Mutational Landscape of the BAP1 Locus Reveals an Intrinsic Control to Regulate the miRNA Network and the Binding of Protein Complexes in Uveal Melanoma

 ,
,  ,
,  , ,
, ,  ,
,  , ,
, ,
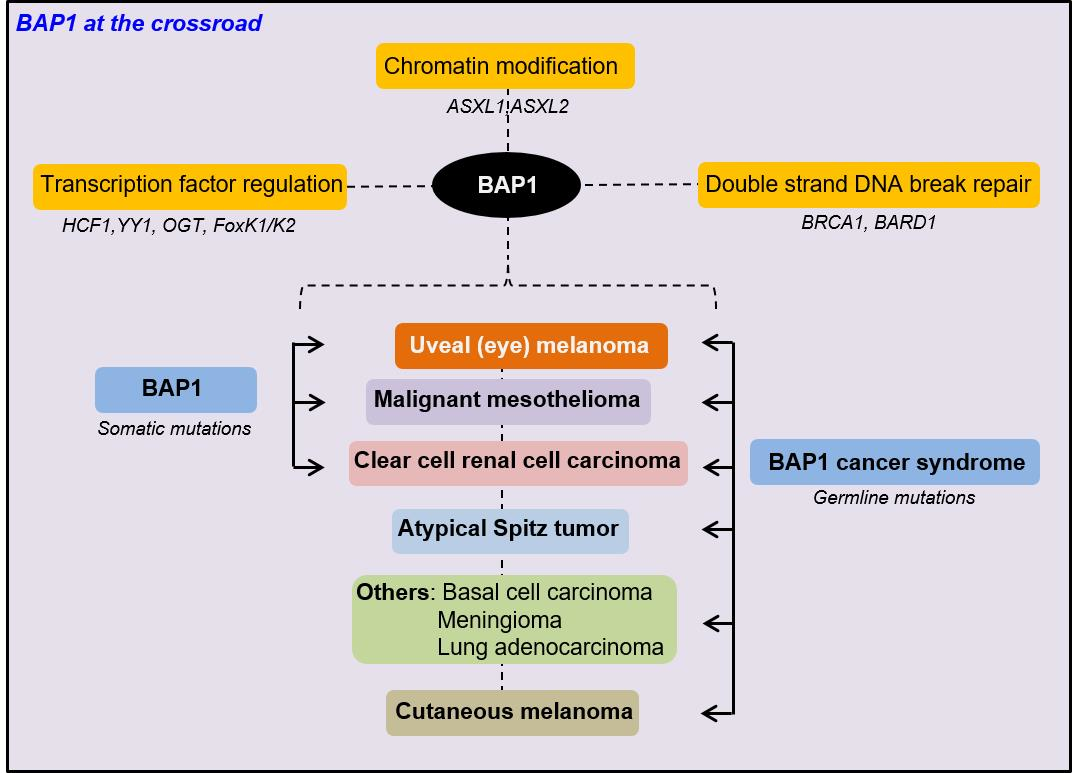
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
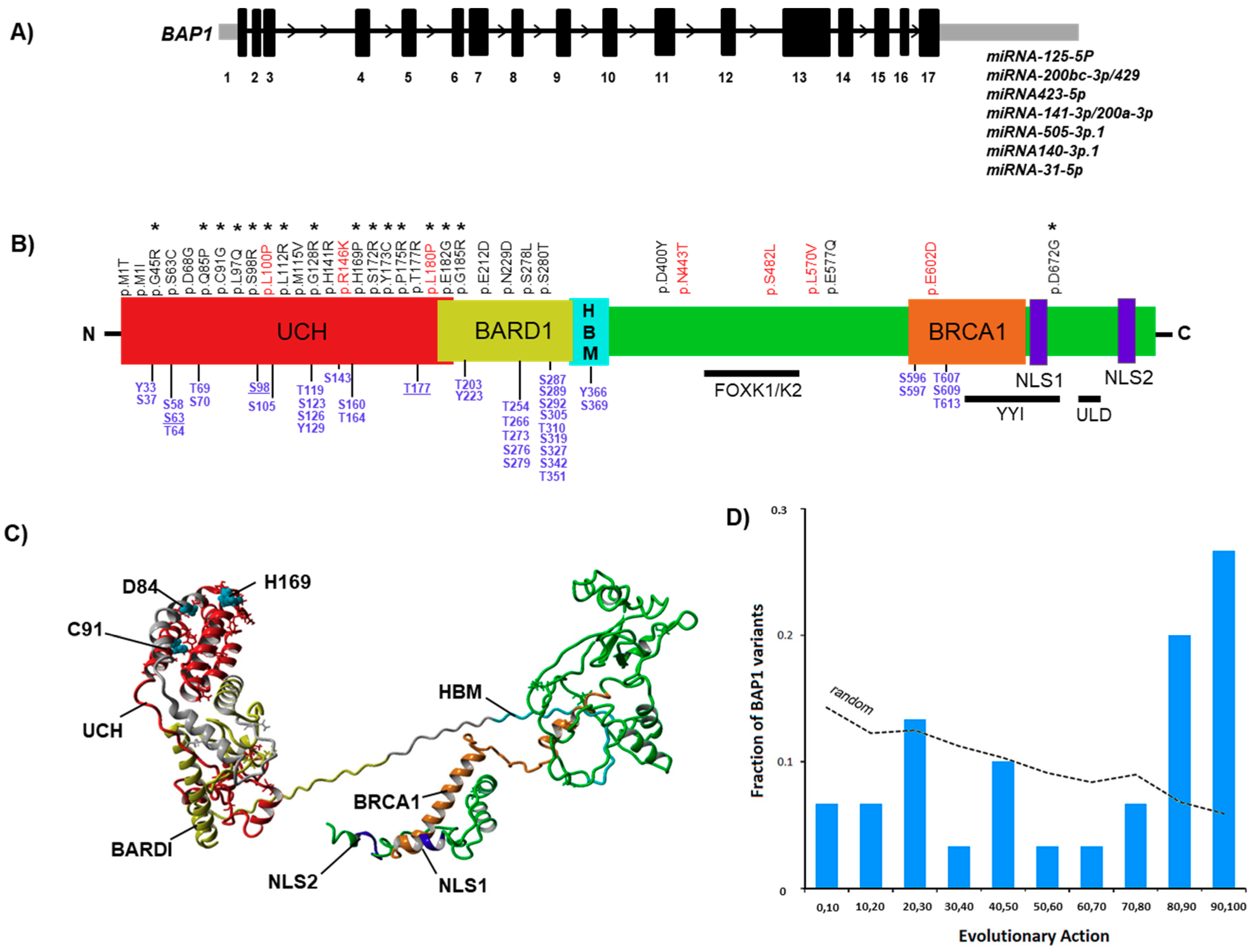
2.1. BAP1 Mutational Landscapes and Structural (Insilco) Analysis
2.2. Evolutionary Action (EA) Score
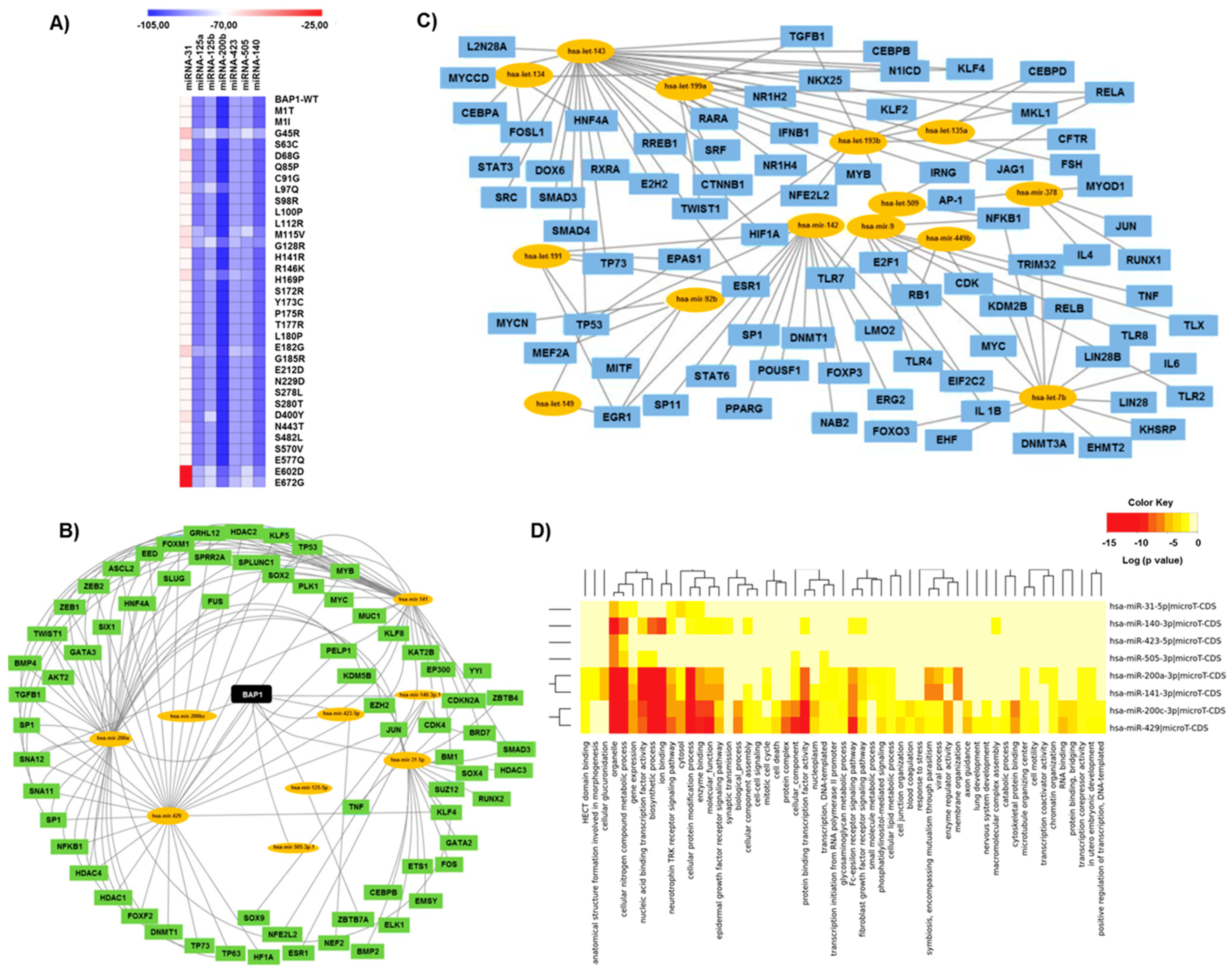
2.3. MiRNA Analysis and Transcription Factor Regulatory Networks
2.4. BAP1 Gene Expression Datasets and Prognostic/Diagnostic Estimations
3. Results
3.1. Interpretation of BAP1 Variants and the Respective Evolutionary Action Score
3.2. In Silico Characterization and Structural Modeling of the BAP1 Protein
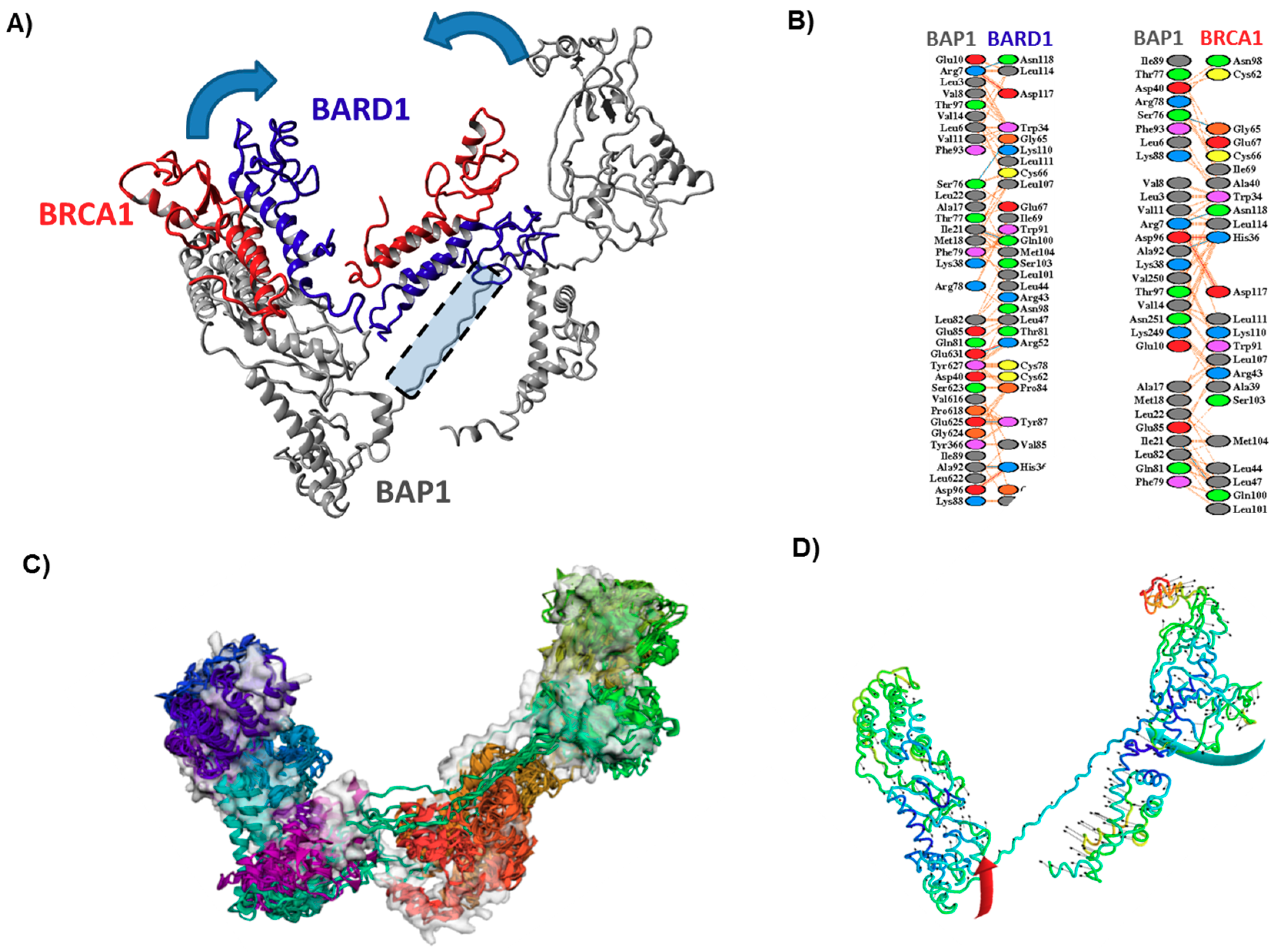
3.3. Effect of Mutations on the Main Fold of BAP1
3.4. Effect of Mutations on the Interaction of BAP1 with Other Proteins
3.5. Impact of Variants on the Binding Affinity of BAP1-Associated miRNAs
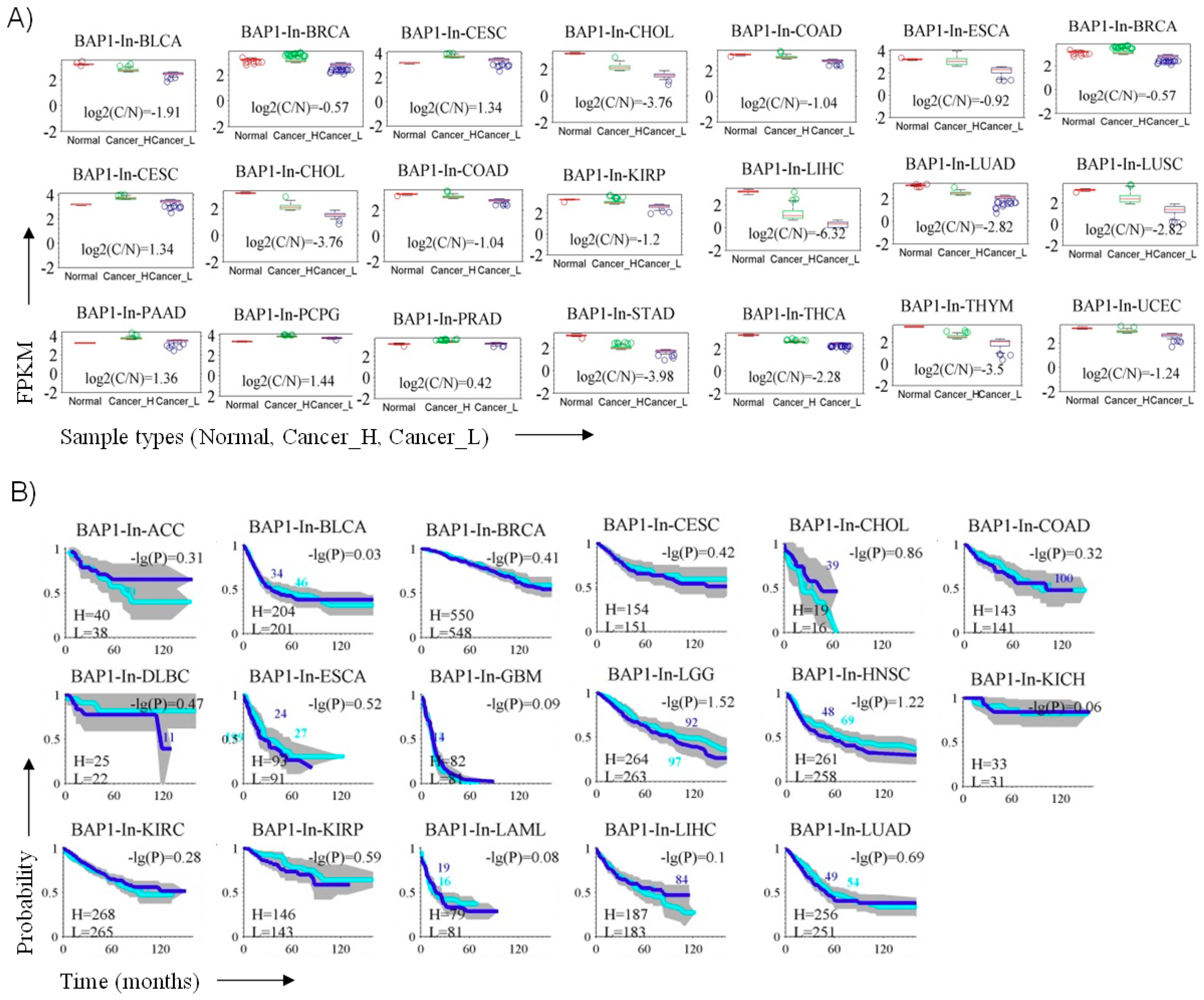
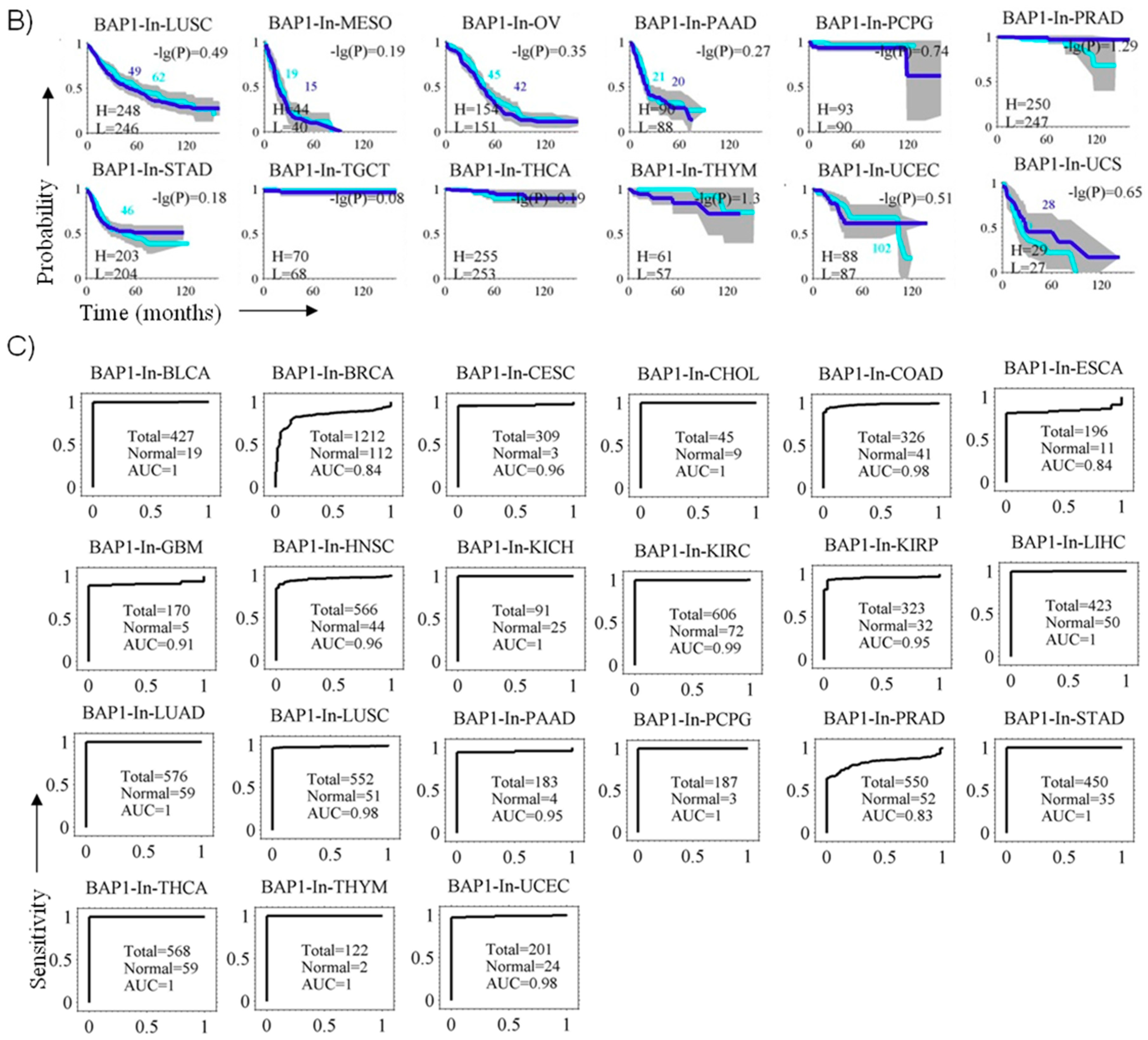
3.6. BAP1 Gene Expression Penetrance and Prevalence in Cancer Spectrum
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviation List
| UCH | Ubiquitin carboxyl hydrolase domain |
| HBM | HCF1 binding motif |
| NLS | Nuclear localization signals |
| LAML | Acute Myeloid Leukemia |
| ACC | Adrenocortical carcinoma |
| BLCA | Bladder Urothelial Carcinoma |
| LGG | Brain Lower Grade Glioma |
| BRCA | Breast invasive carcinoma |
| CESC | Cervical squamous cell carcinoma and endocervical adenocarcinoma |
| CHOL | Cholangiocarcinoma |
| LCML | Chronic Myelogenous Leukemia |
| COAD | Colon adenocarcinoma |
| ESCA | Esophageal carcinoma |
| GBM | Glioblastoma multiforme |
| HNSC | Head and Neck squamous cell carcinoma |
| BAP1 | BRCA1-associated protein 1 |
| KICH | Kidney Chromophobe |
| KIRC | Kidney renal clear cell carcinoma |
