Clinical Application of Next-Generation Sequencing as A Liquid Biopsy Technique in Advanced Colorectal Cancer: A Trick or A Treat?

 ,
, 

Abstract
1. Introduction
2. Background
2.1. Colorectal Cancer: From Molecular Heterogeneity to Optimal Therapy Selection
2.2. Circulating Tumor DNA as A Promising CRC Biomarker
2.3. Next-Generation Sequencing in CRC
3. Clinical Application of ctDNA NGS in Advanced CRC Patients
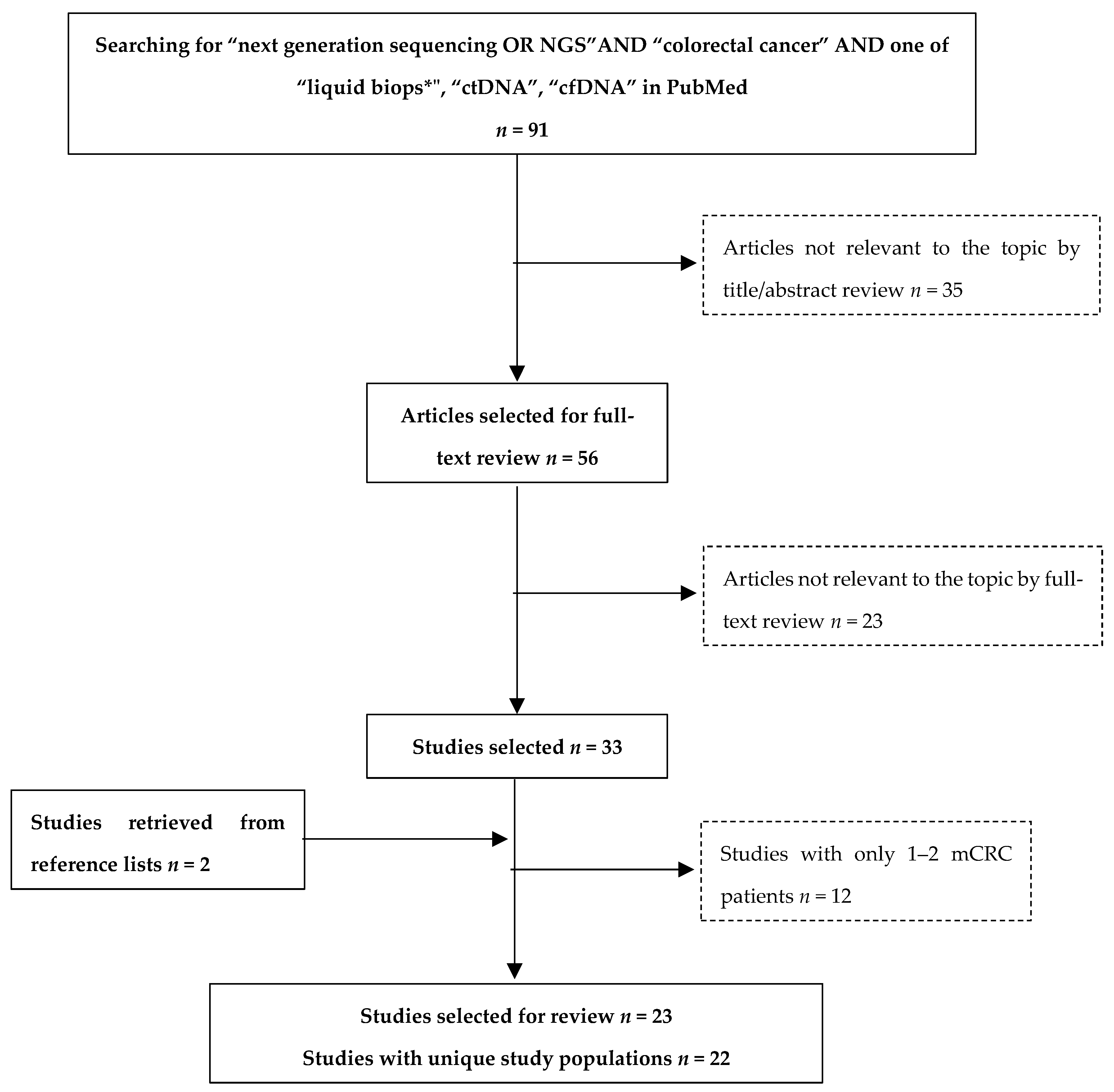
3.1. Review Methods
3.2. Overview of Studies
3.3. Pre-Analytical Parameters
4. Overview of Studies Results
4.1. Prevalence of Mutations in Plasma
4.2. Comparison of Plasma NGS with Tissue Testing
4.3. Comparison of Plasma NGS with Other Liquid Biopsy Techniques
5. Conclusions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aguilar, E.A.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef] [PubMed]
- Signorelli, D.; Giannatempo, P.; Grazia, G.; Aiello, M.M.; Bertolini, F.; Mirabile, A.; Buti, S.; Vasile, E.; Scotti, V.; Pisapia, P.; et al. Patients Selection for Immunotherapy in Solid Tumors: Overcome the Naïve Vision of a Single Biomarker. BioMed Res. Int. 2019, 2019, 9056417-15. [Google Scholar] [CrossRef] [PubMed]
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 2016, 107, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Alix-Panabières, C. Liquid biopsy and minimal residual disease—Latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Wang, Y.; Li, L.; Christie, M.; Simons, K.; Elsaleh, H.; Kosmider, S.; Wong, R.; Yip, D.; et al. Serial circulating tumour DNA analysis during multimodality treatment of locally advanced rectal cancer: A prospective biomarker study. Gut 2019, 68, 663–671. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef]
- Diaz, L.A.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef]
- Hironaka-Mitsuhashi, A.; Calle, A.S.; Ochiya, T.; Takayama, S.; Suto, A. Towards Circulating-Tumor DNA-Based Precision Medicine. J. Clin. Med. 2019, 8, 1365. [Google Scholar] [CrossRef]
- Kyrochristos, I.D.; Roukos, D.H. Comprehensive intra-individual genomic and transcriptional heterogeneity: Evidence-based Colorectal Cancer Precision Medicine. Cancer Treat Rev. 2019, 80, 101894. [Google Scholar] [CrossRef]
