Single-Cell Analysis of Circulating Tumor Cells: Why Heterogeneity Matters


Abstract
1. Introduction
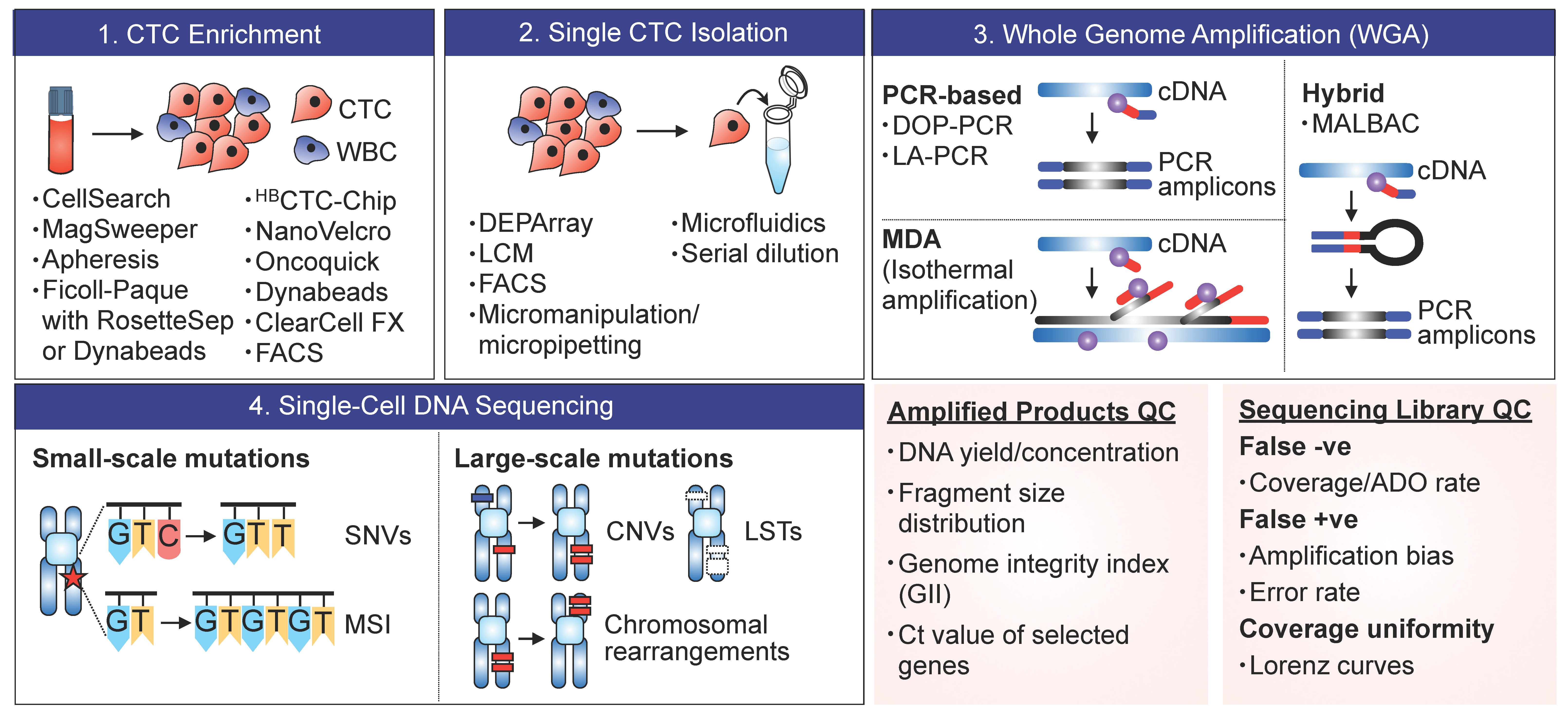
2. Methods and Technologies
2.1. CTC Enrichment
2.2. Single-Cell Isolation
2.3. Whole-Genome Amplification (WGA)
2.4. Sequencing and Profiling
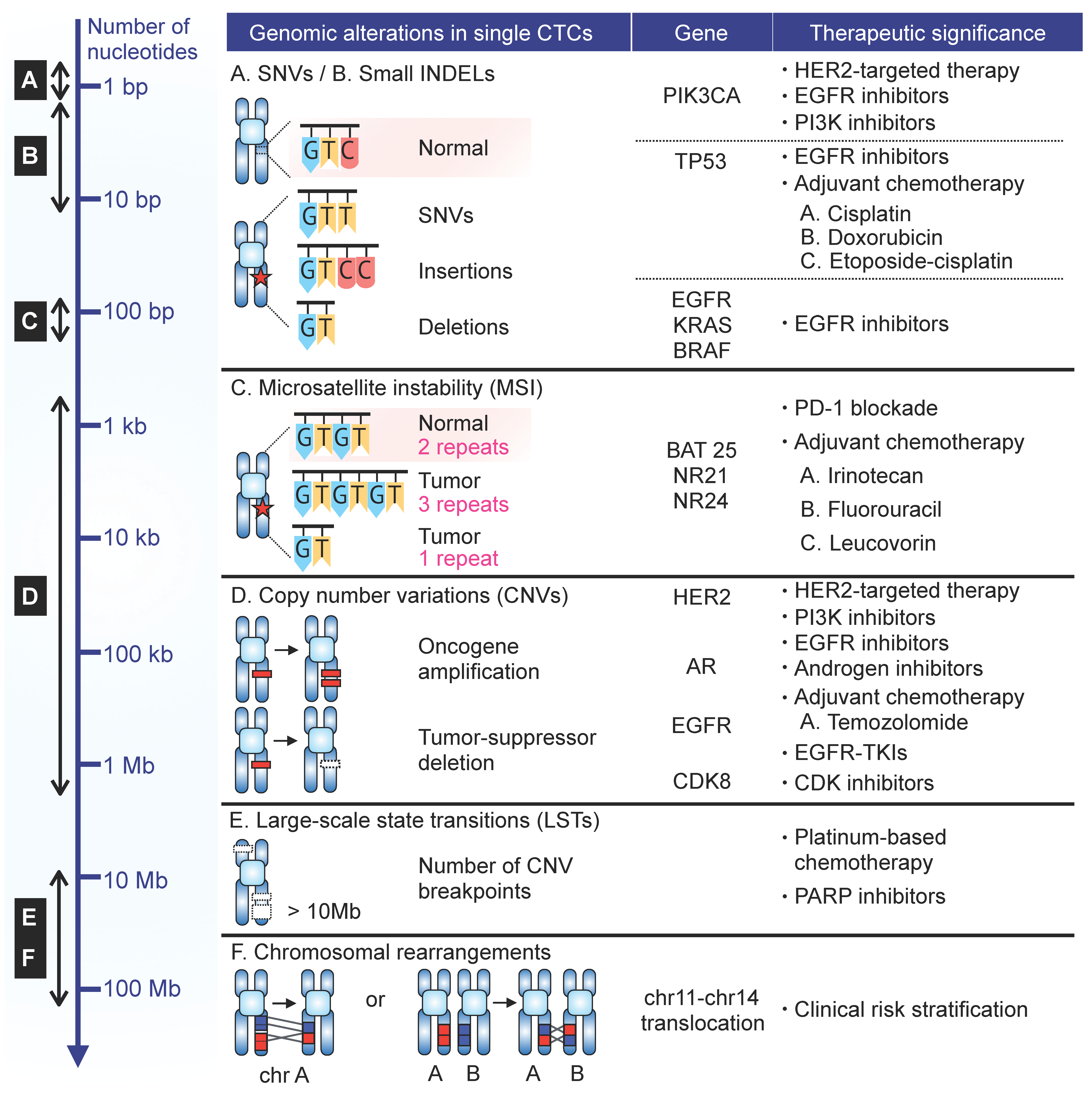
3. CTC Heterogeneity and Clinical Impact
3.1. Single Nucelotide Variation (SNV)
3.1.1. PIK3CA
3.1.2. TP53
3.1.3. EGFR
3.1.4. KRAS
3.1.5. BRAF
3.2. Microsatellite Instability (MSI)
3.3. Copy-Number Variation (CNV)
3.4. Chromosomal Breakpoints
3.5. Chromosomal Rearrangement
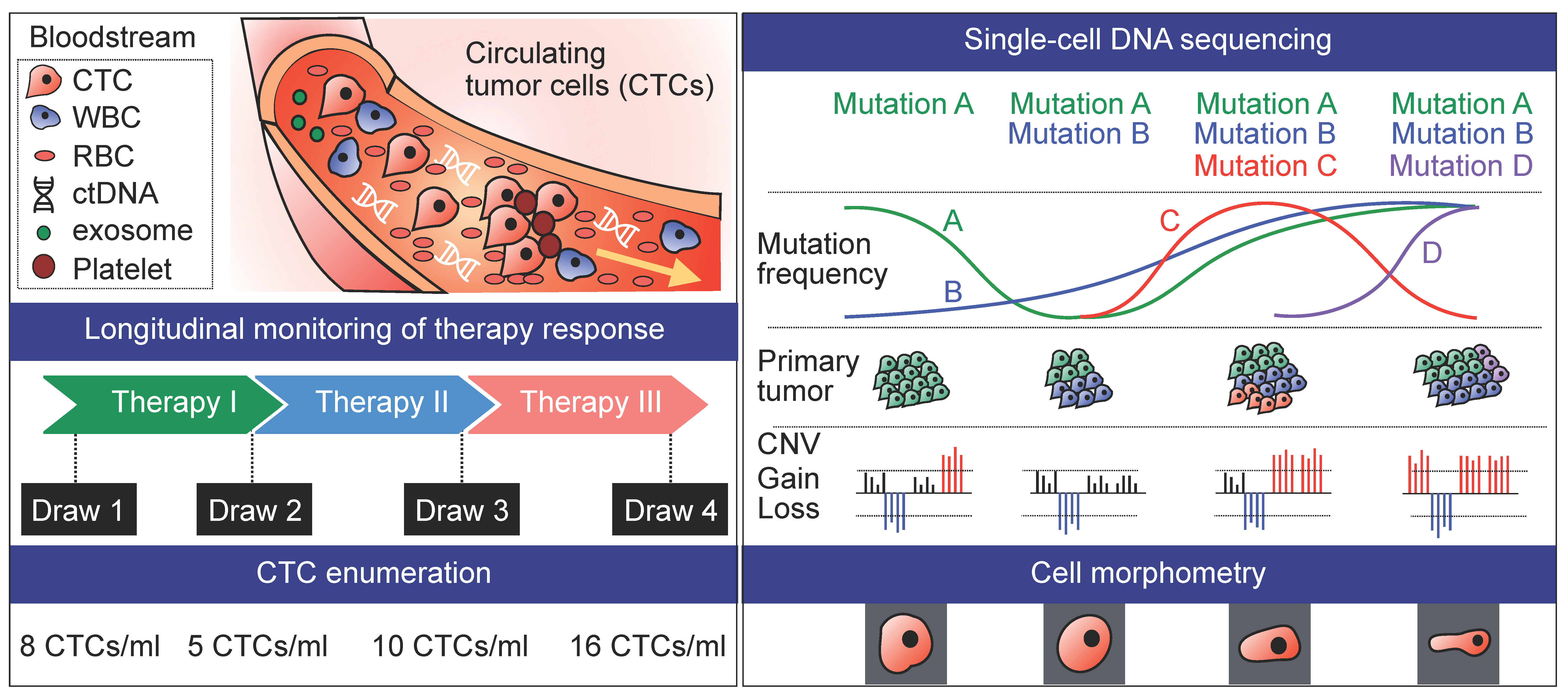
4. Longitudinal Studies
5. Challenges and Emerging Technologies
5.1. Fresh-Frozen Versus Formalin-Fixed
5.2. Increasing Number of CTC Libraries
5.3. Multidimensional Measurements
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fidler, I.J.; Hart, I.R. Biological diversity in metastatic neoplasms: Origins and implications. Science 1982, 217, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Suvà, M.L. Deciphering Human Tumor Biology by Single-Cell Expression Profiling. Annu. Rev. Cancer Biol. 2019, 3, 151–166. [Google Scholar] [CrossRef]
- Shi, X.; Chakraborty, P.; Chaudhuri, A. Unmasking tumor heterogeneity and clonal evolution by single-cell analysis. J. Cancer Metastasis Treat. 2018, 4, 47. [Google Scholar] [CrossRef][Green Version]
- Lim, S.B.; Yeo, T.; Lee, W.D.; Bhagat, A.A.S.; Tan, S.J.; Tan, D.S.W.; Lim, W.T.; Lim, C.T. Addressing cellular heterogeneity in tumor and circulation for refined prognostication. Proc. Natl. Acad. Sci. USA 2019, 116, 17957–17962. [Google Scholar] [CrossRef]
- Lim, S.B.; Tan, S.J.; Lim, W.T.; Lim, C.T. An extracellular matrix-related prognostic and predictive indicator for early-stage non-small cell lung cancer. Nat. Commun. 2017, 8, 1734. [Google Scholar] [CrossRef]
- Lim, S.B.; Lim, C.T.; Lim, D.W.-T. MO2-6-3Matrisomal abnormality: A predictive biomarker for cancer immunotherapy. Ann. Oncol. 2019, 30. [Google Scholar] [CrossRef]
- Lim, S.B.; Chua, M.L.K.; Yeong, J.P.S.; Tan, S.J.; Lim, W.-T.; Lim, C.T. Pan-cancer analysis connects tumor matrisome to immune response. NPJ Precis. Oncol. 2019, 3, 15. [Google Scholar] [CrossRef]
- Francart, M.E.; Lambert, J.; Vanwynsberghe, A.M.; Thompson, E.W.; Bourcy, M.; Polette, M.; Gilles, C. Epithelial-mesenchymal plasticity and circulating tumor cells: Travel companions to metastases. Dev. Dyn. 2018, 247, 432–450. [Google Scholar] [CrossRef]
- Menon, N.V.; Lim, S.B.; Lim, C.T. Microfluidics for personalized drug screening of cancer. Curr. Opin. Pharmacol. 2019, 48, 155–161. [Google Scholar] [CrossRef]
- Koh, Y.; Yagi, S.; Akamatsu, H.; Kanai, K.; Hayata, A.; Tokudome, N.; Akamatsu, K.; Higuchi, M.; Kanbara, H.; Nakanishi, M.; et al. Heterogeneous Expression of Programmed Death Receptor-ligand 1 on Circulating Tumor Cells in Patients with Lung Cancer. Clin. Lung Cancer 2019, 20, 270–277.e271. [Google Scholar] [CrossRef]
- Grillet, F.; Bayet, E.; Villeronce, O.; Zappia, L.; Lagerqvist, E.L.; Lunke, S.; Charafe-Jauffret, E.; Pham, K.; Molck, C.; Rolland, N.; et al. Circulating tumour cells from patients with colorectal cancer have cancer stem cell hallmarks in ex vivo culture. Gut 2017, 66, 1802. [Google Scholar] [CrossRef] [PubMed]
- Jordan, N.V.; Bardia, A.; Wittner, B.S.; Benes, C.; Ligorio, M.; Zheng, Y.; Yu, M.; Sundaresan, T.K.; Licausi, J.A.; Desai, R.; et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature 2016, 537, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Agnoletto, C.; Corra, F.; Minotti, L.; Baldassari, F.; Crudele, F.; Cook, W.J.J.; Di Leva, G.; d’Adamo, A.P.; Gasparini, P.; Volinia, S. Heterogeneity in Circulating Tumor Cells: The Relevance of the Stem-Cell Subset. Cancers 2019, 11, 483. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.B.; Di Lee, W.; Vasudevan, J.; Lim, W.-T.; Lim, C.T. Liquid biopsy: One cell at a time. NPJ Precis. Oncol. 2019, 3, 23. [Google Scholar] [CrossRef]
- Racila, E.; Euhus, D.; Weiss, A.J.; Rao, C.; McConnell, J.; Terstappen, L.W.M.M.; Uhr, J.W. Detection and characterization of carcinoma cells in the blood. Proc. Natl. Acad. Sci. USA 1998, 95, 4589. [Google Scholar] [CrossRef]
- Heitzer, E.; Auer, M.; Gasch, C.; Pichler, M.; Ulz, P.; Hoffmann, E.M.; Lax, S.; Waldispuehl-Geigl, J.; Mauermann, O.; Lackner, C.; et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 2013, 73, 2965–2975. [Google Scholar] [CrossRef]
- Lambros, M.B.; Seed, G.; Sumanasuriya, S.; Gil, V.; Crespo, M.; Fontes, M.; Chandler, R.; Mehra, N.; Fowler, G.; Ebbs, B.; et al. Single-Cell Analyses of Prostate Cancer Liquid Biopsies Acquired by Apheresis. Clin. Cancer Res. 2018, 24, 5635–5644. [Google Scholar] [CrossRef]
- Pestrin, M.; Salvianti, F.; Galardi, F.; De Luca, F.; Turner, N.; Malorni, L.; Pazzagli, M.; Di Leo, A.; Pinzani, P. Heterogeneity of PIK3CA mutational status at the single cell level in circulating tumor cells from metastatic breast cancer patients. Mol. Oncol. 2015, 9, 749–757. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, L.; Feng, L.; Zhang, W.; Xiao, T.; Di, X.; Chen, G.; Zhang, K. Single nucleotide variant profiles of viable single circulating tumour cells reveal CTC behaviours in breast cancer. Oncol. Rep. 2018, 39, 2147–2159. [Google Scholar] [CrossRef]
- Yin, J.; Wang, Z.; Li, G.; Lin, F.; Shao, K.; Cao, B.; Hou, Y. Characterization of circulating tumor cells in breast cancer patients by spiral microfluidics. Cell Biol. Toxicol. 2019, 35, 59–66. [Google Scholar] [CrossRef]
