 30, 180–192. [Google Scholar] [CrossRef]
- Tong, N.; Zhang, L.; Sheng, X.; Wang, M.; Zhang, Z.; Fang, Y.; Xue, Y.; Li, J.; Zhang, Z. Functional polymorphisms in FAS, FASL and CASP8 genes and risk of childhood acute lymphoblastic leukemia: A case-control study. Leuk. Lymphoma 2012, 53, 1360–1366. [Google Scholar] [CrossRef]
- Komada, Y.; Zhou, Y.W.; Zhang, X.L.; Xue, H.L.; Sakai, H.; Tanaka, S.; Sakatoku, H.; Sakurai, M. Fas receptor (CD95)-mediated apoptosis is induced in leukemic cells entering G1B compartment of the cell cycle. Blood 1995, 86, 3848–3860. [Google Scholar] [CrossRef]
- Plumas, J.; Jacob, M.C.; Chaperot, L.; Molens, J.P.; Sotto, J.J.; Bensa, J.C. Tumor B cells from non-Hodgkin’s lymphoma are resistant to CD95 (Fas/Apo-1)-mediated apoptosis. Blood 1998, 91, 2875–2885. [Google Scholar] [CrossRef]
- Friesen, C.; Herr, I.; Krammer, P.H.; Debatin, K.M. Involvement of the CD95 (APO-1/FAS) receptor/ligand system in drug-induced apoptosis in leukemia cells. Nat. Med. 1996, 2, 574–577. [Google Scholar] [CrossRef] [PubMed]
- Friesen, C.; Fulda, S.; Debatin, K.M. Deficient activation of the CD95 (APO-1/Fas) system in drug-resistant cells. Leukemia 1997, 11, 1833–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S.; Los, M.; Friesen, C.; Debatin, K.M. Chemosensitivity of solid tumor cells in vitro is related to activation of the CD95 system. Int. J. Cancer 1998, 76, 105–114. [Google Scholar] [CrossRef]
- Nguyen, P.L.; Harris, N.L.; Ritz, J.; Robertson, M.J. Expression of CD95 antigen and Bcl-2 protein in non-Hodgkin’s lymphomas and Hodgkin’s disease. Am. J. Pathol. 1996, 148, 847–853. [Google Scholar]
- Min, Y.H.; Lee, S.; Lee, J.W.; Chong, S.Y.; Hahn, J.S.; Ko, Y.W. Expression of Fas antigen in acute myeloid leukaemia is associated with therapeutic response to chemotherapy. Br. J. Haematol. 1996, 93, 928–930. [Google Scholar] [CrossRef]
- Friesen, C.; Fulda, S.; Debatin, K.M. Cytotoxic drugs and the CD95 pathway. Leukemia 1999, 13, 1854–1858. [Google Scholar] [CrossRef] [Green Version]
- Debatin, K.M.; Krammer, P.H. Resistance to APO-1 (CD95) induced apoptosis in T-ALL is determined by a BCL-2 independent anti-apoptotic program. Leukemia 1995, 9, 815–820. [Google Scholar]
- Karawajew, L.; Wuchter, C.; Ruppert, V.; Drexler, H.; Gruss, H.J.; Dorken, B.; Ludwig, W.D. Differential CD95 expression and function in T and B lineage acute lymphoblastic leukemia cells. Leukemia 1997, 11, 1245–1252. [Google Scholar] [CrossRef] [Green Version]
- Lucking-Famira, K.M.; Daniel, P.T.; Moller, P.; Krammer, P.H.; Debatin, K.M. APO-1 (CD95) mediated apoptosis in human T-ALL engrafted in SCID mice. Leukemia 1994, 8, 1825–1833. [Google Scholar]
- Cheng, J.; Zhou, T.; Liu, C.; Shapiro, J.P.; Brauer, M.J.; Kiefer, M.C.; Barr, P.J.; Mountz, J.D. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science 1994, 263, 1759–1762. [Google Scholar] [CrossRef]
- Wood, C.M.; Goodman, P.A.; Vassilev, A.O.; Uckun, F.M. CD95 (APO-1/FAS) deficiency in infant acute lymphoblastic leukemia: Detection of novel soluble Fas splice variants. Eur. J. Haematol. 2003, 70, 156–171. [Google Scholar] [CrossRef] [PubMed]
- Knipping, E.; Debatin, K.M.; Stricker, K.; Heilig, B.; Eder, A.; Krammer, P.H. Identification of soluble APO-1 in supernatants of human B- and T-cell lines and increased serum levels in B- and T-cell leukemias. Blood 1995, 85, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Munker, R.; Midis, G.; Owen-Schaub, L.; Andreff, M. Soluble FAS (CD95) is not elevated in the serum of patients with myeloid leukemias, myeloproliferative and myelodysplastic syndromes. Leukemia 1996, 10, 1531–1533. [Google Scholar] [PubMed]
- Cascino, I.; Papoff, G.; De Maria, R.; Testi, R.; Ruberti, G. Fas/Apo-1 (CD95) receptor lacking the intracytoplasmic signaling domain protects tumor cells from Fas-mediated apoptosis. J. Immunol. 1996, 156, 13–17. [Google Scholar]
- Beltinger, C.; Kurz, E.; Bohler, T.; Schrappe, M.; Ludwig, W.D.; Debatin, K.M. CD95 (APO-1/Fas) mutations in childhood T-lineage acute lymphoblastic leukemia. Blood 1998, 91, 3943–3951. [Google Scholar] [CrossRef]
- Sibley, K.; Rollinson, S.; Allan, J.M.; Smith, A.G.; Law, G.R.; Roddam, P.L.; Skibola, C.F.; Smith, M.T.; Morgan, G.J. Functional FAS promoter polymorphisms are associated with increased risk of acute myeloid leukemia. Cancer Res. 2003, 63, 4327–4330. [Google Scholar]
- Muschen, M.; Rajewsky, K.; Kronke, M.; Kuppers, R. The origin of CD95-gene mutations in B-cell lymphoma. Trends Immunol. 2002, 23, 75–80. [Google Scholar] [CrossRef]
- Bazzoni, F.; Beutler, B. The tumor necrosis factor ligand and receptor families. N. Engl. J. Med. 1996, 334, 1717–1725. [Google Scholar] [CrossRef]
- Ogasawara, J.; Watanabe-Fukunaga, R.; Adachi, M.; Matsuzawa, A.; Kasugai, T.; Kitamura, Y.; Itoh, N.; Suda, T.; Nagata, S. Lethal effect of the anti-Fas antibody in mice. Nature 1993, 364, 806–809. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Pai, R.C.; Fong, S.; Leung, S.; Lawrence, D.A.; Marsters, S.A.; Blackie, C.; Chang, L.; McMurtrey, A.E.; Hebert, A.; et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Investig. 1999, 104, 155–162. [Google Scholar] [CrossRef]
- Ehrhardt, H.; Fulda, S.; Schmid, I.; Hiscott, J.; Debatin, K.M.; Jeremias, I. TRAIL induced survival and proliferation in cancer cells resistant towards TRAIL-induced apoptosis mediated by NF-kappaB. Oncogene 2003, 22, 3842–3852. [Google Scholar] [CrossRef] [PubMed]