- Corti, G.; Bartolini, A.; Crisafulli, G.; Novara, L.; Rospo, G.; Montone, M.; Negrino, C.; Mussolin, B.; Buscarino, M.; Isella, C.; et al. A Genomic Analysis Workflow for Colorectal Cancer Precision Oncology. Clin. Color. Cancer 2019, 18, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Karapetis, C.S.; Jonker, D.; Daneshmand, M.; Hanson, J.E.; O’Callaghan, C.J.; Marginean, C.; Zalcberg, J.R.; Simes, J.; Moore, M.J.; Tebbutt, N.C.; et al. PIK3CA, BRAF, and PTEN status and benefit from cetuximab in the treatment of advanced colorectal cancer--results from NCIC CTG/AGITG CO.17. Clin. Cancer Res. 2014, 20, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Kheder, E.S.; Hong, D.S. Emerging Targeted Therapy for Tumors with NTRK Fusion Proteins. Clin. Cancer Res. 2018, 24, 5807–5814. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Yonesaka, K.; Zejnullahu, K.; Okamoto, I.; Satoh, T.; Cappuzzo, F.; Souglakos, J.; Ercan, D.; Rogers, A.; Roncalli, M.; Takeda, M.; et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci. Transl. Med. 2011, 3, 99ra86. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Arena, S.; Lamba, S.; Siravegna, G.; Lallo, A.; Hobor, S.; Russo, M.; Buscarino, M.; Lazzari, L.; Sartore-Bianchi, A.; et al. Blockade of EGFR and MEK Intercepts Heterogeneous Mechanisms of Acquired Resistance to Anti-EGFR Therapies in Colorectal Cancer. Sci. Transl. Med. 2014, 6, 224ra26. [Google Scholar] [CrossRef]
- Morelli, M.P.; Overman, M.J.; Dasari, A.; Kazmi, S.M.A.; Mazard, T.; Vilar, E.; Morris, V.K.; Lee, M.S.; Herron, D.; Eng, C.; et al. Characterizing the patterns of clonal selection in circulating tumor DNA from patients with colorectal cancer refractory to anti-EGFR treatment. Ann. Oncol. 2015, 26, 731–736. [Google Scholar] [CrossRef]
- Crowley, E.; Di Nicolantonio, F.; Loupakis, F.; Bardelli, A. Liquid biopsy: Monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 2013, 10, 472–484. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Reimers, N.; Pantel, K. Liquid biopsy: Novel technologies and clinical applications. Clin. Chem. Lab. Med. 2019, 57, 312–316. [Google Scholar] [CrossRef]
- Thierry, A.R.; El Messaoudi, S.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef] [PubMed]
- Volckmar, A.L.; Sültmann, H.; Riediger, A.; Fioretos, T.; Schirmacher, P.; Endris, V.; Stenzinger, A.; Dietz, S. A field guide for cancer diagnostics using cell-free DNA: From principles to practice and clinical applications. Genes Chromosomes Cancer 2018, 57, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Genetic Testing in Screening Patients with Metastatic or Unresectable Colon or Rectal Cancer for a COLOMATE Trial. Available online: https://clinicaltrials.gov/ct2/show/NCT03765736 (accessed on 15 April 2019).