- Lohr, J.G.; Kim, S.; Gould, J.; Knoechel, B.; Drier, Y.; Cotton, M.J.; Gray, D.; Birrer, N.; Wong, B.; Ha, G.; et al. Genetic interrogation of circulating multiple myeloma cells at single-cell resolution. Sci. Transl. Med. 2016, 8, 363ra147. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Wong, D.J.; Ooi, C.C.; Kurtz, D.M.; Vermesh, O.; Aalipour, A.; Suh, S.; Pian, K.L.; Chabon, J.J.; Lee, S.H.; et al. Molecular profiling of single circulating tumor cells from lung cancer patients. Proc. Natl. Acad. Sci. USA 2016, 113, E8379–E8386. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, N.; Hu, P.; Bedard, P.; Clemons, M.; McCready, D.; Done, S.J. Identification of genomic signatures in circulating tumor cells from breast cancer. Int. J. Cancer 2015, 137, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Adalsteinsson, V.A.; Cibulskis, K.; Choudhury, A.D.; Rosenberg, M.; Cruz-Gordillo, P.; Francis, J.M.; Zhang, C.-Z.; Shalek, A.K.; Satija, R.; et al. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat. Biotechnol. 2014, 32, 479–484. [Google Scholar] [CrossRef]
- Greene, S.B.; Dago, A.E.; Leitz, L.J.; Wang, Y.; Lee, J.; Werner, S.L.; Gendreau, S.; Patel, P.; Jia, S.; Zhang, L.; et al. Chromosomal Instability Estimation Based on Next Generation Sequencing and Single Cell Genome Wide Copy Number Variation Analysis. PLoS ONE 2016, 11, e0165089. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Lu, Y.-T.; Ho, H.; Li, B.; Chen, J.-F.; Lin, M.; Li, F.; Wu, K.; Wu, H.; Lichterman, J.; et al. A comparison of isolated circulating tumor cells and tissue biopsies using whole-genome sequencing in prostate cancer. Oncotarget 2015, 6, 44781–44793. [Google Scholar] [CrossRef]
- Dago, A.E.; Stepansky, A.; Carlsson, A.; Luttgen, M.; Kendall, J.; Baslan, T.; Kolatkar, A.; Wigler, M.; Bethel, K.; Gross, M.E.; et al. Rapid phenotypic and genomic change in response to therapeutic pressure in prostate cancer inferred by high content analysis of single circulating tumor cells. PLoS ONE 2014, 9, e101777. [Google Scholar] [CrossRef]
- Deng, G.; Krishnakumar, S.; Powell, A.A.; Zhang, H.; Mindrinos, M.N.; Telli, M.L.; Davis, R.W.; Jeffrey, S.S. Single cell mutational analysis of PIK3CA in circulating tumor cells and metastases in breast cancer reveals heterogeneity, discordance, and mutation persistence in cultured disseminated tumor cells from bone marrow. BMC Cancer 2014, 14, 456. [Google Scholar] [CrossRef]
- Fernandez, S.V.; Bingham, C.; Fittipaldi, P.; Austin, L.; Palazzo, J.; Palmer, G.; Alpaugh, K.; Cristofanilli, M. TP53 mutations detected in circulating tumor cells present in the blood of metastatic triple negative breast cancer patients. Breast Cancer Res. 2014, 16, 445. [Google Scholar] [CrossRef]
- Polzer, B.; Medoro, G.; Pasch, S.; Fontana, F.; Zorzino, L.; Pestka, A.; Andergassen, U.; Meier-Stiegen, F.; Czyz, Z.T.; Alberter, B.; et al. Molecular profiling of single circulating tumor cells with diagnostic intention. EMBO Mol. Med. 2014, 6, 1371–1386. [Google Scholar] [CrossRef]
- De Luca, F.; Rotunno, G.; Salvianti, F.; Galardi, F.; Pestrin, M.; Gabellini, S.; Simi, L.; Mancini, I.; Vannucchi, A.M.; Pazzagli, M.; et al. Mutational analysis of single circulating tumor cells by next generation sequencing in metastatic breast cancer. Oncotarget 2016, 7, 26107–26119. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.C.; Niederacher, D.; Topp, S.A.; Honisch, E.; Schumacher, S.; Schmitz, N.; Zacarias Fohrding, L.; Vay, C.; Hoffmann, I.; Kasprowicz, N.S.; et al. Diagnostic leukapheresis enables reliable detection of circulating tumor cells of nonmetastatic cancer patients. Proc. Natl. Acad. Sci. USA 2013, 110, 16580–16585. [Google Scholar] [CrossRef] [PubMed]
- Neves, R.P.; Raba, K.; Schmidt, O.; Honisch, E.; Meier-Stiegen, F.; Behrens, B.; Mohlendick, B.; Fehm, T.; Neubauer, H.; Klein, C.A.; et al. Genomic high-resolution profiling of single CKpos/CD45neg flow-sorting purified circulating tumor cells from patients with metastatic breast cancer. Clin. Chem. 2014, 60, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Vishnoi, M.; Peddibhotla, S.; Yin, W.; Scamardo, A.T.; George, G.C.; Hong, D.S.; Marchetti, D. The isolation and characterization of CTC subsets related to breast cancer dormancy. Sci. Rep. 2015, 5, 17533. [Google Scholar] [CrossRef] [PubMed]
- Gasch, C.; Oldopp, T.; Mauermann, O.; Gorges, T.M.; Andreas, A.; Coith, C.; Muller, V.; Fehm, T.; Janni, W.; Pantel, K.; et al. Frequent detection of PIK3CA mutations in single circulating tumor cells of patients suffering from HER2-negative metastatic breast cancer. Mol. Oncol. 2016, 10, 1330–1343. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.A.; Guttery, D.S.; Hills, A.; Fernandez-Garcia, D.; Page, K.; Rosales, B.M.; Goddard, K.S.; Hastings, R.K.; Luo, J.; Ogle, O.; et al. Mutation Analysis of Cell-Free DNA and Single Circulating Tumor Cells in Metastatic Breast Cancer Patients with High Circulating Tumor Cell Counts. Clin. Cancer Res. 2017, 23, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.H.D.; Schneck, H.; Decker, Y.; Schömer, S.; Franken, A.; Endris, V.; Pfarr, N.; Weichert, W.; Niederacher, D.; Fehm, T.; et al. Isolation and characterization of circulating tumor cells using a novel workflow combining the CellSearch® system and the CellCelector™. Biotechnol. Prog. 2017, 33, 125–132. [Google Scholar] [CrossRef]
