Epigenetic Reprogramming for Targeting IDH-Mutant Malignant Gliomas



Abstract
1. Introduction
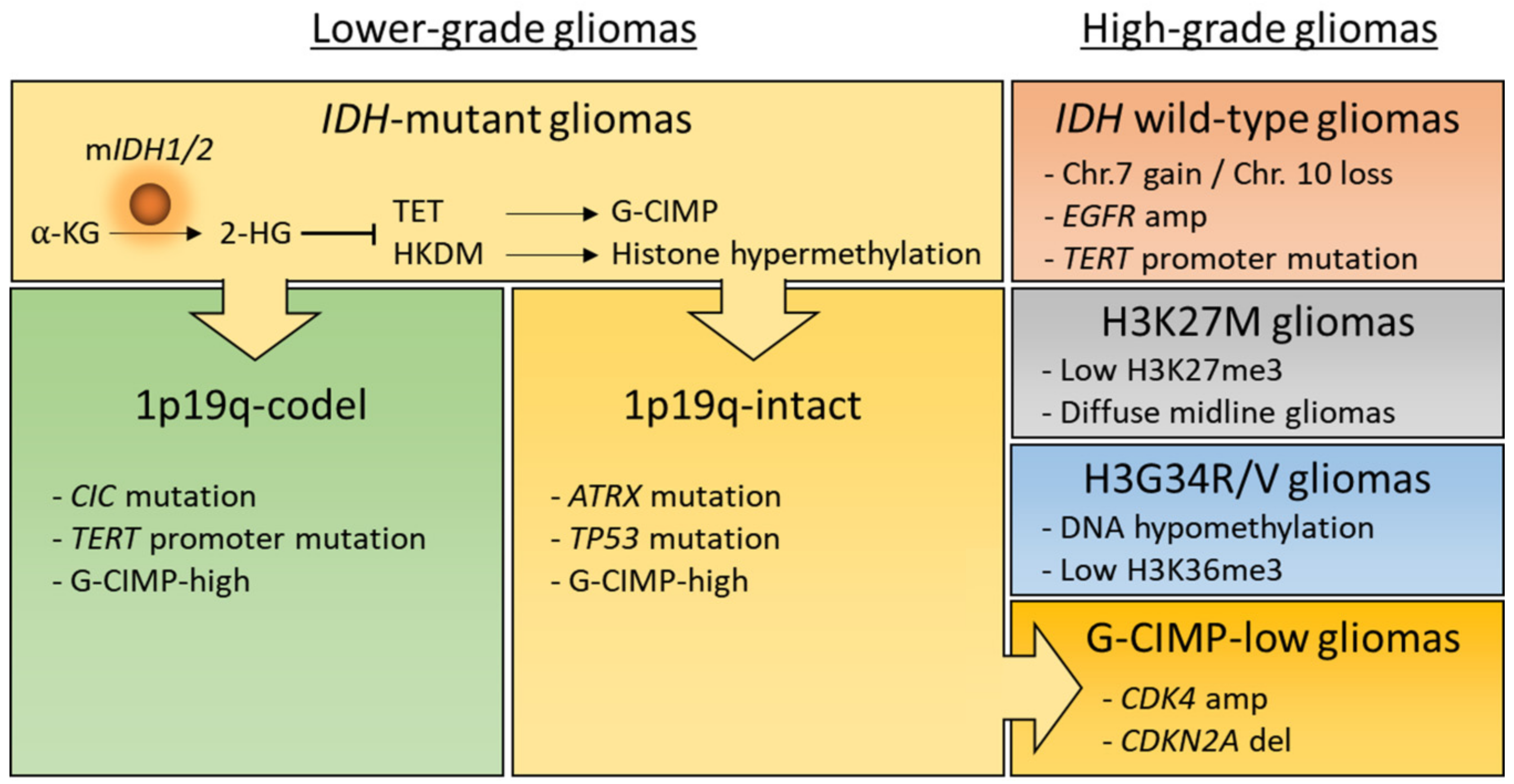
2. Epigenetic Modifications in Gliomas
2.1. IDH-Mutation Induced DNA Hypermethylation
2.2. MGMT Promoter Methylation
2.3. Aberrant Histone Modifications in Gliomas
2.4. Chromatin Remodeling
3. Epigenetic Therapy for Gliomas
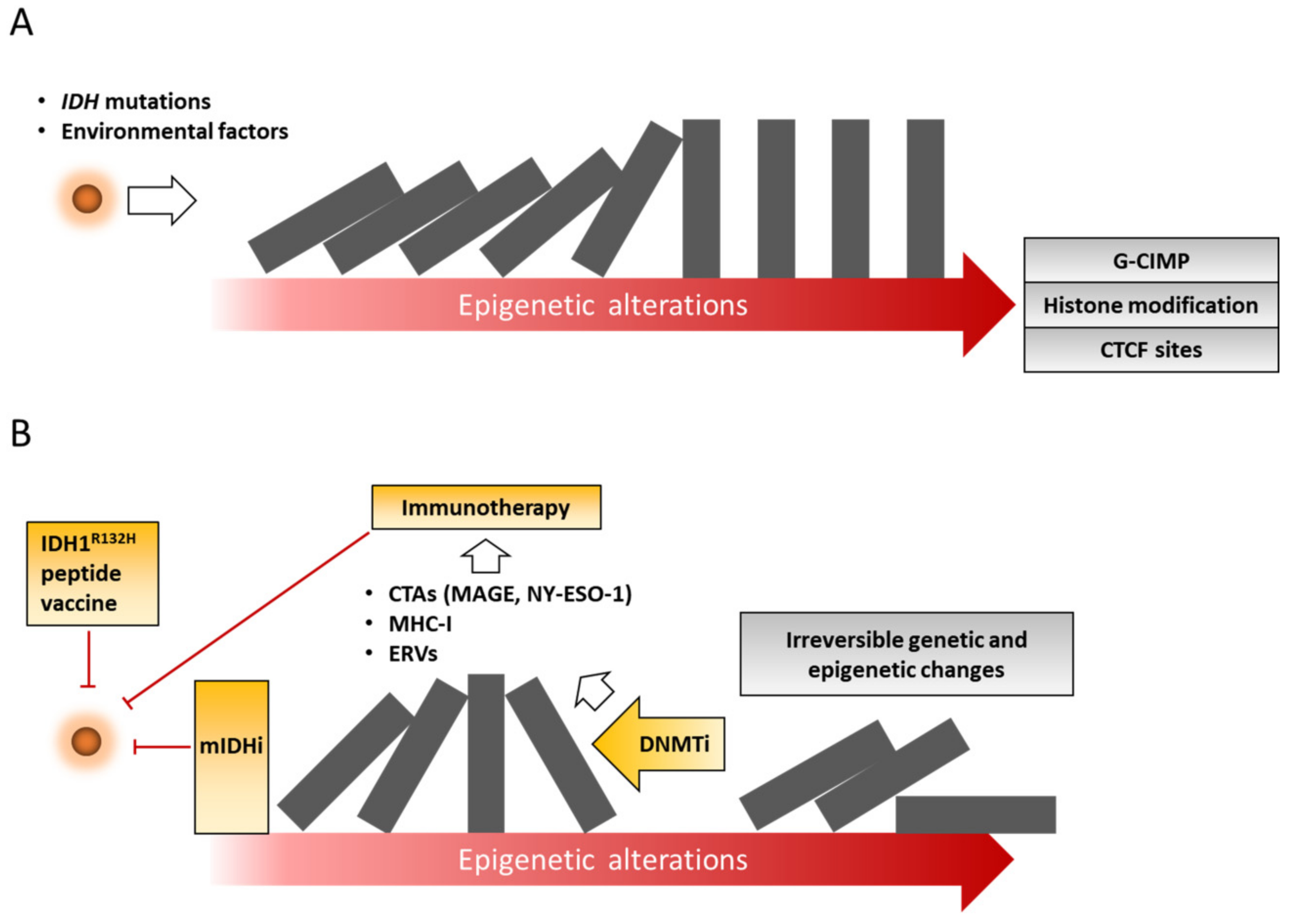
3.1. Mutant IDH Inhibitors
3.2. IDH1 Peptide Vaccination
3.3. DNA Methylation Inhibitors
3.4. Combining DNMTi with Cytotoxic Drugs for Glioma
3.5. Combination of Epigenetic Drugs and Immunotherapy
4. Conclusions
Funding
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro Oncol. 2018, 20, iv1–iv86. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Primers 2015, 1, 15017. [Google Scholar] [CrossRef] [PubMed]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tonjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N.; Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Park, J.W.; Wollmann, G.; Urbiola, C.; Fogli, B.; Florio, T.; Geley, S.; Klimaschewski, L. Sprouty2 enhances the tumorigenic potential of glioblastoma cells. Neuro Oncol. 2018, 20, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Florio, T.; Barbieri, F. The status of the art of human malignant glioma management: The promising role of targeting tumor-initiating cells. Drug Discov. Today 2012, 17, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed]
- Mazor, T.; Pankov, A.; Johnson, B.E.; Hong, C.; Hamilton, E.G.; Bell, R.J.A.; Smirnov, I.V.; Reis, G.F.; Phillips, J.J.; Barnes, M.J.; et al. DNA Methylation and Somatic Mutations Converge on the Cell Cycle and Define Similar Evolutionary Histories in Brain Tumors. Cancer Cell 2015, 28, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.; Kharbanda, S.; Pope, W.B.; Tran, A.; Solis, O.E.; Peale, F.; Forrest, W.F.; Pujara, K.; Carrillo, J.A.; Pandita, A.; et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J. Clin. Oncol. 2011, 29, 4482–4490. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Nobusawa, S.; Kleihues, P.; Ohgaki, H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am. J. Pathol. 2009, 174, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Shen, H.; Laird, P.W. Interplay between the cancer genome and epigenome. Cell 2013, 153, 38–55. [Google Scholar] [CrossRef]
- Ehrlich, M.; Lacey, M. DNA hypomethylation and hemimethylation in cancer. Adv. Exp. Med. Biol. 2013, 754, 31–56. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Hermann, A.; Goyal, R.; Jeltsch, A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem. 2004, 279, 48350–48359. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome-biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef]
- Cairns, R.A.; Mak, T.W. Oncogenic isocitrate dehydrogenase mutations: Mechanisms, models, and clinical opportunities. Cancer Discov. 2013, 3, 730–741. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Amankulor, N.M.; Kim, Y.; Arora, S.; Kargl, J.; Szulzewsky, F.; Hanke, M.; Margineantu, D.H.; Rao, A.; Bolouri, H.; Delrow, J.; et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017, 31, 774–786. [Google Scholar] [CrossRef]
- Bunse, L.; Pusch, S.; Bunse, T.; Sahm, F.; Sanghvi, K.; Friedrich, M.; Alansary, D.; Sonner, J.K.; Green, E.; Deumelandt, K.; et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 2018, 24, 1192–1203. [Google Scholar] [CrossRef]