| KIRP | Kidney renal papillary cell carcinoma |
| LIHC | Liver hepatocellular carcinoma |
| LUAD | Lung adenocarcinoma |
| LUSC | Lung squamous cell carcinoma |
| DLBC | Lymphoid Neoplasm Diffuse Large B-cell Lymphoma |
| MESO | Mesothelioma |
| OV | Ovarian serous cystadenocarcinoma |
| PAAD | Pancreatic adenocarcinoma |
| PCPG | Pheochromocytoma and Paraganglioma |
| PRAD | Prostate adenocarcinoma |
| STAD | Stomach adenocarcinoma |
| TGCT | Testicular Germ Cell Tumors |
| THYM | Thymoma |
| THCA | Thyroid carcinoma |
| UCS | Uterine Carcinosarcoma |
| UCEC | Uterine Corpus Endometrial Carcinoma |
References
- Ventii, K.H.; Devi, N.S.; Friedrich, K.L.; Chernova, T.A.; Tighiouart, M.; Van Meir, E.G.; Wilkinson, K.D. BRCA1-associated protein-1 is a tumor suppressor that requires deubiquitinating activity and nuclear localization. Cancer Res. 2008, 68, 6953–6962. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef] [PubMed]
- Bott, M.; Brevet, M.; Taylor, B.S.; Shimizu, S.; Ito, T.; Wang, L.; Creaney, J.; Lake, R.A.; Zakowski, M.F.; Reva, B.; et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011, 43, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Pena-Llopis, S.; Vega-Rubin-de-Celis, S.; Liao, A.; Leng, N.; Pavia-Jimenez, A.; Wang, S.; Yamasaki, T.; Zhrebker, L.; Sivanand, S.; Spence, P.; et al. BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 2012, 44, 751–759. [Google Scholar] [CrossRef]
- Carbone, M.; Yang, H.; Pass, H.I.; Krausz, T.; Testa, J.R.; Gaudino, G. BAP1 and cancer. Nat. Rev. Cancer 2013, 13, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, N.; Thornton, S.; Coupland, S.E.; Coulson, J.M.; Sacco, J.J.; Krishna, Y.; Heimann, H.; Taktak, A.; Cebulla, C.M.; Abdel-Rahman, M.H.; et al. Patterns of BAP1 protein expression provide insights into prognostic significance and the biology of uveal melanoma. J. Pathol. Clin. Res. 2018, 4, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Herwig-Carl, M.C.; Sharma, A.; Moulin, A.; Strack, C.; Loeffler, K.U. BAP1 Immunostaining in Uveal Melanoma: Potentials and Pitfalls. Ocul. Oncol. Pathol. 2018, 4, 297. [Google Scholar] [CrossRef]
