Musclin, A Myokine Induced by Aerobic Exercise, Retards Muscle Atrophy During Cancer Cachexia in Mice

 , , ,
, , ,
Abstract
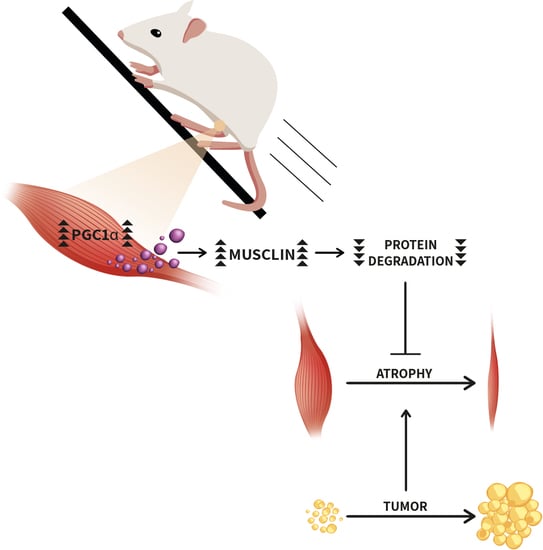
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Peroxisome Proliferator-Activated Receptor γ Coactivator 1α (PGC1α)-Overexpressing Myotubes Secrete Unknown Factors with Anti-Catabolic Action
2.2. Unlike PGC1β, Exogenous PGC1α Induces the Expression of Fibroblast Growth Factor 18 (FGF18), Natriuretic Peptide Precursor B (NppB), Amphiregulin (AREG) and Musclin in Myotubes and Tibialis Anterior (TA) Muscle
2.3. Among the Newly Identified PGC1α-Related Myokines, only Musclin Is Strongly Reduced in TA and Plasma of C26-Bearing Mice
2.4. Mice Deficient of Musclin Undergo more Muscle Depletion during Growth of Two Unrelated Tumors: Lewis Lung Carcinoma (LLC) or Methylcholanthrene-Induced Sarcoma 101 (MCG101)
2.5. Exogenous Expression of Musclin or Its Receptor Npr3 Partially Preserves Fiber Areas during C26 Growth in Mice
2.6. Running Protects Gastrocnemius from C26-Induced Atrophy and Restores PGC1α, Musclin and Npr3 Expression in Mice
2.7. Musclin Expression Does Not Change in Myristoylated AKT-Expressing Myotubes or in Hypertrophied Plantaris
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Adenoviruses and Plasmids
4.3. Microarrays
4.4. Protein Degradation and Synthesis in C2C12 Myotubes
4.5. Luciferase-Based Assays
4.6. Mice and Tumor Model
4.7. Musclin (Ostn)-Knockout (KO) Mouse Model
4.8. RNA Isolation from Cultured Cells or Muscles and Reverse Transcription
4.9. Quantitative Real-Time Polymerase Chain Reaction (PCR)
4.10. Protein Extraction and Western Blot
4.11. Enzyme-Linked Immunosorbent Assay (ELISA)
4.12. Electroporation of the TA with Plasmid DNA
4.13. Compensatory Hypertrophy
4.14. Muscle Sample Processing and Fiber Size Measurements
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Houten, L.; Reilley, A.A. An Investigation of the Cause of Death from Cancer. J. Surg. Oncol. 1980, 13, 111–116. [Google Scholar] [CrossRef]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and Classification of Cancer Cachexia: An International Consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Argiles, J.M.; Busquets, S.; Stemmler, B.; Lopez-Soriano, F.J. Cancer Cachexia: Understanding the Molecular Basis. Nat. Rev. Cancer. 2014, 14, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo Transcription Factors Induce the Atrophy-Related Ubiquitin Ligase Atrogin-1 and Cause Skeletal Muscle Atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Segatto, M.; Fittipaldi, R.; Pin, F.; Sartori, R.; Dae Ko, K.; Zare, H.; Fenizia, C.; Zanchettin, G.; Pierobon, E.S.; Hatakeyama, S.; et al. Epigenetic Targeting of Bromodomain Protein BRD4 Counteracts Cancer Cachexia and Prolongs Survival. Nat. Commun. 2017, 8, 1707. [Google Scholar] [CrossRef]