- Unruh, D.; Zewde, M.; Buss, A.; Drumm, M.R.; Tran, A.N.; Scholtens, D.M.; Horbinski, C. Methylation and transcription patterns are distinct in IDH mutant gliomas compared to other IDH mutant cancers. Sci. Rep. 2019, 9, 8946. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Meyer, J.; Balss, J.; Capper, D.; Mueller, W.; Christians, A.; Felsberg, J.; Wolter, M.; Mawrin, C.; Wick, W.; et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: A study of 1010 diffuse gliomas. Acta Neuropathol. 2009, 118, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009, 324, 261–265. [Google Scholar] [CrossRef]
- Madala, H.R.; Punganuru, S.R.; Arutla, V.; Misra, S.; Thomas, T.J.; Srivenugopal, K.S. Beyond Brooding on Oncometabolic Havoc in IDH-Mutant Gliomas and AML: Current and Future Therapeutic Strategies. Cancers 2018, 10, 49. [Google Scholar] [CrossRef]
- Sasaki, M.; Knobbe, C.B.; Itsumi, M.; Elia, A.J.; Harris, I.S.; Chio, I.I.; Cairns, R.A.; McCracken, S.; Wakeham, A.; Haight, J.; et al. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012, 26, 2038–2049. [Google Scholar] [CrossRef]
- Bardella, C.; Al-Dalahmah, O.; Krell, D.; Brazauskas, P.; Al-Qahtani, K.; Tomkova, M.; Adam, J.; Serres, S.; Lockstone, H.; Freeman-Mills, L.; et al. Expression of Idh1(R132H) in the Murine Subventricular Zone Stem Cell Niche Recapitulates Features of Early Gliomagenesis. Cancer Cell 2016, 30, 578–594. [Google Scholar] [CrossRef]
- Suzuki, H.; Aoki, K.; Chiba, K.; Sato, Y.; Shiozawa, Y.; Shiraishi, Y.; Shimamura, T.; Niida, A.; Motomura, K.; Ohka, F.; et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat. Genet. 2015, 47, 458–468. [Google Scholar] [CrossRef]
- Bettegowda, C.; Agrawal, N.; Jiao, Y.; Sausen, M.; Wood, L.D.; Hruban, R.H.; Rodriguez, F.J.; Cahill, D.P.; McLendon, R.; Riggins, G.; et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science 2011, 333, 1453–1455. [Google Scholar] [CrossRef]
- Jiao, Y.; Killela, P.J.; Reitman, Z.J.; Rasheed, A.B.; Heaphy, C.M.; de Wilde, R.F.; Rodriguez, F.J.; Rosemberg, S.; Oba-Shinjo, S.M.; Nagahashi Marie, S.K.; et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 2012, 3, 709–722. [Google Scholar] [CrossRef]
- Binder, H.; Willscher, E.; Loeffler-Wirth, H.; Hopp, L.; Jones, D.T.W.; Pfister, S.M.; Kreuz, M.; Gramatzki, D.; Fortenbacher, E.; Hentschel, B.; et al. DNA methylation, transcriptome and genetic copy number signatures of diffuse cerebral WHO grade II/III gliomas resolve cancer heterogeneity and development. Acta Neuropathol. Commun. 2019, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- De Souza, C.F.; Sabedot, T.S.; Malta, T.M.; Stetson, L.; Morozova, O.; Sokolov, A.; Laird, P.W.; Wiznerowicz, M.; Iavarone, A.; Snyder, J.; et al. A Distinct DNA Methylation Shift in a Subset of Glioma CpG Island Methylator Phenotypes during Tumor Recurrence. Cell Rep. 2018, 23, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Saito, K.; Aihara, K.; Nagae, G.; Yamamoto, S.; Tatsuno, K.; Ueda, H.; Fukuda, S.; Umeda, T.; Tanaka, S.; et al. DNA demethylation is associated with malignant progression of lower-grade gliomas. Sci. Rep. 2019, 9, 1903. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Baylin, S.B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003, 349, 2042–2054. [Google Scholar] [CrossRef]