- Kontny, H.U.; Hammerle, K.; Klein, R.; Shayan, P.; Mackall, C.L.; Niemeyer, C.M. Sensitivity of Ewing’s sarcoma to TRAIL-induced apoptosis. Cell Death Differ. 2001, 8, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Van Valen, F.; Fulda, S.; Truckenbrod, B.; Eckervogt, V.; Sonnemann, J.; Hillmann, A.; Rodl, R.; Hoffmann, C.; Winkelmann, W.; Schafer, L.; et al. Apoptotic responsiveness of the Ewing’s sarcoma family of tumours to tumour necrosis factor-related apoptosis-inducing ligand (TRAIL). Int. J. Cancer 2000, 88, 252–259. [Google Scholar] [CrossRef]
- Petak, I.; Douglas, L.; Tillman, D.M.; Vernes, R.; Houghton, J.A. Pediatric rhabdomyosarcoma cell lines are resistant to Fas-induced apoptosis and highly sensitive to TRAIL-induced apoptosis. Clin. Cancer Res. 2000, 6, 4119–4127. [Google Scholar]
- Yang, X.; Thiele, C.J. Targeting the tumor necrosis factor-related apoptosis-inducing ligand path in neuroblastoma. Cancer Lett. 2003, 197, 137–143. [Google Scholar] [CrossRef]
- Van Noesel, M.M.; Van Bezouw, S.; Voute, P.A.; Herman, J.G.; Pieters, R.; Versteeg, R. Clustering of hypermethylated genes in neuroblastoma. Genes Chromosomes Cancer 2003, 38, 226–233. [Google Scholar] [CrossRef]
- Van Noesel, M.M.; van Bezouw, S.; Salomons, G.S.; Voute, P.A.; Pieters, R.; Baylin, S.B.; Herman, J.G.; Versteeg, R. Tumor-specific down-regulation of the tumor necrosis factor-related apoptosis-inducing ligand decoy receptors DcR1 and DcR2 is associated with dense promoter hypermethylation. Cancer Res. 2002, 62, 2157–2161. [Google Scholar]
- Fulda, S.; Kufer, M.U.; Meyer, E.; van Valen, F.; Dockhorn-Dworniczak, B.; Debatin, K.M. Sensitization for death receptor- or drug-induced apoptosis by re-expression of caspase-8 through demethylation or gene transfer. Oncogene 2001, 20, 5865–5877. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Kiernan, C.M.; Tian, Y.; Salwen, H.R.; Chlenski, A.; Brumback, B.A.; London, W.B.; Cohn, S.L. Methylation of CASP8, DCR2, and HIN-1 in neuroblastoma is associated with poor outcome. Clin. Cancer Res. 2007, 13, 3191–3197. [Google Scholar] [CrossRef]
- Teitz, T.; Wei, T.; Valentine, M.B.; Vanin, E.F.; Grenet, J.; Valentine, V.A.; Behm, F.G.; Look, A.T.; Lahti, J.M.; Kidd, V.J. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat. Med. 2000, 6, 529–535. [Google Scholar] [CrossRef]
- Fakler, M.; Loeder, S.; Vogler, M.; Schneider, K.; Jeremias, I.; Debatin, K.M.; Fulda, S. Small molecule XIAP inhibitors cooperate with TRAIL to induce apoptosis in childhood acute leukemia cells and overcome Bcl-2-mediated resistance. Blood 2009, 113, 1710–1722. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Shirley, S.; Dufour, F. Death receptors as targets in cancer. Br. J. Pharmacol. 2013, 169, 1723–1744. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Herbst, R.S. To kill a tumor cell: The potential of proapoptotic receptor agonists. J. Clin. Investig. 2008, 118, 1979–1990. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Morton, C.L.; Kolb, E.A.; Gorlick, R.; Keir, S.T.; Carol, H.; Lock, R.; Kang, M.H.; Reynolds, C.P.; Maris, J.M.; et al. Initial testing (stage 1) of mapatumumab (HGS-ETR1) by the pediatric preclinical testing program. Pediatr. Blood Cancer 2010, 54, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.S.; Geller, J.I.; Baird, K.; Chou, A.J.; Galli, S.; Charles, A.; Amaoko, M.; Rhee, E.H.; Price, A.; Wexler, L.H.; et al. Phase I trial and pharmacokinetic study of lexatumumab in pediatric patients with solid tumors. J. Clin. Oncol. 2012, 30, 4141–4147. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.; McDonnell, J.M.; Korsmeyer, S.J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999, 13, 1899–1911. [Google Scholar] [CrossRef] [Green Version]
- Vitagliano, O.; Addeo, R.; D’Angelo, V.; Indolfi, C.; Indolfi, P.; Casale, F. The Bcl-2/Bax and Ras/Raf/MEK/ERK signaling pathways: Implications in pediatric leukemia pathogenesis and new prospects for therapeutic approaches. Expert Rev. Hematol. 2013, 6, 587–597. [Google Scholar] [CrossRef]
- Wei, C.C.; Ball, S.; Lin, L.; Liu, A.; Fuchs, J.R.; Li, P.K.; Li, C.; Lin, J. Two small molecule compounds, LLL12 and FLLL32, exhibit potent inhibitory activity on STAT3 in human rhabdomyosarcoma cells. Int. J. Oncol. 2011, 38, 279–285. [Google Scholar]
- Chang, F.; Lee, J.T.; Navolanic, P.M.; Steelman, L.S.; Shelton, J.G.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: A target for cancer chemotherapy. Leukemia 2003, 17, 590–603. [Google Scholar] [CrossRef]
- Levine, A.J.; Momand, J.; Finlay, C.A. The p53 tumour suppressor gene. Nature 1991, 351, 453–456. [Google Scholar] [CrossRef]
- Goldsmith, K.C.; Lestini, B.J.; Gross, M.; Ip, L.; Bhumbla, A.; Zhang, X.; Zhao, H.; Liu, X.; Hogarty, M.D. BH3 response profiles from neuroblastoma mitochondria predict activity of small molecule Bcl-2 family antagonists. Cell Death Differ. 2010, 17, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Porwit-MacDonald, A.; Ivory, K.; Wilkinson, S.; Wheatley, K.; Wong, L.; Janossy, G. Bcl-2 protein expression in normal human bone marrow precursors and in acute myelogenous leukemia. Leukemia 1995, 9, 1191–1198. [Google Scholar] [PubMed]
- Banker, D.E.; Radich, J.; Becker, A.; Kerkof, K.; Norwood, T.; Willman, C.; Appelbaum, F.R. The t(8;21) translocation is not consistently associated with high Bcl-2 expression in de novo acute myeloid leukemias of adults. Clin. Cancer Res. 1998, 4, 3051–3062. [Google Scholar] [PubMed]
- Coustan-Smith, E.; Kitanaka, A.; Pui, C.H.; McNinch, L.; Evans, W.E.; Raimondi, S.C.; Behm, F.G.; Arico, M.; Campana, D. Clinical relevance of BCL-2 overexpression in childhood acute lymphoblastic leukemia. Blood 1996, 87, 1140–1146. [Google Scholar] [CrossRef] [Green Version]