- Bononi, A.; Giorgi, C.; Patergnani, S.; Larson, D.; Verbruggen, K.; Tanji, M.; Pellegrini, L.; Signorato, V.; Olivetto, F.; Pastorino, S.; et al. BAP1 regulates IP3R3-mediated Ca(2+) flux to mitochondria suppressing cell transformation. Nature 2017, 546, 549–553. [Google Scholar] [CrossRef]
- Berus, T.; Halon, A.; Markiewicz, A.; Orlowska-Heitzman, J.; Romanowska-Dixon, B.; Donizy, P. Clinical, Histopathological and Cytogenetic Prognosticators in Uveal Melanoma—A Comprehensive Review. Anticancer Res. 2017, 37, 6541–6549. [Google Scholar] [CrossRef]
- Mensink, H.W.; Vaarwater, J.; Kilic, E.; Naus, N.C.; Mooy, N.; Luyten, G.; Bruggenwirth, H.T.; Paridaens, D.; de Klein, A. Chromosome 3 intratumor heterogeneity in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2009, 50, 500–504. [Google Scholar] [CrossRef]
- Smit, K.N.; Chang, J.; Derks, K.; Vaarwater, J.; Brands, T.; Verdijk, R.M.; Wiemer, E.A.C.; Mensink, H.W.; Pothof, J.; de Klein, A.; et al. Aberrant MicroRNA Expression and Its Implications for Uveal Melanoma Metastasis. Cancers 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Romano, G.L.; Salemi, R.; Bucolo, C.; Tomasello, B.; Lupo, G.; Anfuso, C.D.; Spandidos, D.A.; Libra, M.; Candido, S. Prognostic significance of deregulated microRNAs in uveal melanomas. Mol. Med. Rep. 2019, 19, 2599–2610. [Google Scholar] [CrossRef] [PubMed]
- Xin, X.; Zhang, Y.; Ling, F.; Wang, L.; Sheng, X.; Qin, L.; Zhao, X. Identification of a nine-miRNA signature for the prognosis of Uveal Melanoma. Exp. Eye Res. 2019, 180, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.; Tang, J.; Han, Y.; Wang, D. Co-expression modules construction by WGCNA and identify potential prognostic markers of uveal melanoma. Exp. Eye Res. 2018, 166, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Brzovic, P.S.; Rajagopal, P.; Hoyt, D.W.; King, M.C.; Klevit, R.E. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat. Struct. Biol. 2001, 8, 833–837. [Google Scholar] [CrossRef]
- Pieper, U.; Webb, B.M.; Dong, G.Q.; Schneidman-Duhovny, D.; Fan, H.; Kim, S.J.; Khuri, N.; Spill, Y.G.; Weinkam, P.; Hammel, M.; et al. ModBase, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 2014, 42, D336–D346. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef]
- Kuriata, A.; Gierut, A.M.; Oleniecki, T.; Ciemny, M.P.; Kolinski, A.; Kurcinski, M.; Kmiecik, S. CABS-flex 2.0: A web server for fast simulations of flexibility of protein structures. Nucleic Acids Res. 2018, 46, W338–W343. [Google Scholar] [CrossRef]
- Lopez-Blanco, J.R.; Aliaga, J.I.; Quintana-Orti, E.S.; Chacon, P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Blom, N.; Sicheritz-Ponten, T.; Gupta, R.; Gammeltoft, S.; Brunak, S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 2004, 4, 1633–1649. [Google Scholar] [CrossRef] [PubMed]
- Katsonis, P.; Lichtarge, O. A formal perturbation equation between genotype and phenotype determines the Evolutionary Action of protein-coding variations on fitness. Genome Res. 2014, 24, 2050–2058. [Google Scholar] [CrossRef] [PubMed]
- Lichtarge, O.; Bourne, H.R.; Cohen, F.E. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 1996, 257, 342–358. [Google Scholar] [CrossRef] [PubMed]
- Mihalek, I.; Res, I.; Lichtarge, O. A family of evolution-entropy hybrid methods for ranking protein residues by importance. J. Mol. Biol. 2004, 336, 1265–1282. [Google Scholar] [CrossRef]
- Rehmsmeier, M.; Steffen, P.; Hochsmann, M.; Giegerich, R. Fast and effective prediction of microRNA/target duplexes. RNA 2004, 10, 1507–1517. [Google Scholar] [CrossRef]