- Esteller, M.; Herman, J.G. Generating mutations but providing chemosensitivity: The role of O6-methylguanine DNA methyltransferase in human cancer. Oncogene 2004, 23, 1–8. [Google Scholar] [CrossRef]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Rivera, A.L.; Pelloski, C.E.; Gilbert, M.R.; Colman, H.; De La Cruz, C.; Sulman, E.P.; Bekele, B.N.; Aldape, K.D. MGMT promoter methylation is predictive of response to radiotherapy and prognostic in the absence of adjuvant alkylating chemotherapy for glioblastoma. Neuro Oncol. 2010, 12, 116–121. [Google Scholar] [CrossRef]
- Baumert, B.G.; Hegi, M.E.; van den Bent, M.J.; von Deimling, A.; Gorlia, T.; Hoang-Xuan, K.; Brandes, A.A.; Kantor, G.; Taphoorn, M.J.B.; Hassel, M.B.; et al. Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033-26033): A randomised, open-label, phase 3 intergroup study. Lancet Oncol. 2016, 17, 1521–1532. [Google Scholar] [CrossRef]
- Leu, S.; von Felten, S.; Frank, S.; Vassella, E.; Vajtai, I.; Taylor, E.; Schulz, M.; Hutter, G.; Hench, J.; Schucht, P.; et al. IDH/MGMT-driven molecular classification of low-grade glioma is a strong predictor for long-term survival. Neuro Oncol. 2013, 15, 469–479. [Google Scholar] [CrossRef]
- Bady, P.; Kurscheid, S.; Delorenzi, M.; Gorlia, T.; van den Bent, M.J.; Hoang-Xuan, K.; Vauleon, E.; Gijtenbeek, A.; Enting, R.; Thiessen, B.; et al. The DNA methylome of DDR genes and benefit from RT or TMZ in IDH mutant low-grade glioma treated in EORTC 22033. Acta Neuropathol. 2018, 135, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Turcan, S.; Makarov, V.; Taranda, J.; Wang, Y.; Fabius, A.W.M.; Wu, W.; Zheng, Y.; El-Amine, N.; Haddock, S.; Nanjangud, G.; et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat. Genet. 2018, 50, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Venneti, S.; Felicella, M.M.; Coyne, T.; Phillips, J.J.; Gorovets, D.; Huse, J.T.; Kofler, J.; Lu, C.; Tihan, T.; Sullivan, L.M.; et al. Histone 3 lysine 9 trimethylation is differentially associated with isocitrate dehydrogenase mutations in oligodendrogliomas and high-grade astrocytomas. J. Neuropathol. Exp. Neurol. 2013, 72, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T. The epigenetic magic of histone lysine methylation. FEBS J. 2006, 273, 3121–3135. [Google Scholar] [CrossRef] [PubMed]
- Shinkai, Y.; Tachibana, M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011, 25, 781–788. [Google Scholar] [CrossRef]
- Kubicek, S.; O’Sullivan, R.J.; August, E.M.; Hickey, E.R.; Zhang, Q.; Teodoro, M.L.; Rea, S.; Mechtler, K.; Kowalski, J.A.; Homon, C.A.; et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 2007, 25, 473–481. [Google Scholar] [CrossRef]
- Scoumanne, A.; Zhang, J.; Chen, X. PRMT5 is required for cell-cycle progression and p53 tumor suppressor function. Nucleic Acids Res. 2009, 37, 4965–4976. [Google Scholar] [CrossRef]
- Yan, F.; Alinari, L.; Lustberg, M.E.; Martin, L.K.; Cordero-Nieves, H.M.; Banasavadi-Siddegowda, Y.; Virk, S.; Barnholtz-Sloan, J.; Bell, E.H.; Wojton, J.; et al. Genetic validation of the protein arginine methyltransferase PRMT5 as a candidate therapeutic target in glioblastoma. Cancer Res. 2014, 74, 1752–1765. [Google Scholar] [CrossRef]