- Stewart, C.M.; Tsui, D.W. Circulating cell-free DNA for non-invasive cancer management. Cancer Genet. 2018, 228, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Barzi, A.; Sartore-Bianchi, A.; Cassingena, A.; Siravegna, G.; Karp, D.D.; Piha-Paul, S.A.; Subbiah, V.; Tsimberidou, A.M.; Huang, H.J.; et al. Mutation-Enrichment Next-Generation Sequencing for Quantitative Detection of KRAS Mutations in Urine Cell-Free DNA from Patients with Advanced Cancers. Clin. Cancer Res. 2017, 23, 3657–3666. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Pantel, K. Liquid Biopsies, What We Do Not Know (Yet). Cancer Cell 2017, 31, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, F.; Mastroiaco, V.; Di Marco, A.; Compagnoni, C.; Capece, D.; Zazzeroni, F.; Capalbo, C.; Alesse, E.; Tessitore, A. Next-generation sequencing: Recent applications to the analysis of colorectal cancer. J. Transl. Med. 2017, 15, 246. [Google Scholar] [CrossRef]
- Vymetalkova, V.; Cervena, K.; Bartu, L.; Vodicka, P. Circulating Cell-Free DNA and Colorectal Cancer: A Systematic Review. Int. J. Mol. Sci. 2018, 19, 3356. [Google Scholar] [CrossRef]
- Fiala, C.; Diamandis, E.P. Utility of circulating tumor DNA in cancer diagnostics with emphasis on early detection. BMC Med. 2018, 16, 166. [Google Scholar] [CrossRef]
- Gonzalez-Garay, M.L. The road from next-generation sequencing to personalized medicine. Pers. Med. 2014, 11, 523–544. [Google Scholar] [CrossRef]
- Rabbani, B.; Nakaoka, H.; Akhondzadeh, S.; Tekin, M.; Mahdieh, N. Next generation sequencing: Implications in personalized medicine and pharmacogenomics. Mol. BioSyst. 2016, 12, 1818–1830. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, B.; Tekin, M.; Mahdieh, N. The promise of whole-exome sequencing in medical genetics. J. Hum. Genet. 2014, 59, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Samorodnitsky, E.; Jewell, B.M.; Hagopian, R.; Miya, J.; Wing, M.R.; Lyon, E.; Damodaran, S.; Bhatt, D.; Reeser, J.W.; Datta, J.; et al. Evaluation of Hybridization Capture Versus Amplicon-Based Methods for Whole-Exome Sequencing. Hum. Mutat. 2015, 36, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhao, H. Next-generation sequencing in liquid biopsy: Cancer screening and early detection. Hum. Genom. 2019, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Satya, R.V.; Lewis, M.; Randad, P.; Dicarlo, J.; Wang, Y. Abstract 4879: Reducing amplification artifacts in highly multiplex amplicon sequencing by using molecular barcodes. Mol. Cell. Biol. 2015, 75, 4879. [Google Scholar]
- Fontanges, Q.; De Mendonca, R.; Salmon, I.; Le Mercier, M.; D’Haene, N. Clinical Application of Targeted Next Generation Sequencing for Colorectal Cancers. Int. J. Mol. Sci. 2016, 17, 2117. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.E.; Myers, R.M. Advancements in Next-Generation Sequencing. Annu. Rev. Genom. Hum. Genet. 2016, 17, 95–115. [Google Scholar] [CrossRef]
- Kamps, R.; Brandão, R.D.; Bosch, B.J.V.D.; Paulussen, A.D.C.; Xanthoulea, S.; Blok, M.J.; Romano, A. Next-Generation Sequencing in Oncology: Genetic Diagnosis, Risk Prediction and Cancer Classification. Int. J. Mol. Sci. 2017, 18, 308. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of Next-Generation Sequencing Systems. J. Biomed. Biotechnol. 2012, 2012, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lanman, R.B.; Mortimer, S.A.; Zill, O.A.; Sebisanovic, D.; Lopez, R.; Blau, S.; Collisson, E.A.; Divers, S.G.; Hoon, D.S.B.; Kopetz, E.S.; et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLoS ONE 2015, 10, e0140712. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.H.; Cunningham, D.; Werner, B.; Vlachogiannis, G.; Spiteri, I.; Heide, T.; Mateos, J.F.; Vatsiou, A.; Lampis, A.; Damavandi, M.D.; et al. Longitudinal Liquid Biopsy and Mathematical Modeling of Clonal Evolution Forecast Time to Treatment Failure in the PROSPECT-C Phase II Colorectal Cancer Clinical Trial. Cancer Discov. 2018, 8, 1270–1285. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Kinde, I.; Wang, Y.; Wong, H.L.; Roebert, J.; Christie, M.; Tacey, M.; Wong, R.; Singh, M.; Karapetis, C.S.; et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann. Oncol. 2015, 26, 1715–1722. [Google Scholar] [CrossRef]
- Yang, Y.-C.; Wang, D.; Jin, L.; Yao, H.-W.; Zhang, J.-H.; Wang, J.; Zhao, X.-M.; Shen, C.-Y.; Chen, W.; Wang, X.-L.; et al. Circulating tumor DNA detectable in early- and late-stage colorectal cancer patients. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef]
- Kim, T.W.; Peeters, M.; Thomas, A.L.; Gibbs, P.; Hool, K.; Zhang, J.; Ang, A.L.; Bach, B.A.; Price, T. Impact of Emergent Circulating Tumor DNA RAS Mutation in Panitumumab-Treated Chemoresistant Metastatic Colorectal Cancer. Clin. Cancer Res. 2018, 24, 5602–5609. [Google Scholar] [CrossRef]
- Peeters, M.; Price, T.; Boedigheimer, M.; Kim, T.W.; Ruff, P.; Gibbs, P.; Thomas, A.; Demonty, G.; Hool, K.; Ang, A. Evaluation of Emergent Mutations in Circulating Cell-Free DNA and Clinical Outcomes in Patients with Metastatic Colorectal Cancer Treated with Panitumumab in the ASPECCT Study. Clin. Cancer Res. 2019, 25, 1216–1225. [Google Scholar] [CrossRef]
- Kim, S.T.; Lee, W.S.; Lanman, R.B.; Mortimer, S.; Zill, O.A.; Kim, K.-M.; Jang, K.T.; Kim, S.-H.; Park, S.H.; Park, J.O.; et al. Prospective blinded study of somatic mutation detection in cell-free DNA utilizing a targeted 54-gene next generation sequencing panel in metastatic solid tumor patients. Oncotarget 2015, 6, 40360–40369. [Google Scholar] [CrossRef]
- Rachiglio, A.M.; Abate, R.E.; Sacco, A.; Pasquale, R.; Fenizia, F.; Lambiase, M.; Morabito, A.; Montanino, A.; Rocco, G.; Romano, C.; et al. Limits and potential of targeted sequencing analysis of liquid biopsy in patients with lung and colon carcinoma. Oncotarget 2016, 7, 66595–66605. [Google Scholar] [CrossRef]
- Onidani, K.; Shoji, H.; Kakizaki, T.; Yoshimoto, S.; Okaya, S.; Miura, N.; Sekikawa, S.; Furuta, K.; Lim, C.T.; Shibahara, T.; et al. Monitoring of cancer patients via next-generation sequencing of patient-derived circulating tumor cells and tumor DNA. Cancer Sci. 2019, 110, 2590–2599. [Google Scholar] [CrossRef]
- Yamauchi, M.; Urabe, Y.; Ono, A.; Miki, D.; Ochi, H.; Chayama, K. Serial profiling of circulating tumor DNA for optimization of anti-VEGF chemotherapy in metastatic colorectal cancer patients. Int. J. Cancer 2018, 142, 1418–1426. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, R.; Yan, C.; Liu, L.; Tong, Z.; Jiang, W.; Yao, M.; Fang, W.; Chen, Z. Advantage of Next-Generation Sequencing in Dynamic Monitoring of Circulating Tumor DNA over Droplet Digital PCR in Cetuximab Treated Colorectal Cancer Patients. Transl. Oncol. 2019, 12, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Beije, N.; Helmijr, J.C.; Weerts, M.J.; Beaufort, C.M.; Wiggin, M.; Marziali, A.; Verhoef, C.; Sleijfer, S.; Jansen, M.P.; Martens, J.W. Somatic mutation detection using various targeted detection assays in paired samples of circulating tumor DNA, primary tumor and metastases from patients undergoing resection of colorectal liver metastases. Mol. Oncol. 2016, 10, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Bachet, J.B.; Bouché, O.; Taieb, J.; Dubreuil, O.; Garcia, M.L.; Meurisse, A.; Normand, C.; Gornet, J.M.; Artru, P.; Louafi, S.; et al. RAS mutation analysis in circulating tumor DNA from patients with metastatic colorectal cancer: The AGEO RASANC prospective multicenter study. Ann. Oncol. 2018, 29, 1211–1219. [Google Scholar] [CrossRef]