- Kruger, J.; Rehmsmeier, M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34, W451–WW454. [Google Scholar] [CrossRef]
- Wang, J.; Lu, M.; Qiu, C.; Cui, Q. TransmiR: A transcription factor-microRNA regulation database. Nucleic Acids Res. 2010, 38, D119–D122. [Google Scholar] [CrossRef]
- Cline, M.S.; Smoot, M.; Cerami, E.; Kuchinsky, A.; Landys, N.; Workman, C.; Christmas, R.; Avila-Campilo, I.; Creech, M.; Gross, B.; et al. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2007, 2, 2366–2382. [Google Scholar] [CrossRef]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef]
- Samur, M.K. RTCGAToolbox: A new tool for exporting TCGA Firehose data. PLoS ONE 2014, 9, e106397. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; Sharma, A.; Peters, S.; Aretz, S.; Biswas, A.; Holz, F.G.; Loeffler, K.U.; Herwig-Carl, M.C. Basal cell carcinomas developing independently from BAP1-tumor predisposition syndrome in a patient with bilateral uveal melanoma: Diagnostic challenges to identify patients with BAP1-TPDS. Genes Chromosomes Cancer 2019, 58, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Stei, M.M.; Frohlich, H.; Holz, F.G.; Loeffler, K.U.; Herwig-Carl, M.C. Genetic and epigenetic insights into uveal melanoma. Clin. Genet. 2018, 93, 952–961. [Google Scholar] [CrossRef]
- Kuznetsov, J.N.; Aguero, T.H.; Owens, D.A.; Kurtenbach, S.; Field, M.G.; Durante, M.A.; Rodriguez, D.A.; King, M.L.; Harbour, J.W. BAP1 regulates epigenetic switch from pluripotency to differentiation in developmental lineages giving rise to BAP1-mutant cancers. Sci. Adv. 2019, 5, eaax1738. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Katsonis, P.; Lichtarge, O. Objective assessment of the evolutionary action equation for the fitness effect of missense mutations across CAGI-blinded contests. Hum. Mutat. 2017, 38, 1072–1084. [Google Scholar] [CrossRef]