- Campana, D.; Coustan-Smith, E.; Manabe, A.; Buschle, M.; Raimondi, S.C.; Behm, F.G.; Ashmun, R.; Arico, M.; Biondi, A.; Pui, C.H. Prolonged survival of B-lineage acute lymphoblastic leukemia cells is accompanied by overexpression of bcl-2 protein. Blood 1993, 81, 1025–1031. [Google Scholar] [CrossRef] [Green Version]
- Prokop, A.; Wieder, T.; Sturm, I.; Essmann, F.; Seeger, K.; Wuchter, C.; Ludwig, W.D.; Henze, G.; Dorken, B.; Daniel, P.T. Relapse in childhood acute lymphoblastic leukemia is associated with a decrease of the Bax/Bcl-2 ratio and loss of spontaneous caspase-3 processing in vivo. Leukemia 2000, 14, 1606–1613. [Google Scholar] [CrossRef] [Green Version]
- Del Poeta, G.; Venditti, A.; Del Principe, M.I.; Maurillo, L.; Buccisano, F.; Tamburini, A.; Cox, M.C.; Franchi, A.; Bruno, A.; Mazzone, C.; et al. Amount of spontaneous apoptosis detected by Bax/Bcl-2 ratio predicts outcome in acute myeloid leukemia (AML). Blood 2003, 101, 2125–2131. [Google Scholar] [CrossRef]
- Nedelcu, T.; Kubista, B.; Koller, A.; Sulzbacher, I.; Mosberger, I.; Arrich, F.; Trieb, K.; Kotz, R.; Toma, C.D. Livin and Bcl-2 expression in high-grade osteosarcoma. J. Cancer Res. Clin. Oncol. 2008, 134, 237–244. [Google Scholar] [CrossRef]
- Ganigi, P.M.; Santosh, V.; Anandh, B.; Chandramouli, B.A.; Sastry Kolluri, V.R. Expression of p53, EGFR, pRb and bcl-2 proteins in pediatric glioblastoma multiforme: A study of 54 patients. Pediatr. Neurosurg. 2005, 41, 292–299. [Google Scholar] [CrossRef]
- Re, G.G.; Hazen-Martin, D.J.; El Bahtimi, R.; Brownlee, N.A.; Willingham, M.C.; Garvin, A.J. Prognostic significance of Bcl-2 in Wilms’ tumor and oncogenic potential of Bcl-X(L) in rare tumor cases. Int. J. Cancer 1999, 84, 192–200. [Google Scholar] [CrossRef]
- Ikeda, H.; Hirato, J.; Akami, M.; Matsuyama, S.; Suzuki, N.; Takahashi, A.; Kuroiwa, M. Bcl-2 oncoprotein expression and apoptosis in neuroblastoma. J. Pediatr. Surg. 1995, 30, 805–808. [Google Scholar] [CrossRef]
- Lasorella, A.; Iavarone, A.; Israel, M.A. Differentiation of neuroblastoma enhances Bcl-2 expression and induces alterations of apoptosis and drug resistance. Cancer Res. 1995, 55, 4711–4716. [Google Scholar] [PubMed]
- Hanada, M.; Krajewski, S.; Tanaka, S.; Cazals-Hatem, D.; Spengler, B.A.; Ross, R.A.; Biedler, J.L.; Reed, J.C. Regulation of Bcl-2 oncoprotein levels with differentiation of human neuroblastoma cells. Cancer Res. 1993, 53, 4978–4986. [Google Scholar] [PubMed]
- Kirkin, V.; Joos, S.; Zornig, M. The role of Bcl-2 family members in tumorigenesis. Biochim. Biophys. Acta 2004, 1644, 229–249. [Google Scholar] [CrossRef] [PubMed]
- Ganjavi, H.; Gee, M.; Narendran, A.; Parkinson, N.; Krishnamoorthy, M.; Freedman, M.H.; Malkin, D. Adenovirus-mediated p53 gene therapy in osteosarcoma cell lines: Sensitization to cisplatin and doxorubicin. Cancer Gene Ther. 2006, 13, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Ganjavi, H.; Gee, M.; Narendran, A.; Freedman, M.H.; Malkin, D. Adenovirus-mediated p53 gene therapy in pediatric soft-tissue sarcoma cell lines: Sensitization to cisplatin and doxorubicin. Cancer Gene Ther. 2005, 12, 397–406. [Google Scholar] [CrossRef]
- Shetty, S.; Taylor, A.C.; Harris, L.C. Selective chemosensitization of rhabdomyosarcoma cell lines following wild-type p53 adenoviral transduction. Anticancer Drugs 2002, 13, 881–889. [Google Scholar] [CrossRef]
- Cai, B.; Chang, S.H.; Becker, E.B.; Bonni, A.; Xia, Z. p38 MAP kinase mediates apoptosis through phosphorylation of BimEL at Ser-65. J. Biol. Chem. 2006, 281, 25215–25222. [Google Scholar] [CrossRef]
- Lu, J.; Quearry, B.; Harada, H. p38-MAP kinase activation followed by BIM induction is essential for glucocorticoid-induced apoptosis in lymphoblastic leukemia cells. FEBS Lett. 2006, 580, 3539–3544. [Google Scholar] [CrossRef] [Green Version]
- Shimo, T.; Matsumura, S.; Ibaragi, S.; Isowa, S.; Kishimoto, K.; Mese, H.; Nishiyama, A.; Sasaki, A. Specific inhibitor of MEK-mediated cross-talk between ERK and p38 MAPK during differentiation of human osteosarcoma cells. J. Cell Commun. Signal. 2007, 1, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Nowicki, M.; Miskowiak, B. Comparison of the cell immunophenotype of metastatic and primary foci in stage IV-S neuroblastoma. Folia Histochem. Cytobiol. 2002, 40, 297–303. [Google Scholar] [PubMed]
- Nowicki, M.; Miskowiak, B.; Ostalska-Nowicka, D. Detection of substance P and its mRNA in human blast cells in childhood lymphoblastic leukaemia using immunocytochemistry and in situ hybridisation. Folia Histochem. Cytobiol. 2003, 41, 33–36. [Google Scholar] [PubMed]
- DeFea, K.A.; Vaughn, Z.D.; O’Bryan, E.M.; Nishijima, D.; Dery, O.; Bunnett, N.W. The proliferative and antiapoptotic effects of substance P are facilitated by formation of a beta -arrestin-dependent scaffolding complex. Proc. Natl. Acad. Sci. USA 2000, 97, 11086–11091. [Google Scholar] [CrossRef] [PubMed]
- Munoz, M.; Covenas, R. Involvement of substance P and the NK-1 receptor in cancer progression. Peptides 2013, 48, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Opel, D.; Naumann, I.; Kappler, R.; Friedman, L.; von Schweinitz, D.; Debatin, K.M.; Fulda, S. PI3K inhibitors prime neuroblastoma cells for chemotherapy by shifting the balance towards pro-apoptotic Bcl-2 proteins and enhanced mitochondrial apoptosis. Oncogene 2011, 30, 494–503. [Google Scholar] [CrossRef]
- Toretsky, J.A.; Thakar, M.; Eskenazi, A.E.; Frantz, C.N. Phosphoinositide 3-hydroxide kinase blockade enhances apoptosis in the Ewing’s sarcoma family of tumors. Cancer Res. 1999, 59, 5745–5750. [Google Scholar]