- Bingham, C.; Fernandez, S.V.; Fittipaldi, P.; Dempsey, P.W.; Ruth, K.J.; Cristofanilli, M.; Katherine Alpaugh, R. Mutational studies on single circulating tumor cells isolated from the blood of inflammatory breast cancer patients. Breast Cancer Res. Treat. 2017, 163, 219–230. [Google Scholar] [CrossRef]
- Babayan, A.; Hannemann, J.; Spotter, J.; Muller, V.; Pantel, K.; Joosse, S.A. Heterogeneity of estrogen receptor expression in circulating tumor cells from metastatic breast cancer patients. PLoS ONE 2013, 8, e75038. [Google Scholar] [CrossRef]
- Heidary, M.; Auer, M.; Ulz, P.; Heitzer, E.; Petru, E.; Gasch, C.; Riethdorf, S.; Mauermann, O.; Lafer, I.; Pristauz, G.; et al. The dynamic range of circulating tumor DNA in metastatic breast cancer. Breast Cancer Res. 2014, 16, 421. [Google Scholar] [CrossRef]
- Mu, Z.; Benali-Furet, N.; Uzan, G.; Znaty, A.; Ye, Z.; Paolillo, C.; Wang, C.; Austin, L.; Rossi, G.; Fortina, P.; et al. Detection and Characterization of Circulating Tumor Associated Cells in Metastatic Breast Cancer. Int. J. Mol. Sci. 2016, 17, 1665. [Google Scholar] [CrossRef] [PubMed]
- Carter, L.; Rothwell, D.G.; Mesquita, B.; Smowton, C.; Leong, H.S.; Fernandez-Gutierrez, F.; Li, Y.; Burt, D.J.; Antonello, J.; Morrow, C.J.; et al. Molecular analysis of circulating tumor cells identifies distinct copy-number profiles in patients with chemosensitive and chemorefractory small-cell lung cancer. Nat. Med. 2017, 23, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Zhuo, M.; Su, Z.; Duan, J.; Gao, Y.; Wang, Z.; Zong, C.; Bai, H.; Chapman, A.R.; Zhao, J.; et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc. Natl. Acad. Sci. USA 2013, 110, 21083–21088. [Google Scholar] [CrossRef] [PubMed]
- Yeo, T.; Tan, S.J.; Lim, C.L.; Lau, D.P.; Chua, Y.W.; Krisna, S.S.; Iyer, G.; Tan, G.S.; Lim, T.K.; Tan, D.S.; et al. Microfluidic enrichment for the single cell analysis of circulating tumor cells. Sci. Rep. 2016, 6, 22076. [Google Scholar] [CrossRef]
- Gao, Y.; Ni, X.; Guo, H.; Su, Z.; Ba, Y.; Tong, Z.; Guo, Z.; Yao, X.; Chen, X.; Yin, J.; et al. Single-cell sequencing deciphers a convergent evolution of copy number alterations from primary to circulating tumor cells. Genome Res. 2017, 27, 1312–1322. [Google Scholar] [CrossRef]
- Su, Z.; Wang, Z.; Ni, X.; Duan, J.; Gao, Y.; Zhuo, M.; Li, R.; Zhao, J.; Ma, Q.; Bai, H.; et al. Inferring the Evolution and Progression of Small-Cell Lung Cancer by Single-Cell Sequencing of Circulating Tumor Cells. Clin. Cancer Res. 2019, 25, 5049–5060. [Google Scholar] [CrossRef]
- Steinert, G.; Schölch, S.; Niemietz, T.; Iwata, N.; García, S.A.; Behrens, B.; Voigt, A.; Kloor, M.; Benner, A.; Bork, U.; et al. Immune Escape and Survival Mechanisms in Circulating Tumor Cells of Colorectal Cancer. Cancer Res. 2014, 74, 1694–1704. [Google Scholar] [CrossRef]
- Gasch, C.; Bauernhofer, T.; Pichler, M.; Langer-Freitag, S.; Reeh, M.; Seifert, A.M.; Mauermann, O.; Izbicki, J.R.; Pantel, K.; Riethdorf, S. Heterogeneity of epidermal growth factor receptor status and mutations of KRAS/PIK3CA in circulating tumor cells of patients with colorectal cancer. Clin. Chem. 2013, 59, 252–260. [Google Scholar] [CrossRef]
- Fabbri, F.; Carloni, S.; Zoli, W.; Ulivi, P.; Gallerani, G.; Fici, P.; Chiadini, E.; Passardi, A.; Frassineti, G.L.; Ragazzini, A.; et al. Detection and recovery of circulating colon cancer cells using a dielectrophoresis-based device: KRAS mutation status in pure CTCs. Cancer Lett. 2013, 335, 225–231. [Google Scholar] [CrossRef]
- Sakaizawa, K.; Goto, Y.; Kiniwa, Y.; Uchiyama, A.; Harada, K.; Shimada, S.; Saida, T.; Ferrone, S.; Takata, M.; Uhara, H.; et al. Mutation analysis of BRAF and KIT in circulating melanoma cells at the single cell level. Br. J. Cancer 2012, 106, 939–946. [Google Scholar] [CrossRef]
- Kiniwa, Y.; Nakamura, K.; Mikoshiba, A.; Akiyama, Y.; Morimoto, A.; Okuyama, R. Diversity of circulating tumor cells in peripheral blood: Detection of heterogeneous BRAF mutations in a patient with advanced melanoma by single-cell analysis. J. Derm. Sci. 2018, 90, 211–213. [Google Scholar] [CrossRef] [PubMed]
- Anantharaman, A.; Friedlander, T.; Lu, D.; Krupa, R.; Premasekharan, G.; Hough, J.; Edwards, M.; Paz, R.; Lindquist, K.; Graf, R.; et al. Programmed death-ligand 1 (PD-L1) characterization of circulating tumor cells (CTCs) in muscle invasive and metastatic bladder cancer patients. BMC Cancer 2016, 16, 744. [Google Scholar] [CrossRef] [PubMed]
- Court, C.M.; Ankeny, J.S.; Sho, S.; Hou, S.; Li, Q.; Hsieh, C.; Song, M.; Liao, X.; Rochefort, M.M.; Wainberg, Z.A.; et al. Reality of Single Circulating Tumor Cell Sequencing for Molecular Diagnostics in Pancreatic Cancer. J. Mol. Diagn. 2016, 18, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Deneve, E.; Nocca, D.; Coffy, A.; Vendrell, J.P.; Maudelonde, T.; Riethdorf, S.; Alix-Panabieres, C. Circulating epithelial cells in patients with benign colon diseases. Clin. Chem. 2012, 58, 936–940. [Google Scholar] [CrossRef]
- Zhang, L.; Ridgway, L.D.; Wetzel, M.D.; Ngo, J.; Yin, W.; Kumar, D.; Goodman, J.C.; Groves, M.D.; Marchetti, D. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci. Transl. Med. 2013, 5, 180ra148. [Google Scholar] [CrossRef]
- Lee, Y.; Guan, G.; Bhagat, A.A. ClearCell® FX, a label-free microfluidics technology for enrichment of viable circulating tumor cells. Cytom. Part. A 2018, 93, 1251–1254. [Google Scholar] [CrossRef]