- Clarke, C.N.; Katsonis, P.; Hsu, T.K.; Koire, A.M.; Silva-Figueroa, A.; Christakis, I.; Williams, M.D.; Kutahyalioglu, M.; Kwatampora, L.; Xi, Y.; et al. Comprehensive Genomic Characterization of Parathyroid Cancer Identifies Novel Candidate Driver Mutations and Core Pathways. J. Endocr. Soc. 2019, 3, 544–559. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Hanpude, P.; Maiti, T.K. Cancer associated missense mutations in BAP1 catalytic domain induce amyloidogenic aggregation: A new insight in enzymatic inactivation. Sci. Rep. 2015, 5, 18462. [Google Scholar] [CrossRef]
- Okino, Y.; Machida, Y.; Frankland-Searby, S.; Machida, Y.J. BRCA1-associated protein 1 (BAP1) deubiquitinase antagonizes the ubiquitin-mediated activation of FoxK2 target genes. J. Biol. Chem. 2015, 290, 1580–1591. [Google Scholar] [CrossRef]
- Yu, H.; Mashtalir, N.; Daou, S.; Hammond-Martel, I.; Ross, J.; Sui, G.; Hart, G.W.; Rauscher, F.J., 3rd; Drobetsky, E.; Milot, E.; et al. The ubiquitin carboxyl hydrolase BAP1 forms a ternary complex with YY1 and HCF-1 and is a critical regulator of gene expression. Mol. Cell Biol. 2010, 30, 5071–5085. [Google Scholar] [CrossRef]
- Sahtoe, D.D.; van Dijk, W.J.; Ekkebus, R.; Ovaa, H.; Sixma, T.K. BAP1/ASXL1 recruitment and activation for H2A deubiquitination. Nat. Commun. 2016, 7, 10292. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, R.A.; Sobhian, B.; Pathania, S.; Cantor, S.B.; Nakatani, Y.; Livingston, D.M. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006, 20, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Wu, W.; Koike, A.; Kojima, R.; Gomi, H.; Fukuda, M.; Ohta, T. BRCA1-associated protein 1 interferes with BRCA1/BARD1 RING heterodimer activity. Cancer Res. 2009, 69, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Reimand, J.; Wagih, O.; Bader, G.D. The mutational landscape of phosphorylation signaling in cancer. Sci. Rep. 2013, 3, 2651. [Google Scholar] [CrossRef]