- Banasavadi-Siddegowda, Y.K.; Welker, A.M.; An, M.; Yang, X.; Zhou, W.; Shi, G.; Imitola, J.; Li, C.; Hsu, S.; Wang, J.; et al. PRMT5 as a druggable target for glioblastoma therapy. Neuro Oncol. 2018, 20, 753–763. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, M.; Buttarelli, F.R.; Arcella, A.; Nobusawa, S.; Donofrio, V.; Oghaki, H.; Giangaspero, F. Prognostic significance of histological grading, p53 status, YKL-40 expression, and IDH1 mutations in pediatric high-grade gliomas. J. Neurooncol. 2010, 99, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Paugh, B.S.; Qu, C.; Jones, C.; Liu, Z.; Adamowicz-Brice, M.; Zhang, J.; Bax, D.A.; Coyle, B.; Barrow, J.; Hargrave, D.; et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J. Clin. Oncol. 2010, 28, 3061–3068. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.W.; Muller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Kondo, Y.; Shen, L.; Cheng, A.S.; Ahmed, S.; Boumber, Y.; Charo, C.; Yamochi, T.; Urano, T.; Furukawa, K.; Kwabi-Addo, B.; et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat. Genet. 2008, 40, 741–750. [Google Scholar] [CrossRef]
- Hashizume, R. Epigenetic Targeted Therapy for Diffuse Intrinsic Pontine Glioma. Neurol. Med. Chir. (Tokyo) 2017, 57, 331–342. [Google Scholar] [CrossRef]
- Bickmore, W.A.; van Steensel, B. Genome architecture: Domain organization of interphase chromosomes. Cell 2013, 152, 1270–1284. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suva, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Danussi, C.; Bose, P.; Parthasarathy, P.T.; Silberman, P.C.; Van Arnam, J.S.; Vitucci, M.; Tang, O.Y.; Heguy, A.; Wang, Y.; Chan, T.A.; et al. Atrx inactivation drives disease-defining phenotypes in glioma cells of origin through global epigenomic remodeling. Nat. Commun. 2018, 9, 1057. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Issa, J.P.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, N.; Sharma, A.R.; Baylin, S.B. Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annu. Rev. Med. 2016, 67, 73–89. [Google Scholar] [CrossRef]
- Dang, L.; Yen, K.; Attar, E.C. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann. Oncol. 2016, 27, 599–608. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Touat, M.; Maher, E.; DeLaFuente, M.; Cloughesy, T.F.; Holdhoff, M.; Cote, G.; Burris, H.; Janku, F.; Huang, R.; et al. ACTR-46. AG120, a first-in-class mutant IDH1 inhibitor in patients with recurrent or progressive IDH1 mutant glioma: Results from the phase 1 glioma expansion cohorts. Neuro Oncol. 2016, 18, vi12. [Google Scholar] [CrossRef][Green Version]
- Yen, K.; Konteatis, Z.; Sui, Z.; Artin, E.; Dang, L.; Straley, K.; Tobin, E.; Campos, C.; Yang, H.; Nagaraja, R.; et al. Abstract B126: AG-881, a brain penetrant, potent, pan-mutant IDH (mIDH) inhibitor for use in mIDH solid and hematologic malignancies. Mol. Cancer Ther. 2018, 17, B126. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Cloughesy, T.F.; Wen, P.Y.; Taylor, J.W.; Maher, E.A.; Arrillaga, I.; Peters, K.B.; Choi, C.; Ellingson, B.M.; Lin, A.P.; et al. A phase I, open label, perioperative study of AG-120 and AG-881 in recurrent IDH1 mutant, low-grade glioma: Results from cohort 1. Int. J. Clin. Oncol. 2019, 37, 2003. [Google Scholar] [CrossRef]
- Mellinghoff, I.; Penas-Prado, M.; Peters, K.; Cloughesy, T.; Burris, H.; Maher, E.; Janku, F.; Cote, G.; De La Fuente, M.; Clarke, J.; et al. ACTR-31. Phase 1 study of AG-881, an inhibitor of mutant IDH1 and IDH2: Results from the recurrent/progressive glioma population. Neuro Oncol. 2018, 20, vi18. [Google Scholar] [CrossRef]