- Ligthart, S.T.; Coumans, F.A.; Bidard, F.C.; Simkens, L.H.; Punt, C.J.; de Groot, M.R.; Attard, G.; de Bono, J.S.; Pierga, J.Y.; Terstappen, L.W. Circulating Tumor Cells Count and Morphological Features in Breast, Colorectal and Prostate Cancer. PLoS ONE 2013, 8, e67148. [Google Scholar] [CrossRef]
- Kallergi, G.; Konstantinidis, G.; Markomanolaki, H.; Papadaki, M.A.; Mavroudis, D.; Stournaras, C.; Georgoulias, V.; Agelaki, S. Apoptotic circulating tumor cells in early and metastatic breast cancer patients. Mol. Cancer Ther. 2013, 12, 1886–1895. [Google Scholar] [CrossRef]
- Blainey, P.C. The future is now: Single-cell genomics of bacteria and archaea. FEMS Microbiol. Rev. 2013, 37, 407–427. [Google Scholar] [CrossRef]
- Salvianti, F.; Pazzagli, M.; Pinzani, P. Single circulating tumor cell sequencing as an advanced tool in cancer management. Expert Rev. Mol. Diagn. 2016, 16, 51–63. [Google Scholar] [CrossRef]
- Swennenhuis, J.F.; Reumers, J.; Thys, K.; Aerssens, J.; Terstappen, L.W. Efficiency of whole genome amplification of single circulating tumor cells enriched by CellSearch and sorted by FACS. Genome Med. 2013, 5, 106. [Google Scholar] [CrossRef] [PubMed]
- Gawad, C.; Koh, W.; Quake, S.R. Single-cell genome sequencing: Current state of the science. Nat. Rev. Genet. 2016, 17, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Girard, N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011, 12, 175–180. [Google Scholar] [CrossRef]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Jhawer, M.; Goel, S.; Wilson, A.J.; Montagna, C.; Ling, Y.H.; Byun, D.S.; Nasser, S.; Arango, D.; Shin, J.; Klampfer, L.; et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res. 2008, 68, 1953–1961. [Google Scholar] [CrossRef] [PubMed]
- Berns, K.; Horlings, H.M.; Hennessy, B.T.; Madiredjo, M.; Hijmans, E.M.; Beelen, K.; Linn, S.C.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Hauptmann, M.; et al. A Functional Genetic Approach Identifies the PI3K Pathway as a Major Determinant of Trastuzumab Resistance in Breast Cancer. Cancer Cell 2007, 12, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Chandarlapaty, S.; Sakr, R.A.; Giri, D.; Patil, S.; Heguy, A.; Morrow, M.; Modi, S.; Norton, L.; Rosen, N.; Hudis, C.; et al. Frequent mutational activation of the PI3K-AKT pathway in trastuzumab-resistant breast cancer. Clin. Cancer Res. 2012, 18, 6784–6791. [Google Scholar] [CrossRef]
- Mukohara, T. Mechanisms of resistance to anti-human epidermal growth factor receptor 2 agents in breast cancer. Cancer Sci. 2011, 102, 1–8. [Google Scholar] [CrossRef]
- Ibrahim, E.M.; Kazkaz, G.A.; Al-Mansour, M.M.; Al-Foheidi, M.E. The predictive and prognostic role of phosphatase phosphoinositol-3 (PI3) kinase (PIK3CA) mutation in HER2-positive breast cancer receiving HER2-targeted therapy: A meta-analysis. Breast Cancer Res. Treat. 2015, 152, 463–476. [Google Scholar] [CrossRef]
- Loibl, S.; von Minckwitz, G.; Schneeweiss, A.; Paepke, S.; Lehmann, A.; Rezai, M.; Zahm, D.M.; Sinn, P.; Khandan, F.; Eidtmann, H.; et al. PIK3CA mutations are associated with lower rates of pathologic complete response to anti-human epidermal growth factor receptor 2 (her2) therapy in primary HER2-overexpressing breast cancer. J. Clin. Oncol. 2014, 32, 3212–3220. [Google Scholar] [CrossRef]
- Neumann, M.; Decker, Y.; Franken, A.; Schömer, S.; Schneck, H.; Fehm, T.; Weichert, W.; Endriss, V.; Neubauer, H.; Niederacher, D. Sequential analysis of circulating tumor cells on genome and protein level: Potential regulation of the invasion marker CapG by PIK3CA. Senologie Zeitschrift für Mammadiagnostik und Herapie 2016, 13, A95. [Google Scholar] [CrossRef]
- Janku, F.; Wheler, J.J.; Westin, S.N.; Moulder, S.L.; Naing, A.; Tsimberidou, A.M.; Fu, S.; Falchook, G.S.; Hong, D.S.; Garrido-Laguna, I.; et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J. Clin. Oncol. 2012, 30, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Eeles, R.; Hollstein, M.; Khan, M.A.; Harris, C.C.; Hainaut, P. The IARC TP53 database: New online mutation analysis and recommendations to users. Hum. Mutat. 2002, 19, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Veronese, A.; Pichiorri, F.; Lee, T.J.; Jeon, Y.-J.; Volinia, S.; Pineau, P.; Marchio, A.; Palatini, J.; Suh, S.-S.; et al. p53 regulates epithelial–mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J. Exp. Med. 2011, 208, 875. [Google Scholar] [CrossRef]
- Chang, C.-J.; Chao, C.-H.; Xia, W.; Yang, J.-Y.; Xiong, Y.; Li, C.-W.; Yu, W.-H.; Rehman, S.K.; Hsu, J.L.; Lee, H.-H.; et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat. Cell Biol. 2011, 13, 317–323. [Google Scholar] [CrossRef]
- Petitjean, A.; Achatz, M.I.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef]
- Li, J.; Yang, L.; Gaur, S.; Zhang, K.; Wu, X.; Yuan, Y.-C.; Li, H.; Hu, S.; Weng, Y.; Yen, Y. Mutants TP53 p.R273H and p.R273C but not p.R273G Enhance Cancer Cell Malignancy. Hum. Mutat. 2014, 35, 575–584. [Google Scholar] [CrossRef]
- Kalemkerian, G.P. Chemotherapy for small-cell lung cancer. Lancet Oncol. 2014, 15, 13–14. [Google Scholar] [CrossRef]
- Sutherland, K.D.; Proost, N.; Brouns, I.; Adriaensen, D.; Song, J.Y.; Berns, A. Cell of origin of small cell lung cancer: Inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 2011, 19, 754–764. [Google Scholar] [CrossRef]
- Nicholson, R.I.; Gee, J.M.; Harper, M.E. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37 (Suppl. S4), S9–S15. [Google Scholar] [CrossRef]