- Jia, N.; Sun, Z.; Gao, X.; Cheng, Y.; Zhou, Y.; Shen, C.; Chen, W.; Wang, X.; Shi, R.; Li, N.; et al. Serial Monitoring of Circulating Tumor DNA in Patients with Metastatic Colorectal Cancer to Predict the Therapeutic Response. Front. Genet. 2019, 10, 470. [Google Scholar] [CrossRef]
- Yao, J.; Zang, W.; Ge, Y.; Weygant, N.; Yu, P.; Li, L.; Rao, G.; Jiang, Z.; Yan, R.; He, L.; et al. RAS/BRAF Circulating Tumor DNA Mutations as a Predictor of Response to First-Line Chemotherapy in Metastatic Colorectal Cancer Patients. Can. J. Gastroenterol. Hepatol. 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Beránek, M.; Sirák, I.; Vošmik, M.; Petera, J.; Drastíková, M.; Palička, V. Carrier molecules and extraction of circulating tumor DNA for next generation sequencing in colorectal cancer. Acta Med. 2016, 59, 54–58. [Google Scholar] [CrossRef][Green Version]
- Furuki, H.; Yamada, T.; Takahashi, G.; Iwai, T.; Koizumi, M.; Shinji, S.; Yokoyama, Y.; Takeda, K.; Taniai, N.; Uchida, E. Evaluation of liquid biopsies for detection of emerging mutated genes in metastatic colorectal cancer. Eur. J. Surg. Oncol. (EJSO) 2018, 44, 975–982. [Google Scholar] [CrossRef]
- Hsu, H.-C.; Lapke, N.; Wang, C.-W.; Lin, P.-Y.; You, J.-F.; Yeh, C.-Y.; Tsai, W.-S.; Hung, H.-Y.; Chang, S.-F.; Chen, H.-C.; et al. Targeted Sequencing of Circulating Tumor DNA to Monitor Genetic Variants and Therapeutic Response in Metastatic Colorectal Cancer. Mol. Cancer Ther. 2018, 17, 2238–2247. [Google Scholar] [CrossRef]
- DeMuth, C.; Spindler, K.-L.G.; Johansen, J.S.; Pallisgaard, N.; Nielsen, D.; Hogdall, E.; Vittrup, B.; Sorensen, B.S. Measuring KRAS Mutations in Circulating Tumor DNA by Droplet Digital PCR and Next-Generation Sequencing. Transl. Oncol. 2018, 11, 1220–1224. [Google Scholar] [CrossRef]
- Osumi, H.; Shinozaki, E.; Takeda, Y.; Wakatsuki, T.; Ichimura, T.; Saiura, A.; Yamaguchi, K.; Takahashi, S.; Noda, T.; Zembutsu, H. Clinical relevance of circulating tumor DNA assessed through deep sequencing in patients with metastatic colorectal cancer. Cancer Med. 2019, 8, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Ghatalia, P.; Smith, C.H.; Winer, A.; Gou, J.; Kiedrowski, L.A.; Slifker, M.; Saltzberg, P.D.; Bubes, N.; Anari, F.M.; Kasireddy, V.; et al. Clinical Utilization Pattern of Liquid Biopsies (LB) to Detect Actionable Driver Mutations, Guide Treatment Decisions and Monitor Disease Burden During Treatment of 33 Metastatic Colorectal Cancer (mCRC) Patients (pts) at a Fox Chase Cancer Center GI Oncology Subspecialty Clinic. Front. Oncol. 2018, 8, 652. [Google Scholar] [PubMed]
- Strickler, J.H.; Loree, J.M.; Ahronian, L.G.; Parikh, A.R.; Niedzwiecki, D.; Pereira, A.A.L.; McKinney, M.; Korn, W.M.; Atreya, C.E.; Banks, K.C.; et al. Genomic Landscape of Cell-Free DNA in Patients with Colorectal Cancer. Cancer Discov. 2018, 8, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Schwaederlé, M.C.; Fanta, P.T.; Okamura, R.; Leichman, L.; Lippman, S.M.; Lanman, R.B.; Raymond, V.M.; Talasaz, A.A.; Kurzrock, R. Genomic Assessment of Blood-Derived Circulating Tumor DNA in Patients with Colorectal Cancers: Correlation with Tissue Sequencing, Therapeutic Response, and Survival. JCO Precis. Oncol. 2019, 3. [Google Scholar] [CrossRef]