- Eichenmuller, M.; Gruner, I.; Hagl, B.; Haberle, B.; Muller-Hocker, J.; Von Schweinitz, D.; Kappler, R. Blocking the hedgehog pathway inhibits hepatoblastoma growth. Hepatology 2009, 49, 482–490. [Google Scholar] [CrossRef]
- Oliver, T.G.; Grasfeder, L.L.; Carroll, A.L.; Kaiser, C.; Gillingham, C.L.; Lin, S.M.; Wickramasinghe, R.; Scott, M.P.; Wechsler-Reya, R.J. Transcriptional profiling of the Sonic hedgehog response: A critical role for N-myc in proliferation of neuronal precursors. Proc. Natl. Acad. Sci. USA 2003, 100, 7331–7336. [Google Scholar] [CrossRef] [Green Version]
- Teglund, S.; Toftgard, R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta 2010, 1805, 181–208. [Google Scholar] [CrossRef] [PubMed]
- Pressey, J.G.; Anderson, J.R.; Crossman, D.K.; Lynch, J.C.; Barr, F.G. Hedgehog pathway activity in pediatric embryonal rhabdomyosarcoma and undifferentiated sarcoma: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2011, 57, 930–938. [Google Scholar] [CrossRef]
- Bigelow, R.L.; Chari, N.S.; Unden, A.B.; Spurgers, K.B.; Lee, S.; Roop, D.R.; Toftgard, R.; McDonnell, T.J. Transcriptional regulation of bcl-2 mediated by the sonic hedgehog signaling pathway through gli-1. J. Biol. Chem. 2004, 279, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Bar, E.E.; Chaudhry, A.; Farah, M.H.; Eberhart, C.G. Hedgehog signaling promotes medulloblastoma survival via Bc/II. Am. J. Pathol. 2007, 170, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Baliko, F.; Bright, T.; Poon, R.; Cohen, B.; Egan, S.E.; Alman, B.A. Inhibition of notch signaling induces neural differentiation in Ewing sarcoma. Am. J. Pathol. 2007, 170, 1686–1694. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Yang, Y.; Zweidler-McKay, P.A.; Hughes, D.P. Critical role of notch signaling in osteosarcoma invasion and metastasis. Clin. Cancer Res. 2008, 14, 2962–2969. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Eberhart, C.G. Medulloblastoma stem cells. J. Clin. Oncol. 2008, 26, 2821–2827. [Google Scholar] [CrossRef] [PubMed]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. Notch signaling in leukemia. Annu. Rev. Pathol. 2008, 3, 587–613. [Google Scholar] [CrossRef]
- Zhang, H.; Cai, X.; Wang, Y.; Tang, H.; Tong, D.; Ji, F. microRNA-143, down-regulated in osteosarcoma, promotes apoptosis and suppresses tumorigenicity by targeting Bcl-2. Oncol. Rep. 2010, 24, 1363–1369. [Google Scholar] [CrossRef]
- Margue, C.M.; Bernasconi, M.; Barr, F.G.; Schafer, B.W. Transcriptional modulation of the anti-apoptotic protein BCL-XL by the paired box transcription factors PAX3 and PAX3/FKHR. Oncogene 2000, 19, 2921–2929. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Liu, Y.; Xu, Z.; Yu, W.; Wang, H.; Li, C.; Lin, J. Novel small molecule, XZH-5, inhibits constitutive and interleukin-6-induced STAT3 phosphorylation in human rhabdomyosarcoma cells. Cancer Sci. 2011, 102, 1381–1387. [Google Scholar] [CrossRef]
- McPake, C.R.; Tillman, D.M.; Poquette, C.A.; George, E.O.; Houghton, J.A.; Harris, L.C. Bax is an important determinant of chemosensitivity in pediatric tumor cell lines independent of Bcl-2 expression and p53 status. Oncol. Res. 1998, 10, 235–244. [Google Scholar]
- Fulda, S.; Meyer, E.; Debatin, K.M. Inhibition of TRAIL-induced apoptosis by Bcl-2 overexpression. Oncogene 2002, 21, 2283–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dole, M.; Nunez, G.; Merchant, A.K.; Maybaum, J.; Rode, C.K.; Bloch, C.A.; Castle, V.P. Bcl-2 inhibits chemotherapy-induced apoptosis in neuroblastoma. Cancer Res. 1994, 54, 3253–3259. [Google Scholar] [PubMed]
- Dole, M.G.; Jasty, R.; Cooper, M.J.; Thompson, C.B.; Nunez, G.; Castle, V.P. Bcl-xL is expressed in neuroblastoma cells and modulates chemotherapy-induced apoptosis. Cancer Res. 1995, 55, 2576–2582. [Google Scholar] [PubMed]
- Campos, L.; Rouault, J.P.; Sabido, O.; Oriol, P.; Roubi, N.; Vasselon, C.; Archimbaud, E.; Magaud, J.P.; Guyotat, D. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 1993, 81, 3091–3096. [Google Scholar] [CrossRef] [Green Version]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Del Gaizo Moore, V.; Schlis, K.D.; Sallan, S.E.; Armstrong, S.A.; Letai, A. BCL-2 dependence and ABT-737 sensitivity in acute lymphoblastic leukemia. Blood 2008, 111, 2300–2309. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.H.; Kang, Y.H.; Szymanska, B.; Wilczynska-Kalak, U.; Sheard, M.A.; Harned, T.M.; Lock, R.B.; Reynolds, C.P. Activity of vincristine, L-ASP, and dexamethasone against acute lymphoblastic leukemia is enhanced by the BH3-mimetic ABT-737 in vitro and in vivo. Blood 2007, 110, 2057–2066. [Google Scholar] [CrossRef] [Green Version]
- High, L.M.; Szymanska, B.; Wilczynska-Kalak, U.; Barber, N.; O’Brien, R.; Khaw, S.L.; Vikstrom, I.B.; Roberts, A.W.; Lock, R.B. The Bcl-2 homology domain 3 mimetic ABT-737 targets the apoptotic machinery in acute lymphoblastic leukemia resulting in synergistic in vitro and in vivo interactions with established drugs. Mol. Pharmacol. 2010, 77, 483–494. [Google Scholar] [CrossRef]
- Goldsmith, K.C.; Gross, M.; Peirce, S.; Luyindula, D.; Liu, X.; Vu, A.; Sliozberg, M.; Guo, R.; Zhao, H.; Reynolds, C.P.; et al. Mitochondrial Bcl-2 family dynamics define therapy response and resistance in neuroblastoma. Cancer Res. 2012, 72, 2565–2577. [Google Scholar] [CrossRef]
- Lin, X.; Morgan-Lappe, S.; Huang, X.; Li, L.; Zakula, D.M.; Vernetti, L.A.; Fesik, S.W.; Shen, Y. ‘Seed’ analysis of off-target siRNAs reveals an essential role of Mcl-1 in resistance to the small-molecule Bcl-2/Bcl-XL inhibitor ABT-737. Oncogene 2007, 26, 3972–3979. [Google Scholar] [CrossRef]