- Pusch, S.; Krausert, S.; Fischer, V.; Balss, J.; Ott, M.; Schrimpf, D.; Capper, D.; Sahm, F.; Eisel, J.; Beck, A.C.; et al. Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo. Acta Neuropathol. 2017, 133, 629–644. [Google Scholar] [CrossRef]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, T.A.; Mukherjee, J.; Viswanath, P.; Ohba, S.; Ronen, S.M.; Bjerkvig, R.; Pieper, R.O. Rapid Conversion of Mutant IDH1 from Driver to Passenger in a Model of Human Gliomagenesis. Mol. Cancer Res. 2016, 14, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Turcan, S.; Fabius, A.W.; Borodovsky, A.; Pedraza, A.; Brennan, C.; Huse, J.; Viale, A.; Riggins, G.J.; Chan, T.A. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget 2013, 4, 1729–1736. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Mazor, T.; Chesnelong, C.; Pankov, A.; Jalbert, L.E.; Hong, C.; Hayes, J.; Smirnov, I.V.; Marshall, R.; Souza, C.F.; Shen, Y.; et al. Clonal expansion and epigenetic reprogramming following deletion or amplification of mutant IDH1. Proc. Natl. Acad. Sci. USA 2017, 114, 10743–10748. [Google Scholar] [CrossRef] [PubMed]
- Moure, C.J.; Diplas, B.H.; Chen, L.H.; Yang, R.; Pirozzi, C.J.; Wang, Z.; Spasojevic, I.; Waitkus, M.S.; He, Y.; Yan, H. CRISPR editing of mutant IDH1 R132H induces a CpG methylation-low state in patient-derived glioma models of G-CIMP. Mol. Cancer Res. 2019, 17, 2042–2050. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef]
- Pellegatta, S.; Valletta, L.; Corbetta, C.; Patane, M.; Zucca, I.; Riccardi Sirtori, F.; Bruzzone, M.G.; Fogliatto, G.; Isacchi, A.; Pollo, B.; et al. Effective immuno-targeting of the IDH1 mutation R132H in a murine model of intracranial glioma. Acta Neuropathol. Commun. 2015, 3, 4. [Google Scholar] [CrossRef]
- Platten, M.; Schilling, D.; Bunse, T.; Sahm, F.; Hueckelhoven, A.; Schenkel, I.; Stevanovic, S.; Schmitt, A.; Laumann, M.; Steinbach, J.P.; et al. A mutation-specific peptide vaccine targeting IDH1R132H in patients with newly diagnosed malignant astrocytomas: A first-in-man multicenter phase I clinical trial of the German Neurooncology Working Group (NOA-16). Int. J. Clin. Oncol. 2016, 34, TPS2082. [Google Scholar] [CrossRef]
- Raynal, N.J.; Si, J.; Taby, R.F.; Gharibyan, V.; Ahmed, S.; Jelinek, J.; Estecio, M.R.; Issa, J.P. DNA methylation does not stably lock gene expression but instead serves as a molecular mark for gene silencing memory. Cancer Res. 2012, 72, 1170–1181. [Google Scholar] [CrossRef]
- Stresemann, C.; Brueckner, B.; Musch, T.; Stopper, H.; Lyko, F. Functional diversity of DNA methyltransferase inhibitors in human cancer cell lines. Cancer Res. 2006, 66, 2794–2800. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Taylor, S.M. Cellular differentiation, cytidine analogs and DNA methylation. Cell 1980, 20, 85–93. [Google Scholar] [CrossRef]
- Issa, J.P. DNA methylation as a therapeutic target in cancer. Clin. Cancer Res. 2007, 13, 1634–1637. [Google Scholar] [CrossRef] [PubMed]
- Bender, C.M.; Pao, M.M.; Jones, P.A. Inhibition of DNA methylation by 5-aza-2′-deoxycytidine suppresses the growth of human tumor cell lines. Cancer Res. 1998, 58, 95–101. [Google Scholar]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D’Souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef]