- Wang, B.; Wu, S.; Huang, F.; Shen, M.; Jiang, H.; Yu, Y.; Yu, Q.; Yang, Y.; Zhao, Y.; Zhou, Y.; et al. Analytical and clinical validation of a novel amplicon-based NGS assay for the evaluation of circulating tumor DNA in metastatic colorectal cancer patients. Clin. Chem. Lab. Med. 2019, 57, 1501–1510. [Google Scholar] [CrossRef]
- Shi, X.; Duose, D.Y.; Mehrotra, M.; Harmon, M.A.; Hu, P.; Wistuba, I.I.; Kopetz, S.; Luthra, R. Non-invasive genotyping of metastatic colorectal cancer using circulating cell free DNA. Cancer Genet. 2019, 237, 82–89. [Google Scholar] [CrossRef]
- Kato, S.; Okamura, R.; Mareboina, M.; Lee, S.; Goodman, A.; Patel, S.P.; Fanta, P.T.; Schwab, R.B.; Vu, P.; Raymond, V.M.; et al. Revisiting Epidermal Growth Factor Receptor (EGFR) Amplification as a Target for Anti-EGFR Therapy: Analysis of Cell-Free Circulating Tumor DNA in Patients with Advanced Malignancies. JCO Precis. Oncol. 2019, 3, 1–14. [Google Scholar] [CrossRef]
- El Messaoudi, S.; Rolet, F.; Mouliere, F.; Thierry, A.R. Circulating cell free DNA: Preanalytical considerations. Clin. Chim. Acta 2013, 424, 222–230. [Google Scholar] [CrossRef]
- Gorgannezhad, L.; Umer, M.; Islam, M.N.; Nguyen, N.-T.; Shiddiky, M.J.A. Circulating tumor DNA and liquid biopsy: Opportunities, challenges, and recent advances in detection technologies. Lab Chip 2018, 18, 1174–1196. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Lee, M.; Jeffrey, S.S. Circulating Tumor Cells and Circulating Tumor DNA: Challenges and Opportunities on the Path to Clinical Utility. Clin. Cancer Res. 2015, 21, 4786–4800. [Google Scholar] [CrossRef]
- Meddeb, R.; Pisareva, E.; Thierry, A.R. Guidelines for the Preanalytical Conditions for Analyzing Circulating Cell-Free DNA. Clin. Chem. 2019, 65, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Cremolini, C.; Rossini, D.; Dell’Aquila, E.; Lonardi, S.; Conca, E.; Del Re, M.; Busico, A.; Pietrantonio, F.; Danesi, R.; Aprile, G.; et al. Rechallenge for Patients with RAS and BRAF Wild-Type Metastatic Colorectal Cancer with Acquired Resistance to First-line Cetuximab and Irinotecan: A Phase 2 Single-Arm Clinical Trial. JAMA Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Anti-EGFR Therapy Rechallenge in Combination with Chemotherapy in Patients with Advanced Colorectal Cancer (A-REPEAT). Available online: https://clinicaltrials.gov/ct2/show/NCT03311750 (accessed on 5 August 2019).
- Overman, M.J.; Morris, V.; Kee, B.; Fogelman, D.; Xiao, L.; Eng, C.; Dasari, A.; Shroff, R.; Mazard, T.; Shaw, K.; et al. Utility of a molecular prescreening program in advanced colorectal cancer for enrollment on biomarker-selected clinical trials†. Ann. Oncol. 2016, 27, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Denis, J.A.; Guillerm, E.; Coulet, F.; Larsen, A.K.; Lacorte, J.-M. The Role of BEAMing and Digital PCR for Multiplexed Analysis in Molecular Oncology in the Era of Next-Generation Sequencing. Mol. Diagn. Ther. 2017, 21, 587–600. [Google Scholar] [CrossRef]
- García-Foncillas, J.; Alba, E.; Aranda, E.; Díaz-Rubio, E.; López-López, R.; Tabernero, J.; Vivancos, A. Incorporating BEAMing technology as a liquid biopsy into clinical practice for the management of colorectal cancer patients: An expert taskforce review. Ann. Oncol. 2017, 28, 2943–2949. [Google Scholar] [CrossRef]


{kind=link}
| First Author, Year of Publication | Study Population | Number of Advanced CRC Patients with Plasma NGS | Study Aim | Ref. |
|---|---|---|---|---|
| Beránek et al., 2016 | mCRC patients | 32 | Comparison of cfDNA extraction methods | [58] |
| Furuki et al., 2018 | 22 | Study of CRC heterogeneity | [59] | |
| Hsu et al., 2018 | 32 | Monitoring response to treatment | [60] | |
| Demuth et al., 2018 | 28 | Comparison of genotyping methods | [61] | |