- Radivojac, P.; Baenziger, P.H.; Kann, M.G.; Mort, M.E.; Hahn, M.W.; Mooney, S.D. Gain and loss of phosphorylation sites in human cancer. Bioinformatics 2008, 24, i241–i247. [Google Scholar] [CrossRef]
- Kappelmann, M.; Kuphal, S.; Meister, G.; Vardimon, L.; Bosserhoff, A.K. MicroRNA miR-125b controls melanoma progression by direct regulation of c-Jun protein expression. Oncogene 2013, 32, 2984–2991. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, Q.; Chang, H.; Qiu, X.; Wang, E. [hsa-miR-125a-5p Enhances Invasion in Non-small Cell Lung Carcinoma Cell Lines by Upregulating Rock-1.]. Zhongguo Fei Ai Za Zhi 2009, 12, 1069–1073. [Google Scholar] [CrossRef]
- Ding, M.; Sun, X.; Zhong, J.; Zhang, C.; Tian, Y.; Ge, J.; Zhang, C.Y.; Zen, K.; Wang, J.J.; Wang, C. Decreased miR-200a-3p is a key regulator of renal carcinoma growth and migration by directly targeting CBL. J. Cell Biochem. 2018, 119, 9974–9985. [Google Scholar] [CrossRef]
- Gui, Z.; Luo, F.; Yang, Y.; Shen, C.; Li, S.; Xu, J. Oridonin inhibition and miR200b3p/ZEB1 axis in human pancreatic cancer. Int. J. Oncol. 2017, 50, 111–120. [Google Scholar] [CrossRef]
- Yang, H.; Fu, H.; Wang, B.; Zhang, X.; Mao, J.; Li, X.; Wang, M.; Sun, Z.; Qian, H.; Xu, W. Exosomal miR-423-5p targets SUFU to promote cancer growth and metastasis and serves as a novel marker for gastric cancer. Mol. Carcinog. 2018, 57, 1223–1236. [Google Scholar] [CrossRef]
- Lu, Z.; He, Q.; Liang, J.; Li, W.; Su, Q.; Chen, Z.; Wan, Q.; Zhou, X.; Cao, L.; Sun, J.; et al. miR-31-5p Is a Potential Circulating Biomarker and Therapeutic Target for Oral Cancer. Mol. Ther. Nucleic Acids 2019, 16, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, S.; Bjornetro, T.; Lyckander, L.G.; Flatmark, K.; Dueland, S.; Samiappan, R.; Johansen, C.; Kalanxhi, E.; Ree, A.H.; Redalen, K.R. Circulating Exosomal miR-141-3p and miR-375 in Metastatic Progression of Rectal Cancer. Transl. Oncol. 