- Shenker, N.; Flanagan, J.M. Intragenic DNA methylation: Implications of this epigenetic mechanism for cancer research. Br. J. Cancer 2012, 106, 248–253. [Google Scholar] [CrossRef]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef]
- Qin, T.; Jelinek, J.; Si, J.; Shu, J.; Issa, J.P. Mechanisms of resistance to 5-aza-2′-deoxycytidine in human cancer cell lines. Blood 2009, 113, 659–667. [Google Scholar] [CrossRef]
- Kelly, T.K.; De Carvalho, D.D.; Jones, P.A. Epigenetic modifications as therapeutic targets. Nat. Biotechnol. 2010, 28, 1069–1078. [Google Scholar] [CrossRef]
- Tsai, H.C.; Li, H.; Van Neste, L.; Cai, Y.; Robert, C.; Rassool, F.V.; Shin, J.J.; Harbom, K.M.; Beaty, R.; Pappou, E.; et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell 2012, 21, 430–446. [Google Scholar] [CrossRef]
- Issa, J.P.; Kantarjian, H.M. Targeting DNA methylation. Clin. Cancer Res. 2009, 15, 3938–3946. [Google Scholar] [CrossRef] [PubMed]
- Borodovsky, A.; Salmasi, V.; Turcan, S.; Fabius, A.W.; Baia, G.S.; Eberhart, C.G.; Weingart, J.D.; Gallia, G.L.; Baylin, S.B.; Chan, T.A.; et al. 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget 2013, 4, 1737–1747. [Google Scholar] [CrossRef] [PubMed]
- Camiener, G.W.; Smith, C.G. Studies of the enzymatic deamination of cytosine arabinoside. I. Enzyme distribution and species specificity. Biochem. Pharmacol. 1965, 14, 1405–1416. [Google Scholar] [CrossRef]
- Neil, G.L.; Moxley, T.E.; Kuentzel, S.L.; Manak, R.C.; Hanka, L.J. Enhancement by tetrahydrouridine (NSC-112907) of the oral activity of 5-azacytidine (NSC-102816) in L1210 leukemic mice. Cancer Chemother. Rep. 1975, 59, 459–465. [Google Scholar]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef]
- Luo, N.; Nixon, M.J.; Gonzalez-Ericsson, P.I.; Sanchez, V.; Opalenik, S.R.; Li, H.; Zahnow, C.A.; Nickels, M.L.; Liu, F.; Tantawy, M.N.; et al. DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nat. Commun. 2018, 9, 248. [Google Scholar] [CrossRef]
- Zhang, X.; Rao, A.; Sette, P.; Deibert, C.; Pomerantz, A.; Kim, W.J.; Kohanbash, G.; Chang, Y.; Park, Y.; Engh, J.; et al. IDH mutant gliomas escape natural killer cell immune surveillance by downregulation of NKG2D ligand expression. Neuro Oncol. 2016, 18, 1402–1412. [Google Scholar] [CrossRef]
- Riccadonna, C.; Yacoub Maroun, C.; Vuillefroy de Silly, R.; Boehler, M.; Calvo Tardon, M.; Jueliger, S.; Taverna, P.; Barba, L.; Marinari, E.; Pellegatta, S.; et al. Decitabine Treatment of Glioma-Initiating Cells Enhances Immune Recognition and Killing. PLoS ONE 2016, 11, e0162105. [Google Scholar] [CrossRef]
- Bao, L.; Dunham, K.; Lucas, K. MAGE-A1, MAGE-A3, and NY-ESO-1 can be upregulated on neuroblastoma cells to facilitate cytotoxic T lymphocyte-mediated tumor cell killing. Cancer Immunol. Immunother. 2011, 60, 1299–1307. [Google Scholar] [CrossRef]
- Van Nifterik, K.A.; van den Berg, J.; van der Meide, W.F.; Ameziane, N.; Wedekind, L.E.; Steenbergen, R.D.; Leenstra, S.; Lafleur, M.V.; Slotman, B.J.; Stalpers, L.J.; et al. Absence of the MGMT protein as well as methylation of the MGMT promoter predict the sensitivity for temozolomide. Br. J. Cancer 2010, 103, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Moen, E.L.; Stark, A.L.; Zhang, W.; Dolan, M.E.; Godley, L.A. The role of gene body cytosine modifications in MGMT expression and sensitivity to temozolomide. Mol. Cancer Ther. 2014, 13, 1334–1344. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cui, Y.; Naz, A.; Thompson, D.H.; Irudayaraj, J. Decitabine nanoconjugate sensitizes human glioblastoma cells to temozolomide. Mol. Pharm. 2015, 12, 1279–1288. [Google Scholar] [CrossRef]