| Osumi et al., 2018 | 101 | Assessment of feasibility and clinical relevance | [62] | |
| Ghatalia et al., 2019 | 33 | Assessment of feasibility and clinical relevance | [63] | |
| Strickler et al., 2018 | CRC patients, advanced | 1397 | Study of CRC heterogeneity | [64] |
| Kato et al., 2019 | CRC patients, 96% mCRC | 94 | Monitoring response to treatment | [65] |
| Yamauchi et al., 2017 | mCRC patients, bevacizumab-treated at first line | 21 | Study of the development of resistance | [52] |
| Peeters et al., 2018 | mCRC patients, panitumumab-treated at first line (ASPECCT trial) | 261 | Study of the development of resistance | [48] |
| Zhang et al., 2019 | mCRC, cetuximab-treated at first line | 15 | Comparison of genotyping methods | [53] |
| Khan et al., 2018 | mCRC, RAS wild-type, chemotherapy-refractory (Prospect-C trial) | 23 | Study of the development of resistance | [44] |
| Beije et al., 2016 | mCRC patients, resection of liver metastases | 12 | Comparison of genotyping methods | [54] |
| Tie et al., 2015 | mCRC patients, chemotherapy-naïve | 54 | Monitoring response to treatment | [45] |
| Bachet et al., 2018 | 412 | Comparison of genotyping methods | [55] | |
| Yao et al., 2018 | 76 | Monitoring response to treatment | [57] | |
| Jia et al., 2019 | 41 | Monitoring response to treatment | [56] | |
| Rachiglio et al., 2016 | Metastatic NSCLC and CRC patients (mCRC cohort) | 35 | Assessment of feasibility and clinical relevance | [50] |
| Kim et al., 2015 | Solid tumor patients (mCRC cohort) | 32 | Assessment of feasibility and clinical relevance | [49] |
| Onidani et al., 2019 | Head and neck cancer and gastrointestinal cancers (second phase with mCRC) | 7 | Study of the development of resistance | [51] |
| Wang et al., 2019 | mCRC | 184 | Analytical validation and assessment of clinical relevance | [66] |
| Shi et al., 2019 | Metastatic or locally advanced unresectable CRC | 34 | Assessment of feasibility and clinical relevance | [67] |
| Ref. | Company | Platform | Gene Panel | Number of Tested Genes | Library Preparation Protocols |
|---|---|---|---|---|---|
| [60] | Life Technologies | Ion PGM | Custom panel | 12 | NR |
| [54] | 21 | Ion AmpliSeq Library Kit 2.0 | |||
| [56] | SV-CA50-ctDNA panel | 50 | Ion AmpliSeq Library Kit 2.0 | ||
| [59] | Ion AmpliSeq Cancer Hotspot panel v2 | 50 | Ion AmpliSeq Kit | ||
| [51] | Ion AmpliSeq Cancer Hotspot panel v2 | 50 | Ion AmpliSeq Library Kit Plus | ||
| [50] | Not reported | 22 | Oncomine Solid Tumor DNA Kit | ||
| [61] | Ion AmpliSeq Colon & Lung v2 | 22 (KRAS) * | Oncomine Solid Tumor DNA Kit | ||
| [55] | Ion Proton | 22 | NR | ||
| [62] | Ion S5 | Custom panel | 14 | Oncomine Colon cfDNA | |
| [45] | Illumina | MiSeq | Custom panel | 15 (single mutation) * | NR |
| [58] | SOMATIC 1 MASTRTM v2 | BRAF, KRAS, NRAS | NR | ||
| [66] | MiSeq | Accu-Act Panel | 61 | CRC01 | |
| [52] | HiSeq 2500 | NCC Oncopanel | 90 | KAPA Hyper-prep | |
| [57] | Custom assay | 40 (KRAS, NRAS, HRAS, BRAF) * | SureSelect QTX | ||
| [44] | NextSeq 500 | AVENIO ctDNA Expanded Kit | 77 | Custom | |
| [53] | Custom assay | 18 | NR | ||
| [48] | NR | PlasmaSelect-R | 63 | NR | |
| [49] | Guardant Health | Guardant360 | 54 | Hybrid capture | |
| [64] | 54, 68, or 70 | ||||
| [65] | 54–73 | ||||
| [63] | 73 | ||||
| [67] | Guardant Health | Guardant360 | Ion AmpliSeq 2.0 | 68 | |
| Thermo Fisher Scientific | Ion Torrent | Oncomine Comprehensive Cancer Panel | 143 | ||
| Ref. | Whole Blood Input (mL) | Tubes | Time to Centrifugation | Centrifugation 1 | Centrifugation 2 | Plasma Input (mL) | DNA Input (ng) |
|---|---|---|---|---|---|---|---|