- Ciardiello, F.; Tortora, G. Epidermal growth factor receptor (EGFR) as a target in cancer therapy: Understanding the role of receptor expression and other molecular determinants that could influence the response to anti-EGFR drugs. Eur. J. Cancer 2003, 39, 1348–1354. [Google Scholar] [CrossRef]
- Moroni, M.; Veronese, S.; Benvenuti, S.; Marrapese, G.; Sartore-Bianchi, A.; Di Nicolantonio, F.; Gambacorta, M.; Siena, S.; Bardelli, A. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: A cohort study. Lancet Oncol. 2005, 6, 279–286. [Google Scholar] [CrossRef]
- Lièvre, A.; Bachet, J.-B.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.-F.; Côté, J.-F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS Mutation Status Is Predictive of Response to Cetuximab Therapy in Colorectal Cancer. Cancer Res. 2006, 66, 3992. [Google Scholar] [CrossRef]
- Amado, R.G.; Wolf, M.; Peeters, M.; Van Cutsem, E.; Siena, S.; Freeman, D.J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; et al. Wild-Type KRAS Is Required for Panitumumab Efficacy in Patients with Metastatic Colorectal Cancer. J. Clin. Oncol. 2008, 26, 1626–1634. [Google Scholar] [CrossRef]
- Zhu, C.-Q.; da Cunha Santos, G.; Ding, K.; Sakurada, A.; Cutz, J.-C.; Liu, N.; Zhang, T.; Marrano, P.; Whitehead, M.; Squire, J.A.; et al. Role of KRAS and EGFR as Biomarkers of Response to Erlotinib in National Cancer Institute of Canada Clinical Trials Group Study BR.21. J. Clin. Oncol. 2008, 26, 4268–4275. [Google Scholar] [CrossRef]
- Watanabe, T.; Kobunai, T.; Yamamoto, Y.; Matsuda, K.; Ishihara, S.; Nozawa, K.; Iinuma, H.; Shibuya, H.; Eshima, K. Heterogeneity of KRAS Status May Explain the Subset of Discordant KRAS Status Between Primary and Metastatic Colorectal Cancer. Dis. Colon Rectum 2011, 54, 1170–1178. [Google Scholar] [CrossRef]
- Ducreux, M.; Chamseddine, A.; Laurent-Puig, P.; Smolenschi, C.; Hollebecque, A.; Dartigues, P.; Samallin, E.; Boige, V.; Malka, D.; Gelli, M. Molecular targeted therapy of BRAF-mutant colorectal cancer. Ther. Adv. Med. Oncol. 2019, 11. [Google Scholar] [CrossRef]
- Marconcini, R.; Galli, L.; Antonuzzo, A.; Bursi, S.; Roncella, C.; Fontanini, G.; Sensi, E.; Falcone, A. Metastatic BRAF K601E-mutated melanoma reaches complete response to MEK inhibitor trametinib administered for over 36 months. Exp. Hematol. Oncol. 2017, 6, 6. [Google Scholar] [CrossRef]
- Mostert, B.; Jiang, Y.; Sieuwerts, A.M.; Wang, H.; Bolt-de Vries, J.; Biermann, K.; Kraan, J.; Lalmahomed, Z.; van Galen, A.; de Weerd, V.; et al. KRAS and BRAF mutation status in circulating colorectal tumor cells and their correlation with primary and metastatic tumor tissue. Int. J. Cancer 2013, 133, 130–141. [Google Scholar] [CrossRef]
- Wheeler, J.M.; Bodmer, W.F.; Mortensen, N.J. DNA mismatch repair genes and colorectal cancer. Gut 2000, 47, 148–153. [Google Scholar] [CrossRef]
- Mackay, H.J.; Gallinger, S.; Tsao, M.S.; McLachlin, C.M.; Tu, D.; Keiser, K.; Eisenhauer, E.A.; Oza, A.M. Prognostic value of microsatellite instability (MSI) and PTEN expression in women with endometrial cancer: Results from studies of the NCIC Clinical Trials Group (NCIC CTG). Eur. J. Cancer 2010, 46, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic Review of Microsatellite Instability and Colorectal Cancer Prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Thibodeau, S.N.; Bren, G.; Schaid, D. Microsatellite instability in cancer of the proximal colon. Science 1993, 260, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.P.; Colangelo, L.H.; Wieand, H.S.; Paik, S.; Kirsch, I.R.; Wolmark, N.; Allegra, C.J.; National Cancer, I. Prognostic and predictive roles of high-degree microsatellite instability in colon cancer: A National Cancer Institute-National Surgical Adjuvant Breast and Bowel Project Collaborative Study. J. Clin. Oncol. 2007, 25, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Ribic, C.M.; Sargent, D.J.; Moore, M.J.; Thibodeau, S.N.; French, A.J.; Goldberg, R.M.; Hamilton, S.R.; Laurent-Puig, P.; Gryfe, R.; Shepherd, L.E.; et al. Tumor Microsatellite-Instability Status as a Predictor of Benefit from Fluorouracil-Based Adjuvant Chemotherapy for Colon Cancer. N. Engl. J. Med. 2003, 349, 247–257. [Google Scholar] [CrossRef]
- Goldstein, J.; Tran, B.; Ensor, J.; Gibbs, P.; Wong, H.L.; Wong, S.F.; Vilar, E.; Tie, J.; Broaddus, R.; Kopetz, S.; et al. Multicenter retrospective analysis of metastatic colorectal cancer (CRC) with high-level microsatellite instability (MSI-H). Ann. Oncol. 2014, 25, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Findeisen, P.; Kloor, M.; Merx, S.; Sutter, C.; Woerner, S.M.; Dostmann, N.; Benner, A.; Dondog, B.; Pawlita, M.; Dippold, W.; et al. T25 repeat in the 3’ untranslated region of the CASP2 gene: A sensitive and specific marker for microsatellite instability in colorectal cancer. Cancer Res. 2005, 65, 8072–8078. [Google Scholar] [CrossRef]
- Carethers, J.M.; Smith, E.J.; Behling, C.A.; Nguyen, L.; Tajima, A.; Doctolero, R.T.; Cabrera, B.L.; Goel, A.; Arnold, C.A.; Miyai, K.; et al. Use of 5-fluorouracil and survival in patients with microsatellite-unstable colorectal cancer. Gastroenterology 2004, 126, 394–401. [Google Scholar] [CrossRef]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar]
- Bergamaschi, A.; Kim, Y.H.; Wang, P.; Sorlie, T.; Hernandez-Boussard, T.; Lonning, P.E.; Tibshirani, R.; Borresen-Dale, A.L.; Pollack, J.R. Distinct patterns of DNA copy number alteration are associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes Chromosomes Cancer 2006, 45, 1033–1040. [Google Scholar] [CrossRef]