- Loumaye, A.; Thissen, J.-P. Biomarkers of Cancer Cachexia. Clin. Biochem. 2017, 50, 1281–1288. [Google Scholar] [CrossRef]
- Mathur, N.; Pedersen, B.K. Exercise as a Mean to Control Low-Grade Systemic Inflammation. Mediat. Inflamm. 2008, 2008, 109502. [Google Scholar] [CrossRef] [PubMed]
- Lira, F.; de Antunes, B.; Seelaender, M.; Neto, J.C. The Therapeutic Potential of Exercise to Treat Cachexia. Curr. Opin. Support. Palliat. Care 2015, 9, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Atherton, P.J.; Babraj, J.; Smith, K.; Singh, J.; Rennie, M.J.; Wackerhage, H. Selective Activation of AMPK-PGC-1alpha or PKB-TSC2-MTOR Signaling Can Explain Specific Adaptive Responses to Endurance or Resistance Training-like Electrical Muscle Stimulation. FASEB J. 2005, 19, 786–788. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.-M.V.; Gonzalez, M.; Poueymirou, W.T.; Kline, W.O.; Na, E.; Zlotchenko, E.; Stitt, T.N.; Economides, A.N.; Yancopoulos, G.D.; Glass, D.J. Conditional Activation of Akt in Adult Skeletal Muscle Induces Rapid Hypertrophy. Mol. Cell. Biol. 2004, 24, 9295–9304. [Google Scholar] [CrossRef]
- Terada, S.; Goto, M.; Kato, M.; Kawanaka, K.; Shimokawa, T.; Tabata, I. Effects of Low-Intensity Prolonged Exercise on PGC-1 MRNA Expression in Rat Epitrochlearis Muscle. Biochem. Biophys. Res. Commun. 2002, 296, 350–354. [Google Scholar] [CrossRef]
- Pilegaard, H.; Saltin, B.; Neufer, P.D. Exercise Induces Transient Transcriptional Activation of the PGC-1alpha Gene in Human Skeletal Muscle. J. Physiol. 2003, 546, 851–858. [Google Scholar] [CrossRef]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a Nodal Regulator of Mitochondrial Biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef]
- Handschin, C.; Chin, S.; Li, P.; Liu, F.; Maratos-Flier, E.; Lebrasseur, N.K.; Yan, Z.; Spiegelman, B.M. Skeletal Muscle Fiber-Type Switching, Exercise Intolerance, and Myopathy in PGC-1alpha Muscle-Specific Knock-out Animals. J. Biol. Chem. 2007, 282, 30014–30021. [Google Scholar] [CrossRef]
- Ciciliot, S.; Rossi, A.C.; Dyar, K.A.; Blaauw, B.; Schiaffino, S. Muscle Type and Fiber Type Specificity in Muscle Wasting. Int. J. Biochem. Cell Biol. 2013, 45, 2191–2199. [Google Scholar] [CrossRef]
- Sandri, M.; Lin, J.; Handschin, C.; Yang, W.; Arany, Z.P.; Lecker, S.H.; Goldberg, A.L.; Spiegelman, B.M. PGC-1alpha Protects Skeletal Muscle from Atrophy by Suppressing FoxO3 Action and Atrophy-Specific Gene Transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 16260–16265. [Google Scholar] [CrossRef] [PubMed]
- Brault, J.J.; Jespersen, J.G.; Goldberg, A.L. Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1alpha or 1beta Overexpression Inhibits Muscle Protein Degradation, Induction of Ubiquitin Ligases, and Disuse Atrophy. J. Biol. Chem. 2010, 285, 19460–19471. [Google Scholar] [CrossRef] [PubMed]