| [58] | 9-10 | EDTA | 1 h | 1300 rcf | 12,000 rcf | 0.75 | 0.35–4 |
| [59] | NR | EDTA | 3 h | 1900 rcf | 16,000 rcf | 1 | 10 |
| [60] | NR | Streck | NR | NR | - | 2.5–4 | NR |
| [61] | NR | NR | NR | 2300 | - | 0.2–2 | 1.1–10 |
| [62] | NR | EDTA | NR | 1600 rcf | 16,000 rcf | 2 | NR |
| [65] | 10 | NR | NR | NR | - | NR | NR |
| [52] | 10 | EDTA | Immediately | 3500 rpm | 12,000 rpm | 3 | 40 |
| [48] | 5 | EDTA | 30 min | 1500 rcf | - | NR | NR |
| [44] | NR | EDTA | 1 h | 1500 rcf | 1500 rcf | 4 | 25 |
| [54] | 30 | EDTA | 24 h | 800 rcf | - | 1 | 3–10 |
| [45] | 10 | NR | 3 h | NR | - | NR | 3 |
| [55] | 30 | Streck | Upon receival | 1600 rcf | 6000 rcf | NR | NR |
| [56] | NR | EDTA | NR | 1600 rcf | 16,000 rcf | NR | NR |
| [50] | 10 | EDTA | NR | 1600 rcf | 3000 rcf | 2 | 10 |
| [49] | NR | EDTA | Immediately | 1600 rcf | - | 1 | NR |
| [51] | 5 | EDTA | 24 h | NR | - | 2 | 20 |
| [66] | 20 | EDTA | 2 h | 1900 rcf | 16,000 rcf | 8 | >20 |
| [67] | 20 | Streck | NR | NR | - | 2*1.4–1.8 | 6–20 |
| [63] | 20 | Streck | NR | NR | - | NR | 5–30 |
| [53] | NR | NR | NR | NR | - | NR | 2–60 |
| Ref. | Prevalence of any Mutation | Prevalence of RAS/RAF Mutations in ctDNA | Method of Tissue Testing | Overall Tissue Concordance | RAS/RAF Tissue Concordance | Sensitivity of ctDNA NGS to Detect Known Tumor Tissue Variants | Cross-Platform Comparison |
|---|---|---|---|---|---|---|---|
| [61] | NR | NR | Routine PCR-based method | - | KRAS, 79% | NR | ddPCR |
| [62] | 87.1% | KRAS, 38.6% NRAS, 4.9% BRAF, 7.9% | - | RAS, 77.2% | NR | - | |
| [55] | 73% (est.) | KRAS, 42% NRAS, 4% | - | RAS, 85.2% | RAS, 76% | - | |
| [57] | NR | KRAS, 32.9% | - | KRAS, 81.25% | KRAS, 66.67% | - | |
| [56] | 95.7% | KRAS, 53.66% NRAS, 2.44% BRAF, 7.32% | - | RAS, 96% | RAS, 93.3% | - | |
| [50] | NR | NR | - | NR | 63.2% | ddPCR | |
| [66] | KRAS/NRAS BRAF/PIK3CA, 40.76% | NR | 79.89% (93.33% pre-treatment) | NR | 100% | - | |
| [49] | NR | NR | NGS | - | KRAS, 86.2% BRAFV600E, 100% | NR | - |
| [63] | NR | NR | - | KRAS, 66.67% | NR | - | |
| [65] | 79% | KRAS, 34% | - | KRAS, 75% BRAFV600E, 100% | NR | - | |
| [45] | 98.1% | NR | 90.57% (est.) | NR | NR | - | |
| [58] | NR | KRAS, 16% NRAS, 0% BRAF, 3% | 86% | NR | NR | - | |
| [60] | 72% | KRAS, 41% | 81% | NR | 85% | - | |
| [67] | NR | NR | 85.29% | NR | 92% | Digital sequencing | |
| [54] | NR | NR | - | NR | KRAS, 39–55% (primary tumor-liver metastases) | OnTarget enrichment, dPCR | |
| [59] | NR | NR | - | NR | 64% (liver metastases) | dPCR |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kastrisiou, M.; Zarkavelis, G.; Pentheroudakis, G.; Magklara, A. Clinical Application of Next-Generation Sequencing as A Liquid Biopsy Technique in Advanced Colorectal Cancer: A Trick or A Treat? Cancers 2019, 11, 1573. https://doi.org/10.3390/cancers11101573
Kastrisiou M, Zarkavelis G, Pentheroudakis G, Magklara A. Clinical Application of Next-Generation Sequencing as A Liquid Biopsy Technique in Advanced Colorectal Cancer: A Trick or A Treat? Cancers. 2019; 11(10):1573. https://doi.org/10.3390/cancers11101573
Chicago/Turabian StyleKastrisiou, Myrto, George Zarkavelis, George Pentheroudakis, and Angeliki Magklara. 2019. "Clinical Application of Next-Generation Sequencing as A Liquid Biopsy Technique in Advanced Colorectal Cancer: A Trick or A Treat?" Cancers 11, no. 10: 1573. https://doi.org/10.3390/cancers11101573
APA StyleKastrisiou, M., Zarkavelis, G., Pentheroudakis, G., & Magklara, A. (2019). Clinical Application of Next-Generation Sequencing as A Liquid Biopsy Technique in Advanced Colorectal Cancer: A Trick or A Treat? Cancers, 11(10), 1573. https://doi.org/10.3390/cancers11101573