- Chin, K.; DeVries, S.; Fridlyand, J.; Spellman, P.T.; Roydasgupta, R.; Kuo, W.L.; Lapuk, A.; Neve, R.M.; Qian, Z.; Ryder, T.; et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 2006, 10, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, C.; Paez, J.G.; Chin, K.; Jänne, P.A.; Chen, T.-H.; Girard, L.; Minna, J.; Christiani, D.; Leo, C.; et al. An Integrated View of Copy Number and Allelic Alterations in the Cancer Genome Using Single Nucleotide Polymorphism Arrays. Cancer Res. 2004, 64, 3060. [Google Scholar] [CrossRef] [PubMed]
- Cappuzzo, F.; Varella-Garcia, M.; Shigematsu, H.; Domenichini, I.; Bartolini, S.; Ceresoli, G.L.; Rossi, E.; Ludovini, V.; Gregorc, V.; Toschi, L.; et al. Increased HER2 Gene Copy Number Is Associated with Response to Gefitinib Therapy in Epidermal Growth Factor Receptor–Positive Non–Small-Cell Lung Cancer Patients. J. Clin. Oncol. 2005, 23, 5007–5018. [Google Scholar] [CrossRef]
- Watkins, J.A.; Irshad, S.; Grigoriadis, A.; Tutt, A.N. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. 2014, 16, 211. [Google Scholar] [CrossRef]
- Humar, B.; Blair, V.; Charlton, A.; More, H.; Martin, I.; Guilford, P. E-cadherin deficiency initiates gastric signet-ring cell carcinoma in mice and man. Cancer Res. 2009, 69, 2050–2056. [Google Scholar] [CrossRef]
- Takeuchi, T.; Adachi, Y.; Nagayama, T. A WWOX-binding molecule, transmembrane protein 207, is related to the invasiveness of gastric signet-ring cell carcinoma. Carcinogenesis 2012, 33, 548–554. [Google Scholar] [CrossRef]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The genomic complexity of primary human prostate cancer. Nature 2011, 470, 214–220. [Google Scholar] [CrossRef]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated Evolution of Prostate Cancer Genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef]
- Budd, G.T.; Cristofanilli, M.; Ellis, M.J.; Stopeck, A.; Borden, E.; Miller, M.C.; Matera, J.; Repollet, M.; Doyle, G.V.; Terstappen, L.W.M.M.; et al. Circulating Tumor Cells versus Imaging—Predicting Overall Survival in Metastatic Breast Cancer. Clin. Cancer Res. 2006, 12, 6403. [Google Scholar] [CrossRef]
- Cohen, S.J.; Punt, C.J.A.; Iannotti, N.; Saidman, B.H.; Sabbath, K.D.; Gabrail, N.Y.; Picus, J.; Morse, M.; Mitchell, E.; Miller, M.C.; et al. Relationship of Circulating Tumor Cells to Tumor Response, Progression-Free Survival, and Overall Survival in Patients with Metastatic Colorectal Cancer. J. Clin. Oncol. 2008, 26, 3213–3221. [Google Scholar] [CrossRef]
- De Bono, J.S.; Scher, H.I.; Montgomery, R.B.; Parker, C.; Miller, M.C.; Tissing, H.; Doyle, G.V.; Terstappen, L.W.; Pienta, K.J.; Raghavan, D. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2008, 14, 6302–6309. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D.F.; Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Miller, M.C.; Matera, J.; Allard, W.J.; Doyle, G.V.; Terstappen, L.W.W.M. Circulating Tumor Cells at Each Follow-up Time Point during Therapy of Metastatic Breast Cancer Patients Predict Progression-Free and Overall Survival. Clin. Cancer Res. 2006, 12, 4218. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, D.T.; Lee, R.J.; Stott, S.L.; Ting, D.T.; Wittner, B.S.; Ulman, M.; Smas, M.E.; Lord, J.B.; Brannigan, B.W.; Trautwein, J.; et al. Androgen Receptor Signaling in Circulating Tumor Cells as a Marker of Hormonally Responsive Prostate Cancer. Cancer Discov. 2012, 2, 995. [Google Scholar] [CrossRef] [PubMed]
- McArthur, G.A.; Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Dummer, R.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3): Extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014, 15, 323–332. [Google Scholar] [CrossRef]
- Cortés-Funes, H.; Mendiola, C.; Manso, L.; Ciruelos, E. Neratinib, an irreversible pan erB receptor tyrosine kinase inhibitor active for advanced HER2+breast cancer. Breast Cancer Res. 2009, 11, S19. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Baselga, J.; Gelmon, K.A.; Verma, S.; Wardley, A.; Conte, P.; Miles, D.; Bianchi, G.; Cortes, J.; McNally, V.A.; Ross, G.A.; et al. Phase II Trial of Pertuzumab and Trastuzumab in Patients with Human Epidermal Growth Factor Receptor 2–Positive Metastatic Breast Cancer That Progressed During Prior Trastuzumab Therapy. J. Clin. Oncol. 2010, 28, 1138–1144. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.-L.; Chen, G.; Feng, J.; Liu, X.-Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Bailey, R.; Kris, M.; Wolf, M.; Kay, A.; Averbuch, S.; Askaa, J.; Janas, M.; Schmidt, K.; Fukuoka, M. O-242 Gefitinib (‘Iressa’, ZD1839) monotherapy for pretreated advanced non-small-cell lung cancer in IDEAL 1 and 2: Tumor response is not clinically relevantly predictable from tumor EGFR membrane staining alone. Lung Cancer 2003, 41, S71. [Google Scholar] [CrossRef]
- Amir, E.; Miller, N.; Geddie, W.; Freedman, O.; Kassam, F.; Simmons, C.; Oldfield, M.; Dranitsaris, G.; Tomlinson, G.; Laupacis, A.; et al. Prospective study evaluating the impact of tissue confirmation of metastatic disease in patients with breast cancer. J. Clin. Oncol. 2012, 30, 587–592. [Google Scholar] [CrossRef]