2019, 12, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Singh, H.; Trinidad, C.M.; Albarracin, C.T.; Hunt, K.K.; Bedrosian, I. The isomiR-140-3p-regulated mevalonic acid pathway as a potential target for prevention of triple negative breast cancer. Breast Cancer Res. 2018, 20, 150. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, A.; Biswas, A.; Liu, H.; Sen, S.; Paruchuri, A.; Katsonis, P.; Lichtarge, O.; Chand Dakal, T.; Maulik, U.; Gromiha, M.M.; et al. Mutational Landscape of the BAP1 Locus Reveals an Intrinsic Control to Regulate the miRNA Network and the Binding of Protein Complexes in Uveal Melanoma. Cancers 2019, 11, 1600. https://doi.org/10.3390/cancers11101600
Sharma A, Biswas A, Liu H, Sen S, Paruchuri A, Katsonis P, Lichtarge O, Chand Dakal T, Maulik U, Gromiha MM, et al. Mutational Landscape of the BAP1 Locus Reveals an Intrinsic Control to Regulate the miRNA Network and the Binding of Protein Complexes in Uveal Melanoma. Cancers. 2019; 11(10):1600. https://doi.org/10.3390/cancers11101600
Chicago/Turabian StyleSharma, Amit, Arijit Biswas, Hongde Liu, Sagnik Sen, Anoosha Paruchuri, Panagiotis Katsonis, Olivier Lichtarge, Tikam Chand Dakal, Ujjwal Maulik, M. Michael Gromiha, and et al. 2019. "Mutational Landscape of the BAP1 Locus Reveals an Intrinsic Control to Regulate the miRNA Network and the Binding of Protein Complexes in Uveal Melanoma" Cancers 11, no. 10: 1600. https://doi.org/10.3390/cancers11101600
APA StyleSharma, A., Biswas, A., Liu, H., Sen, S., Paruchuri, A., Katsonis, P., Lichtarge, O., Chand Dakal, T., Maulik, U., Gromiha, M. M., Bandyopadhyay, S., Ludwig, M., Holz, F. G., Loeffler, K. U., & Herwig-Carl, M. C. (2019). Mutational Landscape of the BAP1 Locus Reveals an Intrinsic Control to Regulate the miRNA Network and the Binding of Protein Complexes in Uveal Melanoma. Cancers, 11(10), 1600. https://doi.org/10.3390/cancers11101600