- Chen, S.; Dai, Y.; Harada, H.; Dent, P.; Grant, S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007, 67, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Harned, T.M.; Kalous, O.; Maldonado, V.; DeClerck, Y.A.; Reynolds, C.P. Synergistic activity of fenretinide and the Bcl-2 family protein inhibitor ABT-737 against human neuroblastoma. Clin. Cancer Res. 2011, 17, 7093–7104. [Google Scholar] [CrossRef] [PubMed]
- Lestini, B.J.; Goldsmith, K.C.; Fluchel, M.N.; Liu, X.; Chen, N.L.; Goyal, B.; Pawel, B.R.; Hogarty, M.D. Mcl1 downregulation sensitizes neuroblastoma to cytotoxic chemotherapy and small molecule Bcl2-family antagonists. Cancer Biol. Ther. 2009, 8, 1587–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preuss, E.; Hugle, M.; Reimann, R.; Schlecht, M.; Fulda, S. Pan-mammalian target of rapamycin (mTOR) inhibitor AZD8055 primes rhabdomyosarcoma cells for ABT-737-induced apoptosis by down-regulating Mcl-1 protein. J. Biol. Chem. 2013, 288, 35287–35296. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.H.; Wan, Z.; Kang, Y.H.; Sposto, R.; Reynolds, C.P. Mechanism of synergy of N-(4-hydroxyphenyl)retinamide and ABT-737 in acute lymphoblastic leukemia cell lines: Mcl-1 inactivation. J. Natl. Cancer Inst. 2008, 100, 580–595. [Google Scholar] [CrossRef]
- Shore, G.C.; Viallet, J. Modulating the bcl-2 family of apoptosis suppressors for potential therapeutic benefit in cancer. Hematol. Am. Soc. Hematol. Educ. Program 2005, 1, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Contractor, R.; Tsao, T.; Samudio, I.; Ruvolo, P.P.; Kitada, S.; Deng, X.; Zhai, D.; Shi, Y.X.; Sneed, T.; et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006, 10, 375–388. [Google Scholar] [CrossRef] [Green Version]
- Kojima, K.; Konopleva, M.; Samudio, I.J.; Schober, W.D.; Bornmann, W.G.; Andreeff, M. Concomitant inhibition of MDM2 and Bcl-2 protein function synergistically induce mitochondrial apoptosis in AML. Cell Cycle 2006, 5, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Van Delft, M.F.; Wei, A.H.; Mason, K.D.; Vandenberg, C.J.; Chen, L.; Czabotar, P.E.; Willis, S.N.; Scott, C.L.; Day, C.L.; Cory, S.; et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006, 10, 389–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef]
- Lock, R.; Carol, H.; Houghton, P.J.; Morton, C.L.; Kolb, E.A.; Gorlick, R.; Reynolds, C.P.; Maris, J.M.; Keir, S.T.; Wu, J.; et al. Initial testing (stage 1) of the BH3 mimetic ABT-263 by the pediatric preclinical testing program. Pediatr. Blood Cancer 2008, 50, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Suryani, S.; Carol, H.; Chonghaile, T.N.; Frismantas, V.; Sarmah, C.; High, L.; Bornhauser, B.; Cowley, M.J.; Szymanska, B.; Evans, K.; et al. Cell and molecular determinants of in vivo efficacy of the BH3 mimetic ABT-263 against pediatric acute lymphoblastic leukemia xenografts. Clin. Cancer Res. 2014, 20, 4520–4531. [Google Scholar] [CrossRef] [PubMed]
- Faqar-Uz-Zaman, S.F.; Heinicke, U.; Meister, M.T.; Vogler, M.; Fulda, S. BCL-xL-selective BH3 mimetic sensitizes rhabdomyosarcoma cells to chemotherapeutics by activation of the mitochondrial pathway of apoptosis. Cancer Lett. 2018, 412, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Klenke, S.; Akdeli, N.; Stelmach, P.; Heukamp, L.; Schulte, J.H.; Bachmann, H.S. The small molecule Bcl-2/Mcl-1 inhibitor TW-37 shows single-agent cytotoxicity in neuroblastoma cell lines. BMC Cancer 2019, 19, 243. [Google Scholar] [CrossRef]
- Rheingold, S.R.; Hogarty, M.D.; Blaney, S.M.; Zwiebel, J.A.; Sauk-Schubert, C.; Chandula, R.; Krailo, M.D.; Adamson, P.C.; Children’s Oncology Group, S. Phase I Trial of G3139, a bcl-2 antisense oligonucleotide, combined with doxorubicin and cyclophosphamide in children with relapsed solid tumors: A Children’s Oncology Group Study. J. Clin. Oncol. 2007, 25, 1512–1518. [Google Scholar] [CrossRef]
- Marzo, I.; Naval, J. Bcl-2 family members as molecular targets in cancer therapy. Biochem. Pharmacol. 2008, 76, 939–946. [Google Scholar] [CrossRef]
- Salvesen, G.S.; Duckett, C.S. IAP proteins: Blocking the road to death’s door. Nat. Rev. Mol. Cell Biol. 2002, 3, 401–410. [Google Scholar] [CrossRef]
- Wei, Y.; Fan, T.; Yu, M. Inhibitor of apoptosis proteins and apoptosis. Acta Biochim. Biophys. Sin. 2008, 40, 278–288. [Google Scholar] [CrossRef] [Green Version]
- Sung, K.W.; Choi, J.; Hwang, Y.K.; Lee, S.J.; Kim, H.J.; Kim, J.Y.; Cho, E.J.; Yoo, K.H.; Koo, H.H. Overexpression of X-linked inhibitor of apoptosis protein (XIAP) is an independent unfavorable prognostic factor in childhood de novo acute myeloid leukemia. J. Korean Med. Sci. 2009, 24, 605–613. [Google Scholar] [CrossRef]
- Tamm, I.; Richter, S.; Oltersdorf, D.; Creutzig, U.; Harbott, J.; Scholz, F.; Karawajew, L.; Ludwig, W.D.; Wuchter, C. High expression levels of x-linked inhibitor of apoptosis protein and survivin correlate with poor overall survival in childhood de novo acute myeloid leukemia. Clin. Cancer Res. 2004, 10, 3737–3744. [Google Scholar] [CrossRef]
- Hundsdoerfer, P.; Dietrich, I.; Schmelz, K.; Eckert, C.; Henze, G. XIAP expression is post-transcriptionally upregulated in childhood ALL and is associated with glucocorticoid response in T-cell ALL. Pediatr. Blood Cancer 2010, 55, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Holcik, M.; Lefebvre, C.; Yeh, C.; Chow, T.; Korneluk, R.G. A new internal-ribosome-entry-site motif potentiates XIAP-mediated cytoprotection. Nat. Cell Biol. 1999, 1, 190–192. [Google Scholar] [CrossRef] [PubMed]