- Lindström, L.S.; Karlsson, E.; Wilking, U.M.; Johansson, U.; Hartman, J.; Lidbrink, E.K.; Hatschek, T.; Skoog, L.; Bergh, J. Clinically Used Breast Cancer Markers Such as Estrogen Receptor, Progesterone Receptor, and Human Epidermal Growth Factor Receptor 2 Are Unstable Throughout Tumor Progression. J. Clin. Oncol. 2012, 30, 2601–2608. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.H.; Di Leo, A. HER2 discordance between primary and metastatic breast cancer: Assessing the clinical impact. Cancer Treat. Rev. 2013, 39, 947–957. [Google Scholar] [CrossRef]
- Pusztai, L.; Viale, G.; Kelly, C.M.; Hudis, C.A. Estrogen and HER-2 Receptor Discordance Between Primary Breast Cancer and Metastasis. Oncologist 2010, 15, 1164–1168. [Google Scholar] [CrossRef] [PubMed]
- Fehm, T.; Muller, V.; Aktas, B.; Janni, W.; Schneeweiss, A.; Stickeler, E.; Lattrich, C.; Lohberg, C.R.; Solomayer, E.; Rack, B.; et al. HER2 status of circulating tumor cells in patients with metastatic breast cancer: A prospective, multicenter trial. Breast Cancer Res. Treat. 2010, 124, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Wallwiener, M.; Hartkopf, A.D.; Riethdorf, S.; Nees, J.; Sprick, M.R.; Schonfisch, B.; Taran, F.A.; Heil, J.; Sohn, C.; Pantel, K.; et al. The impact of HER2 phenotype of circulating tumor cells in metastatic breast cancer: A retrospective study in 107 patients. BMC Cancer 2015, 15, 403. [Google Scholar] [CrossRef]
- Georgoulias, V.; Bozionelou, V.; Agelaki, S.; Perraki, M.; Apostolaki, S.; Kallergi, G.; Kalbakis, K.; Xyrafas, A.; Mavroudis, D. Trastuzumab decreases the incidence of clinical relapses in patients with early breast cancer presenting chemotherapy-resistant CK-19mRNA-positive circulating tumor cells: Results of a randomized phase II study. Ann. Oncol. 2012, 23, 1744–1750. [Google Scholar] [CrossRef]
- Schramm, A.; Friedl, T.W.P.; Schochter, F.; Scholz, C.; de Gregorio, N.; Huober, J.; Rack, B.; Trapp, E.; Alunni-Fabbroni, M.; Müller, V.; et al. Therapeutic intervention based on circulating tumor cell phenotype in metastatic breast cancer: Concept of the DETECT study program. Arch. Gynecol. Obstet. 2016, 293, 271–281. [Google Scholar] [CrossRef]
- Wong, K.H.K.; Tessier, S.N.; Miyamoto, D.T.; Miller, K.L.; Bookstaver, L.D.; Carey, T.R.; Stannard, C.J.; Thapar, V.; Tai, E.C.; Vo, K.D.; et al. Whole blood stabilization for the microfluidic isolation and molecular characterization of circulating tumor cells. Nat. Commun. 2017, 8, 1733. [Google Scholar] [CrossRef]
- Lake, B.B.; Codeluppi, S.; Yung, Y.C.; Gao, D.; Chun, J.; Kharchenko, P.V.; Linnarsson, S.; Zhang, K. A comparative strategy for single-nucleus and single-cell transcriptomes confirms accuracy in predicted cell-type expression from nuclear RNA. Sci. Rep. 2017, 7, 6031. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Gierahn, T.M.; Wadsworth, M.H., 2nd; Hughes, T.K.; Bryson, B.D.; Butler, A.; Satija, R.; Fortune, S.; Love, J.C.; Shalek, A.K. Seq-Well: Portable, low-cost RNA sequencing of single cells at high throughput. Nat. Methods 2017, 14, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.A.; Kirschner, M.W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015, 161, 1187–1201. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.C.; Fu, G.K.; Fodor, S.P. Expression profiling. Combinatorial labeling of single cells for gene expression cytometry. Science 2015, 347, 1258367. [Google Scholar] [CrossRef]
- Cheng, Y.H.; Chen, Y.C.; Lin, E.; Brien, R.; Jung, S.; Chen, Y.T.; Lee, W.; Hao, Z.; Sahoo, S.; Min Kang, H.; et al. Hydro-Seq enables contamination-free high-throughput single-cell RNA-sequencing for circulating tumor cells. Nat. Commun. 2019, 10, 2163. [Google Scholar] [CrossRef] [PubMed]
- Stott, S.L.; Hsu, C.H.; Tsukrov, D.I.; Yu, M.; Miyamoto, D.T.; Waltman, B.A.; Rothenberg, S.M.; Shah, A.M.; Smas, M.E.; Korir, G.K.; et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc. Natl. Acad. Sci. USA 2010, 107, 18392–18397. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Liquid biopsy and minimal residual disease—Latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Sinkala, E.; Sollier-Christen, E.; Renier, C.; Rosàs-Canyelles, E.; Che, J.; Heirich, K.; Duncombe, T.A.; Vlassakis, J.; Yamauchi, K.A.; Huang, H.; et al. Profiling protein expression in circulating tumour cells using microfluidic western blotting. Nat. Commun. 2017, 8, 14622. [Google Scholar] [CrossRef]
- Abouleila, Y.; Onidani, K.; Ali, A.; Shoji, H.; Kawai, T.; Lim, C.T.; Kumar, V.; Okaya, S.; Kato, K.; Hiyama, E.; et al. Live single cell mass spectrometry reveals cancer-specific metabolic profiles of circulating tumor cells. Cancer Sci. 2019, 110, 697–706. [Google Scholar] [CrossRef]
- Deng, Y.; Zhang, Y.; Sun, S.; Wang, Z.; Wang, M.; Yu, B.; Czajkowsky, D.M.; Liu, B.; Li, Y.; Wei, W.; et al. An Integrated Microfluidic Chip System for Single-Cell Secretion Profiling of Rare Circulating Tumor Cells. Sci. Rep. 2014, 4, 7499. [Google Scholar] [CrossRef]
- Lim, S.B.; Tan, S.J.; Lim, W.-T.; Lim, C.T. Compendiums of cancer transcriptomes for machine learning applications. Sci. Data 2019, 6, 194. [Google Scholar] [CrossRef]
- Lim, S.B.; Tan, S.J.; Lim, W.-T.; Lim, C.T. A merged lung cancer transcriptome dataset for clinical predictive modeling. Sci. Data 2018, 5, 180136. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| CTC Enrichment | Single-Cell Isolation | CTC Criteria | WGA | Profiling | Investigated Genes | Genomic Analysis | Number of Single CTCs (Patients) 1 | Ref. |
|---|---|---|---|---|---|---|---|---|
| Prostate cancer | ||||||||
| MagSweeper | CellCelector | DAPI− CD45− EpCAM+ | MDA | NGS | All | SNVs | 42 (5) | [24] |