- Yamashita, A.S.; da Costa Rosa, M.; Borodovsky, A.; Festuccia, W.T.; Chan, T.; Riggins, G.J. Demethylation and epigenetic modification with 5-azacytidine reduces IDH1 mutant glioma growth in combination with temozolomide. Neuro Oncol. 2019, 21, 189–200. [Google Scholar] [CrossRef]
- Robertson, K.D.; Keyomarsi, K.; Gonzales, F.A.; Velicescu, M.; Jones, P.A. Differential mRNA expression of the human DNA methyltransferases (DNMTs) 1, 3a and 3b during the G(0)/G(1) to S phase transition in normal and tumor cells. Nucleic Acids Res. 2000, 28, 2108–2113. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef]
- Kalinina, J.; Ahn, J.; Devi, N.S.; Wang, L.; Li, Y.; Olson, J.J.; Glantz, M.; Smith, T.; Kim, E.L.; Giese, A.; et al. Selective Detection of the D-enantiomer of 2-Hydroxyglutarate in the CSF of Glioma Patients with Mutated Isocitrate Dehydrogenase. Clin. Cancer Res. 2016, 22, 6256–6265. [Google Scholar] [CrossRef]
- Bottcher, M.; Renner, K.; Berger, R.; Mentz, K.; Thomas, S.; Cardenas-Conejo, Z.E.; Dettmer, K.; Oefner, P.J.; Mackensen, A.; Kreutz, M.; et al. D-2-hydroxyglutarate interferes with HIF-1alpha stability skewing T-cell metabolism towards oxidative phosphorylation and impairing Th17 polarization. Oncoimmunology 2018, 7, e1445454. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Wilhelm, D.; Rajky, O.; Kurscheid, S.; Kresl, P.; Wohrer, A.; Marosi, C.; Hegi, M.E.; et al. Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro Oncol. 2017, 19, 1460–1468. [Google Scholar] [CrossRef]
- Kohanbash, G.; Carrera, D.A.; Shrivastav, S.; Ahn, B.J.; Jahan, N.; Mazor, T.; Chheda, Z.S.; Downey, K.M.; Watchmaker, P.B.; Beppler, C.; et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Invest. 2017, 127, 1425–1437. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Zahnow, C.A.; Ahuja, N.; Baylin, S.B. Combining Epigenetic and Immunotherapy to Combat Cancer. Cancer Res. 2016, 76, 1683–1689. [Google Scholar] [CrossRef]
- Wrangle, J.; Wang, W.; Koch, A.; Easwaran, H.; Mohammad, H.P.; Vendetti, F.; Vancriekinge, W.; Demeyer, T.; Du, Z.; Parsana, P.; et al. Alterations of immune response of Non-Small Cell Lung Cancer with Azacytidine. Oncotarget 2013, 4, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.M.; Shen, N.; Xie, S.; Bi, S.Q.; Luo, B.; Lin, Y.D.; Fu, J.; Zhou, S.F.; Luo, G.R.; Xie, X.X.; et al. MAGED4 expression in glioma and upregulation in glioma cell lines with 5-aza-2′-deoxycytidine treatment. Asian Pac. J. Cancer Prev. 2014, 15, 3495–3501. [Google Scholar] [CrossRef] [PubMed]
- Natsume, A.; Wakabayashi, T.; Tsujimura, K.; Shimato, S.; Ito, M.; Kuzushima, K.; Kondo, Y.; Sekido, Y.; Kawatsura, H.; Narita, Y.; et al. The DNA demethylating agent 5-aza-2′-deoxycytidine activates NY-ESO-1 antigenicity in orthotopic human glioma. Int. J. Cancer 2008, 122, 2542–2553. [Google Scholar] [CrossRef] [PubMed]
- Everson, R.G.; Antonios, J.P.; Lisiero, D.N.; Soto, H.; Scharnweber, R.; Garrett, M.C.; Yong, W.H.; Li, N.; Li, G.; Kruse, C.A.; et al. Efficacy of systemic adoptive transfer immunotherapy targeting NY-ESO-1 for glioblastoma. Neuro Oncol. 2016, 18, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Ganji, S.K.; DeBerardinis, R.J.; Hatanpaa, K.J.; Rakheja, D.; Kovacs, Z.; Yang, X.L.; Mashimo, T.; Raisanen, J.M.; Marin-Valencia, I.; et al. 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat. Med. 2012, 18, 624–629. [Google Scholar] [CrossRef]