- Saraei, R.; Soleimani, M.; Movassaghpour Akbari, A.A.; Farshdousti Hagh, M.; Hassanzadeh, A.; Solali, S. The role of XIAP in resistance to TNF-related apoptosis-inducing ligand (TRAIL) in Leukemia. Biomed. Pharmacother. 2018, 107, 1010–1019. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Validating survivin as a cancer therapeutic target. Nat. Rev. Cancer 2003, 3, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; de Bruin, A.; Caldas, H.; Fangusaro, J.; Hayes, J.; Conway, E.M.; Robinson, M.L.; Altura, R.A. Essential role for survivin in early brain development. J. Neurosci. 2005, 25, 6962–6970. [Google Scholar] [CrossRef] [PubMed]
- Xing, Z.; Conway, E.M.; Kang, C.; Winoto, A. Essential role of survivin, an inhibitor of apoptosis protein, in T cell development, maturation, and homeostasis. J. Exp. Med. 2004, 199, 69–80. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, L.; Linehan, R.; Clynes, M. Survivin: Role in normal cells and in pathological conditions. Curr. Cancer Drug Targets 2003, 3, 131–152. [Google Scholar] [CrossRef]
- Bai, H.; Gayyed, M.F.; Lam-Himlin, D.M.; Klein, A.P.; Nayar, S.K.; Xu, Y.; Khan, M.; Argani, P.; Pan, D.; Anders, R.A. Expression of Yes-associated protein modulates Survivin expression in primary liver malignancies. Hum. Pathol. 2012, 43, 1376–1385. [Google Scholar] [CrossRef] [Green Version]
- Cottini, F.; Anderson, K.C.; Tonon, G. Awakening the Hippo co-activator YAP1, a mercurial cancer gene, in hematologic cancers. Mol. Cell. Oncol. 2014, 1, e970055. [Google Scholar] [CrossRef] [Green Version]
- Bouvier, C.; Macagno, N.; Nguyen, Q.; Loundou, A.; Jiguet-Jiglaire, C.; Gentet, J.C.; Jouve, J.L.; Rochwerger, A.; Mattei, J.C.; Bouvard, D.; et al. Prognostic value of the Hippo pathway transcriptional coactivators YAP/TAZ and beta1-integrin in conventional osteosarcoma. Oncotarget 2016, 7, 64702–64710. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Abedalthagafi, M.; Anwar, A.E.; Bui, M.M. Akt and hippo pathways in Ewing’s sarcoma tumors and their prognostic significance. J. Cancer 2015, 6, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Slemmons, K.K.; Crose, L.E.; Rudzinski, E.; Bentley, R.C.; Linardic, C.M. Role of the YAP Oncoprotein in Priming Ras-Driven Rhabdomyosarcoma. PLoS ONE 2015, 10, e0140781. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Alonzo, T.A.; Gerbing, R.B.; Lange, B.J.; Heerema, N.A.; Franklin, J.; Raimondi, S.C.; Hirsch, B.A.; Gamis, A.S.; Meshinchi, S. BIRC5 (survivin) splice variant expression correlates with refractory disease and poor outcome in pediatric acute myeloid leukemia: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2014, 61, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Troeger, A.; Siepermann, M.; Escherich, G.; Meisel, R.; Willers, R.; Gudowius, S.; Moritz, T.; Laws, H.J.; Hanenberg, H.; Goebel, U.; et al. Survivin and its prognostic significance in pediatric acute B-cell precursor lymphoblastic leukemia. Haematologica 2007, 92, 1043–1050. [Google Scholar] [CrossRef] [Green Version]
- Islam, A.; Kageyama, H.; Takada, N.; Kawamoto, T.; Takayasu, H.; Isogai, E.; Ohira, M.; Hashizume, K.; Kobayashi, H.; Kaneko, Y.; et al. High expression of Survivin, mapped to 17q25, is significantly associated with poor prognostic factors and promotes cell survival in human neuroblastoma. Oncogene 2000, 19, 617–623. [Google Scholar] [CrossRef] [Green Version]
- Takamizawa, S.; Scott, D.; Wen, J.; Grundy, P.; Bishop, W.; Kimura, K.; Sandler, A. The survivin:fas ratio in pediatric renal tumors. J. Pediatr. Surg. 2001, 36, 37–42. [Google Scholar] [CrossRef]
- Zou, J.; Gan, M.; Mao, N.; Zhu, X.; Shi, Q.; Yang, H. Sensitization of osteosarcoma cell line SaOS-2 to chemotherapy by downregulating survivin. Arch. Med. Res. 2010, 41, 162–169. [Google Scholar] [CrossRef]
- Caldas, H.; Holloway, M.P.; Hall, B.M.; Qualman, S.J.; Altura, R.A. Survivin-directed RNA interference cocktail is a potent suppressor of tumour growth in vivo. J. Med. Genet. 2006, 43, 119–128. [Google Scholar] [CrossRef]
- Hingorani, P.; Dickman, P.; Garcia-Filion, P.; White-Collins, A.; Kolb, E.A.; Azorsa, D.O. BIRC5 expression is a poor prognostic marker in Ewing sarcoma. Pediatr. Blood Cancer 2013, 60, 35–40. [Google Scholar] [CrossRef]
- Ito, R.; Asami, S.; Motohashi, S.; Ootsuka, S.; Yamaguchi, Y.; Chin, M.; Shichino, H.; Yoshida, Y.; Nemoto, N.; Mugishima, H.; et al. Significance of survivin mRNA expression in prognosis of neuroblastoma. Biol. Pharm. Bull. 2005, 28, 565–568. [Google Scholar] [CrossRef]
- Miller, M.A.; Ohashi, K.; Zhu, X.; McGrady, P.; London, W.B.; Hogarty, M.; Sandler, A.D. Survivin mRNA levels are associated with biology of disease and patient survival in neuroblastoma: A report from the children’s oncology group. J. Pediatr. Hematol. Oncol. 2006, 28, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Adida, C.; Berrebi, D.; Peuchmaur, M.; Reyes-Mugica, M.; Altieri, D.C. Anti-apoptosis gene, survivin, and prognosis of neuroblastoma. Lancet 1998, 351, 882–883. [Google Scholar] [CrossRef]
- Azuhata, T.; Scott, D.; Takamizawa, S.; Wen, J.; Davidoff, A.; Fukuzawa, M.; Sandler, A. The inhibitor of apoptosis protein survivin is associated with high-risk behavior of neuroblastoma. J. Pediatr. Surg. 2001, 36, 1785–1791. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Role of survivin and its splice variants in tumorigenesis. Br. J. Cancer 2005, 92, 212–216. [Google Scholar] [CrossRef]
- Troger, A.; Siepermann, M.; Mahotka, C.; Wethkamp, N.; Bulle, H.; Laws, H.J.; Escherich, G.; Janka-Schaub, G.; Gobel, U.; Dilloo, D. Role of survivin splice variants in pediatric acute precursor B lymphoblastic leukemia. Klin. Padiatr. 2007, 219, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Trieb, K.; Lehner, R.; Stulnig, T.; Sulzbacher, I.; Shroyer, K.R. Survivin expression in human osteosarcoma is a marker for survival. Eur. J. Surg. Oncol. 2003, 29, 379–382. [Google Scholar] [CrossRef]