| Epic Sciences CTC Platform | Micromanipulation | CD45− CK+/− | DOP-PCR | NGS | All | CNVs, LSTs | 67 (7) | [25] |
| NanoVelcro CTC Chip | LCM | CD45− CK+ | MDA | NGS, Sanger, aCGH | All | SNVs, SVs, CNVs | 12 (1) | [26] |
| HD-CTC Assay | Micromanipulation | DAPI+ CK+ CD45− | LA-PCR | NGS | All | CNVs | 41 (1) | [27] |
| CellSearch, Spectra Optia Apheresis System | FACS | DAPI+ CK+ CD45− | LA-PCR | aCGH | All | CNVs | 205 (14) | [17] |
| Breast cancer | ||||||||
| MagSweeper | Micromanipulation | DAPI+ CK+ CD45− | No WGA | Sanger | PIK3CA | SNVs | 185 (11) | [28] |
| CellSearch | DEPArray | DAPI+ CK+ CD45− | LA-PCR | Sanger | TP53 | SNVs | 11 (2) | [29] |
| CellSearch | DEPArray | DAPI+ CK+ CD45− | LA-PCR | Sanger | PIK3CA | SNVs | 241 (43) | [30] |
| qPCR | HER2 | CNVs | 192 (42) | |||||
| aCGH | All | CNVs | 37 (15) | |||||
| CellSearch | DEPArray | DAPI+ CK+ CD45− CD34− | LA-PCR | Sanger | PIK3CA | SNVs | 115 (18) | [18] |
| CellSearch | DEPArray | DAPI+ CK+ CD45− | LA-PCR | Targeted NGS | 50 cancer-related genes | SNVs | 14 (4) | [31] |
| Leukapheresis, CellSearch | Micromanipulation | CK+ CD45− | DOP-PCR | CGH | All | CNVs | 65 (19) | [32] |
| CellSearch | MoFlo XDP flow-sorting | DAPI+ CK+ CD45− | LA-PCR | aCGH | All | CNVs | 26 (12) | [33] |
| qPCR | CCND1 locus | CNVs | ||||||
| Sanger | PIK3CA, TP53 | SNVs | ||||||
| FACS | DEPArray | DAPI− CD45− EpCAM− CD44+ CD24− uPAR+/− intβ1+/− | LA-PCR | MassARRAY | >200 hallmark cancer genes | SNVs | 7 (-) | [34] |
| CellSearch | Micromanipulation | DAPI+ CK+ CD45− | MDA, LA-PCR | Sanger | PIK3CA | SNVs | 114 (33) | [35] |
| CellSearch | DEPArray | DAPI+ CK+ CD45− | LA-PCR | Targeted NGS, ddPCR | 50 COSMIC genes | SNVs | 40 (5) | [36] |
| CellSearch | CellCelector | DAPI+ CK+ CD45− | LA-PCR | Targeted NGS | 50 COSMIC genes | SNVs | 7 (2) | [37] |
| CellSearch | DEPArray | DAPI+ CK+ CD45− | LA-PCR | Sanger | TP53, HER2, PIK3CA, RB1 | SNVs | 24 (6) | [38] |
| Ficoll Separation | Micromanipulation | DAPI+ CK+ CD45− | LA-PCR | Sanger | ESR1 | SNVs | 8 (4) | [39] |
| AutoMACS Classic Separator | LCM | CK+ CD45− | LA-PCR | SNP Array | All | CNVs | 17 (17) | [23] |
| CellSearch, Oncoquick | CellCelector | EpCAM+ CD45− | MDA | aCGH, targeted NGS | All | SNVs, CNVs | 31 (1) | [40] |
| oHSV-hTERT-GFP method | FACS | CD45− hTERT+ | MALBAC | NGS | All | SNVs, CNVs | 11 (8) | [19] |
| ScreenCell | DEPArray | DAPI+ CK+ CD45− | LA-PCR | Sanger | TP53, ESR1 | SNVs | 7 (1) | [41] |
| ClearCell FX System | Manipulation | DAPI+ CK+ CD45− | MALBAC | NGS | All | SNVs | 3 (1) | [20] |
| Lung cancer | ||||||||
| CellSearch | DEPArray | DAPI+ CK+ CD45− | LA-PCR | NGS | All | CNVs | 72 (13) | [42] |
| CellSearch | Micromanipulation | DAPI+ CK+ CD45− | MALBAC | NGS, digital PCR, Sanger | All | SNVs, INDELs | 24 (4) | [43] |
| CNVs | 61 (11) | |||||||
| ClearCell FX System | Microfluidic chip | DAPI+ CK+ CD45− | No WGA | Sanger | EGFR | SNVs | 26 (7) | [44] |
| CellSearch | Micromanipulation | DAPI+ CK+ CD45− | MALBAC | NGS | All | SNVs, INDELs, CNVs, SVs | 97 (23) | [45] |
| CellSearch | Micromanipulation | DAPI+ CK+ CD45− | MALBAC | NGS | All | SNVs, INDELs, CNVs, SVs | 91 (10) | [46] |
| MagSifter | Single-Nanowell Assay | DAPI+ CK+ CD45− TERT+ MET+ | No WGA | Multiplex PCR | EGFR | SNVs | 202 (7) | [22] |
| Colorectal cancer | ||||||||
| CellSearch | Micromanipulation | EpCAM+ CD45− CK+ | LA-PCR | aCGH | All | CNVs | 8 (8) | [47] |
| Sanger | KRAS, BRAF, TP53 | SNVs | 126 (31) | |||||
| Multiplex PCR | NCI/ICG-HNPCC marker panel | MSI | 122 (30) | |||||
| CellSearch | Micromanipulation | DAPI+ CK+ CD45− | LA-PCR, MDA | aCGH | All | CNVs | 37 (6) | [16] |
| Targeted NGS | 68 colorectal cancer-associated genes | SNVs | 8 (2) | |||||
| CellSearch | Micromanipulation | DAPI+ CK+ CD45− | LA-PCR, MDA | qPCR | EGFR | CNVs | 26 (3) | [48] |
| Sanger | KRAS, BRAF, PIK3CA | SNVs | 69 (5) | |||||
| Oncoquick | DEPArray | HOECHST+ CK+ CD45− | LA-PCR | Sanger, pyrosequencing | KRAS | SNVs | - (16) | [49] |
| Melanoma | ||||||||
| Dynabeads | LCM | HMW− MAA+ CD45− MART-1/gp100+ | No WGA | Sanger | BRAF | SNVs | 14 (9) | [50] |
| KIT | SNVs | 4 (4) | ||||||
| Dielectrophoretic microwell array | Micromanipulation | CD45− MART-1/gp100+ | No WGA | Sanger | BRAF | SNVs | 33 (1) | [51] |
| Multiple myeloma | ||||||||
| MACS beads | Micromanipulation | CD45−CD138+ | MDA | Targeted NGS | 35 most commonly mutated loci | SNVs | 203 (10) | [21] |
| Epic Sciences CTC Platform | Micromanipulation | CK+/− CD45− | DOP-PCR | NGS | All | CNVs | 9 (1) | [52] |
| Pancreatic cancer | ||||||||
| NanoVelcro Chip | LCM | HOECHST+ CD45− CK/CEA+ | MDA | Sanger | KRAS | SNVs | 60 (12) | [53] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, S.B.; Lim, C.T.; Lim, W.-T. Single-Cell Analysis of Circulating Tumor Cells: Why Heterogeneity Matters. Cancers 2019, 11, 1595. https://doi.org/10.3390/cancers11101595
Lim SB, Lim CT, Lim W-T. Single-Cell Analysis of Circulating Tumor Cells: Why Heterogeneity Matters. Cancers. 2019; 11(10):1595. https://doi.org/10.3390/cancers11101595
Chicago/Turabian StyleLim, Su Bin, Chwee Teck Lim, and Wan-Teck Lim. 2019. "Single-Cell Analysis of Circulating Tumor Cells: Why Heterogeneity Matters" Cancers 11, no. 10: 1595. https://doi.org/10.3390/cancers11101595
APA StyleLim, S. B., Lim, C. T., & Lim, W.-T. (2019). Single-Cell Analysis of Circulating Tumor Cells: Why Heterogeneity Matters. Cancers, 11(10), 1595. https://doi.org/10.3390/cancers11101595