- Andronesi, O.C.; Kim, G.S.; Gerstner, E.; Batchelor, T.; Tzika, A.A.; Fantin, V.R.; Vander Heiden, M.G.; Sorensen, A.G. Detection of 2-hydroxyglutarate in IDH-mutated glioma patients by in vivo spectral-editing and 2D correlation magnetic resonance spectroscopy. Sci. Transl. Med. 2012, 4, 116ra114. [Google Scholar] [CrossRef]
- Emir, U.E.; Larkin, S.J.; de Pennington, N.; Voets, N.; Plaha, P.; Stacey, R.; Al-Qahtani, K.; McCullagh, J.; Schofield, C.J.; Clare, S.; et al. Noninvasive Quantification of 2-Hydroxyglutarate in Human Gliomas with IDH1 and IDH2 Mutations. Cancer Res. 2016, 76, 43–49. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| IDH inhibitor | ||||||
| Drug | Clinical Trials gov identifier | Phase | Cancer Type | Enrollment | Primary Endpoint(s) | Sponsor(s) |
| AG-120 (Ivosidenib) | NCT02073994 | 1 | IDH1 mutant malignancies | 170 | Safety and tolerability, MTD and/or RP2D | Agios |
| AG-881 | NCT02481154 | 1 | IDH1 and/or IDH2 mutant malignancies | 150 | Safety and tolerability, MTD and/or RP2D | Agios |
| AG-120 AG-881 | NCT03343197 | 1 | IDH1 mutant gliomas | 45 | 2-HG concentration | Agios |
| DS-1001b | NCT03030066 | 1 | IDH1 mutant gliomas | 60 | DLTs | Daiichi Sankyo Co. |
| IDH305 | NCT02381886 | 1 | IDH1 mutant malignancies | 166 | DLTs | Novartis |
| FT-2102 | NCT03684811 | 1b/2 | IDH1 mutant malignancies | 200 | DLTs, RP2D (phase 1) ORR of FT-2102 single agent or in combination with azacytidine (phase 2) | Forma |
| BAY1436032 | NCT02746081 | 1 | IDH1 mutant malignancies | 81 | Safety and tolerability, MTD and/or RP2D | Bayer |
| IDH1 peptide vaccine | ||||||
| Vaccine | Clinical Trials gov identifier | Phase | Cancer Type | Enrollment | Primary Endpoint(s) | Sponsor (s) |
| PEPIDH1M | NCT02193347 | 1 | IDH1 mutant gliomas | 24 | Safety and tolerability | Gary Archer |
| IDH1R132H- DC | NCT02771301 | NA | IDH1 mutant gliomas | 30 | Safety and efficacy | Hebei Yanda Hospital |
| IDH1R132H peptide vaccine | NCT03893903 | 1 | Malignant glioma | 60 | Safety and tolerability | CGRC |
| DNMT inhibitor | ||||||
| Drug | Clinical Trials gov identifier | Phase | Cancer Type | Enrollment | Primary Endpoint(s) | Sponsor (s) |
| Azacitidine (Vidaza) | NCT03666559 | 2 | IDH mutant gliomas | 63 | Progression-Free Survival at 6 months (PFS-6) | AP-HP |
| ASTX727 | NCT03922555 | 1 | IDH mutant gliomas | 18 | MTD | MGH |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.-W.; Turcan, Ş. Epigenetic Reprogramming for Targeting IDH-Mutant Malignant Gliomas. Cancers 2019, 11, 1616. https://doi.org/10.3390/cancers11101616
Park J-W, Turcan Ş. Epigenetic Reprogramming for Targeting IDH-Mutant Malignant Gliomas. Cancers. 2019; 11(10):1616. https://doi.org/10.3390/cancers11101616
Chicago/Turabian StylePark, Jong-Whi, and Şevin Turcan. 2019. "Epigenetic Reprogramming for Targeting IDH-Mutant Malignant Gliomas" Cancers 11, no. 10: 1616. https://doi.org/10.3390/cancers11101616
APA StylePark, J.-W., & Turcan, Ş. (2019). Epigenetic Reprogramming for Targeting IDH-Mutant Malignant Gliomas. Cancers, 11(10), 1616. https://doi.org/10.3390/cancers11101616