- Sung, K.W.; Choi, J.; Hwang, Y.K.; Lee, S.J.; Kim, H.J.; Lee, S.H.; Yoo, K.H.; Jung, H.L.; Koo, H.H. Overexpression of Apollon, an antiapoptotic protein, is associated with poor prognosis in childhood de novo acute myeloid leukemia. Clin. Cancer Res. 2007, 13, 5109–5114. [Google Scholar] [CrossRef]
- Ismail, E.A.; Mahmoud, H.M.; Tawfik, L.M.; Habashy, D.M.; Adly, A.A.; El-Sherif, N.H.; Abdelwahab, M.A. BIRC6/Apollon gene expression in childhood acute leukemia: Impact on therapeutic response and prognosis. Eur. J. Haematol. 2012, 88, 118–127. [Google Scholar] [CrossRef]
- Ibrahim, L.; Aladle, D.; Mansour, A.; Hammad, A.; Al Wakeel, A.A.; Abd El-Hameed, S.A. Expression and prognostic significance of livin/BIRC7 in childhood acute lymphoblastic leukemia. Med. Oncol. 2014, 31, 941. [Google Scholar] [CrossRef]
- Choi, J.; Hwang, Y.K.; Sung, K.W.; Lee, S.H.; Yoo, K.H.; Jung, H.L.; Koo, H.H.; Kim, H.J.; Kang, H.J.; Shin, H.Y.; et al. Expression of Livin, an antiapoptotic protein, is an independent favorable prognostic factor in childhood acute lymphoblastic leukemia. Blood 2007, 109, 471–477. [Google Scholar] [CrossRef]
- Nachmias, B.; Ashhab, Y.; Bucholtz, V.; Drize, O.; Kadouri, L.; Lotem, M.; Peretz, T.; Mandelboim, O.; Ben-Yehuda, D. Caspase-mediated cleavage converts Livin from an antiapoptotic to a proapoptotic factor: Implications for drug-resistant melanoma. Cancer Res. 2003, 63, 6340–6349. [Google Scholar] [PubMed]
- Chen, D.J.; Huerta, S. Smac mimetics as new cancer therapeutics. Anticancer Drugs 2009, 20, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Pluta, A.; Wrzesien-Kus, A.; Cebula-Obrzut, B.; Wolska, A.; Szmigielska-Kaplon, A.; Czemerska, M.; Pluta, P.; Robak, T.; Smolewski, P.; Wierzbowska, A. Influence of high expression of Smac/DIABLO protein on the clinical outcome in acute myeloid leukemia patients. Leuk. Res. 2010, 34, 1308–1313. [Google Scholar] [CrossRef] [PubMed]
- Hotta, T.; Suzuki, H.; Nagai, S.; Yamamoto, K.; Imakiire, A.; Takada, E.; Itoh, M.; Mizuguchi, J. Chemotherapeutic agents sensitize sarcoma cell lines to tumor necrosis factor-related apoptosis-inducing ligand-induced caspase-8 activation, apoptosis and loss of mitochondrial membrane potential. J. Orthop. Res. 2003, 21, 949–957. [Google Scholar] [CrossRef]
- Arora, V.; Cheung, H.H.; Plenchette, S.; Micali, O.C.; Liston, P.; Korneluk, R.G. Degradation of survivin by the X-linked inhibitor of apoptosis (XIAP)-XAF1 complex. J. Biol. Chem. 2007, 282, 26202–26209. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.X.; Wallis, K.; Fell, S.M.; Sobrado, V.R.; Hemmer, M.C.; Ramskold, D.; Hellman, U.; Sandberg, R.; Kenchappa, R.S.; Martinson, T.; et al. RNA helicase A is a downstream mediator of KIF1Bbeta tumor-suppressor function in neuroblastoma. Cancer Discov. 2014, 4, 434–451. [Google Scholar] [CrossRef]
- Wright, K.M.; Vaughn, A.E.; Deshmukh, M. Apoptosome dependent caspase-3 activation pathway is non-redundant and necessary for apoptosis in sympathetic neurons. Cell Death Differ. 2007, 14, 625–633. [Google Scholar] [CrossRef]
- Deshmukh, M.; Johnson, E.M., Jr. Evidence of a novel event during neuronal death: Development of competence-to-die in response to cytoplasmic cytochrome c. Neuron 1998, 21, 695–705. [Google Scholar] [CrossRef]
- Edwards, S.N.; Tolkovsky, A.M. Characterization of apoptosis in cultured rat sympathetic neurons after nerve growth factor withdrawal. J. Cell Biol. 1994, 124, 537–546. [Google Scholar] [CrossRef] [Green Version]
- Deckwerth, T.L.; Johnson, E.M., Jr. Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J. Cell Biol. 1993, 123, 1207–1222. [Google Scholar] [CrossRef]
- Schlisio, S. Neuronal apoptosis by prolyl hydroxylation: Implication in nervous system tumours and the Warburg conundrum. J. Cell. Mol. Med. 2009, 13, 4104–4112. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Parada, L.F. The molecular and genetic basis of neurological tumours. Nat. Rev. Cancer 2002, 2, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Choo, Z.; Koh, R.Y.; Wallis, K.; Koh, T.J.; Kuick, C.H.; Sobrado, V.; Kenchappa, R.S.; Loh, A.H.; Soh, S.Y.; Schlisio, S.; et al. XAF1 promotes neuroblastoma tumor suppression and is required for KIF1Bbeta-mediated apoptosis. Oncotarget 2016, 7, 34229–34239. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J.; et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007, 131, 669–681. [Google Scholar] [CrossRef]
- Fulda, S.; Vucic, D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012, 11, 109–124. [Google Scholar] [CrossRef]
- Langemann, D.; Trochimiuk, M.; Appl, B.; Hundsdoerfer, P.; Reinshagen, K.; Eschenburg, G. Sensitization of neuroblastoma for vincristine-induced apoptosis by Smac mimetic LCL161 is attended by G2 cell cycle arrest but is independent of NFkappaB, RIP1 and TNF-alpha. Oncotarget 2017, 8, 87763–87772. [Google Scholar] [CrossRef]
- Eschenburg, G.; Eggert, A.; Schramm, A.; Lode, H.N.; Hundsdoerfer, P. Smac mimetic LBW242 sensitizes XIAP-overexpressing neuroblastoma cells for TNF-alpha-independent apoptosis. Cancer Res. 2012, 72, 2645–2656. [Google Scholar] [CrossRef]
- Fulda, S.; Wick, W.; Weller, M.; Debatin, K.M. Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat. Med. 2002, 8, 808–815. [Google Scholar] [CrossRef]
- Giagkousiklidis, S.; Vogler, M.; Westhoff, M.A.; Kasperczyk, H.; Debatin, K.M.; Fulda, S. Sensitization for gamma-irradiation-induced apoptosis by second mitochondria-derived activator of caspase. Cancer Res. 2005, 65, 10502–10513. [Google Scholar] [CrossRef]
- Vogler, M.; Giagkousiklidis, S.; Genze, F.; Gschwend, J.E.; Debatin, K.M.; Fulda, S. Inhibition of clonogenic tumor growth: A novel function of Smac contributing to its antitumor activity. Oncogene 2005, 24, 7190–7202. [Google Scholar] [CrossRef]
- Houghton, P.J.; Kang, M.H.; Reynolds, C.P.; Morton, C.L.; Kolb, E.A.; Gorlick, R.; Keir, S.T.; Carol, H.; Lock, R.; Maris, J.M.; et al. Initial testing (stage 1) of LCL161, a SMAC mimetic, by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2012, 58, 636–639. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, M.; Trentin, L.; Queudeville, M.; Seyfried, F.; Demir, S.; Tausch, E.; Stilgenbauer, S.; Eckhoff, S.M.; Meyer, L.H.; Debatin, K.M. Intrinsic and chemo-sensitizing activity of SMAC-mimetics on high-risk childhood acute lymphoblastic leukemia. Cell Death Dis. 2016, 7, e2052. [Google Scholar] [CrossRef] [PubMed]
- Gerges, S.; Rohde, K.; Fulda, S. Cotreatment with Smac mimetics and demethylating agents induces both apoptotic and necroptotic cell death pathways in acute lymphoblastic leukemia cells. Cancer Lett. 2016, 375, 127–132. [Google Scholar] [CrossRef] [PubMed]
- LaCasse, E.C.; Cherton-Horvat, G.G.; Hewitt, K.E.; Jerome, L.J.; Morris, S.J.; Kandimalla, E.R.; Yu, D.; Wang, H.; Wang, W.; Zhang, R.; et al. Preclinical characterization of AEG35156/GEM 640, a second-generation antisense oligonucleotide targeting X-linked inhibitor of apoptosis. Clin. Cancer Res. 2006, 12, 5231–5241. [Google Scholar] [CrossRef] [PubMed]
- Lacasse, E.C.; Kandimalla, E.R.; Winocour, P.; Sullivan, T.; Agrawal, S.; Gillard, J.W.; Durkin, J. Application of XIAP antisense to cancer and other proliferative disorders: Development of AEG35156/GEM640. Ann. NY Acad. Sci. 2005, 1058, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Holt, S.V.; Brookes, K.E.; Dive, C.; Makin, G.W. Down-regulation of XIAP by AEG35156 in paediatric tumour cells induces apoptosis and sensitises cells to cytotoxic agents. Oncol. Rep. 2011, 25, 1177–1181. [Google Scholar] [CrossRef]
- Schimmer, A.D.; Estey, E.H.; Borthakur, G.; Carter, B.Z.; Schiller, G.J.; Tallman, M.S.; Altman, J.K.; Karp, J.E.; Kassis, J.; Hedley, D.W.; et al. Phase I/II trial of AEG35156 X-linked inhibitor of apoptosis protein antisense oligonucleotide combined with idarubicin and cytarabine in patients with relapsed or primary refractory acute myeloid leukemia. J. Clin. Oncol. 2009, 27, 4741–4746. [Google Scholar] [CrossRef]
- Loder, S.; Fakler, M.; Schoeneberger, H.; Cristofanon, S.; Leibacher, J.; Vanlangenakker, N.; Bertrand, M.J.; Vandenabeele, P.; Jeremias, I.; Debatin, K.M.; et al. RIP1 is required for IAP inhibitor-mediated sensitization of childhood acute leukemia cells to chemotherapy-induced apoptosis. Leukemia 2012, 26, 1020–1029. [Google Scholar] [CrossRef]
- Loeder, S.; Drensek, A.; Jeremias, I.; Debatin, K.M.; Fulda, S. Small molecule XIAP inhibitors sensitize childhood acute leukemia cells for CD95-induced apoptosis. Int. J. Cancer 2010, 126, 2216–2228. [Google Scholar] [CrossRef]
- Yang, T.; Lan, J.; Huang, Q.; Chen, X.; Sun, X.; Liu, X.; Yang, P.; Jin, T.; Wang, S.; Mou, X. Embelin sensitizes acute myeloid leukemia cells to TRAIL through XIAP inhibition and NF-kappaB inactivation. Cell Biochem. Biophys. 2015, 71, 291–297. [Google Scholar] [CrossRef]
- Moreno-Martinez, D.; Nomdedeu, M.; Lara-Castillo, M.C.; Etxabe, A.; Pratcorona, M.; Tesi, N.; Diaz-Beya, M.; Rozman, M.; Montserrat, E.; Urbano-Ispizua, A.; et al. XIAP inhibitors induce differentiation and impair clonogenic capacity of acute myeloid leukemia stem cells. Oncotarget 2014, 5, 4337–4346. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, X.; Gu, Z.; Wang, L. Small molecule survivin inhibitor YM155 displays potent activity against human osteosarcoma cells. Cancer Investig. 2016, 34, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Schultze, K.; Bock, B.; Eckert, A.; Oevermann, L.; Ramacher, D.; Wiestler, O.; Roth, W. Troglitazone sensitizes tumor cells to TRAIL-induced apoptosis via down-regulation of FLIP and Survivin. Apoptosis 2006, 11, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kang, J.; Qiao, J.; Thomas, R.P.; Evers, B.M.; Chung, D.H. Phosphatidylinositol 3-kinase inhibition down-regulates survivin and facilitates TRAIL-mediated apoptosis in neuroblastomas. J. Pediatr. Surg. 2004, 39, 516–521. [Google Scholar] [CrossRef]
- Shankar, S.L.; Mani, S.; O’Guin, K.N.; Kandimalla, E.R.; Agrawal, S.; Shafit-Zagardo, B. Survivin inhibition induces human neural tumor cell death through caspase-independent and -dependent pathways. J. Neurochem. 2001, 79, 426–436. [Google Scholar] [CrossRef]
- Fest, S.; Huebener, N.; Bleeke, M.; Durmus, T.; Stermann, A.; Woehler, A.; Baykan, B.; Zenclussen, A.C.; Michalsky, E.; Jaeger, I.S.; et al. Survivin minigene DNA vaccination is effective against neuroblastoma. Int. J. Cancer 2009, 125, 104–114. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choo, Z.; Loh, A.H.P.; Chen, Z.X. Destined to Die: Apoptosis and Pediatric Cancers. Cancers 2019, 11, 1623. https://doi.org/10.3390/cancers11111623
Choo Z, Loh AHP, Chen ZX. Destined to Die: Apoptosis and Pediatric Cancers. Cancers. 2019; 11(11):1623. https://doi.org/10.3390/cancers11111623
Chicago/Turabian StyleChoo, Zhang’e, Amos Hong Pheng Loh, and Zhi Xiong Chen. 2019. "Destined to Die: Apoptosis and Pediatric Cancers" Cancers 11, no. 11: 1623. https://doi.org/10.3390/cancers11111623
APA StyleChoo, Z., Loh, A. H. P., & Chen, Z. X. (2019). Destined to Die: Apoptosis and Pediatric Cancers. Cancers, 11(11), 1623. https://doi.org/10.3390/cancers11111623