- Demontis, F.; Piccirillo, R.; Goldberg, A.L.; Perrimon, N. The Influence of Skeletal Muscle on Systemic Aging and Lifespan. Aging. Cell 2013, 12, 943–949. [Google Scholar] [CrossRef]
- Piccirillo, R. Exercise-Induced Myokines With Therapeutic Potential for Muscle Wasting. Front. Physiol. 2019, 10, 287. [Google Scholar] [CrossRef] [PubMed]
- Bostrom, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Bostrom, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-Alpha-Dependent Myokine That Drives Brown-Fat-like Development of White Fat and Thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Bina, H.A.; Ouchi, N.; Akasaki, Y.; Kharitonenkov, A.; Walsh, K. FGF21 Is an Akt-Regulated Myokine. FEBS Lett 2008, 582, 3805–3810. [Google Scholar] [CrossRef]
- Gould, D.W.; Lahart, I.; Carmichael, A.R.; Koutedakis, Y.; Metsios, G.S. Cancer Cachexia Prevention via Physical Exercise: Molecular Mechanisms. J. Cachexia Sarcopenia Muscle 2013, 4, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Bonetto, A.; Muscaritoli, M.; Costamagna, D.; Minero, V.G.; Bonelli, G.; Rossi Fanelli, F.; Baccino, F.M.; Costelli, P. Muscle Atrophy in Experimental Cancer Cachexia: Is the IGF-1 Signaling Pathway Involved? Int. J. Cancer 2010, 127, 1706–1717. [Google Scholar] [CrossRef] [PubMed]
- Subbotina, E.; Sierra, A.; Zhu, Z.; Gao, Z.; Koganti, S.R.; Reyes, S.; Stepniak, E.; Walsh, S.A.; Acevedo, M.R.; Perez-Terzic, C.M.; et al. Musclin Is an Activity-Stimulated Myokine That Enhances Physical Endurance. Proc. Natl. Acad. Sci. USA 2015, 112, 16042–16047. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, H.; Matsuda, M.; Yamada, Y.; Kawai, K.; Suzuki, E.; Makishima, M.; Kitamura, T.; Shimomura, I. Musclin, a Novel Skeletal Muscle-Derived Secretory Factor. J. Biol. Chem. 2004, 279, 19391–19395. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.; Moffatt, P.; Salois, P.; Gaumond, M.-H.; Gingras, R.; Godin, E.; Miao, D.; Goltzman, D.; Lanctôt, C. Osteocrin, a Novel Bone-Specific Secreted Protein That Modulates the Osteoblast Phenotype. J. Biol. Chem. 2003, 278, 50563–50571. [Google Scholar] [CrossRef] [PubMed]
- Potter, L.R.; Abbey-Hosch, S.; Dickey, D.M. Natriuretic Peptides, Their Receptors, and Cyclic Guanosine Monophosphate-Dependent Signaling Functions. Endocr. Rev. 2006, 27, 47–72. [Google Scholar] [CrossRef] [PubMed]
- Kita, S.; Nishizawa, H.; Okuno, Y.; Tanaka, M.; Yasui, A.; Matsuda, M.; Yamada, Y.; Shimomura, I. Competitive Binding of Musclin to Natriuretic Peptide Receptor 3 with Atrial Natriuretic Peptide. J Endocrinol. 2009, 201, 287–295. [Google Scholar] [CrossRef]
- Miyazaki, T.; Otani, K.; Chiba, A.; Nishimura, H.; Tokudome, T.; Takano-Watanabe, H.; Matsuo, A.; Ishikawa, H.; Shimamoto, K.; Fukui, H.; et al. A New Secretory Peptide of Natriuretic Peptide Family, Osteocrin, Suppresses the Progression of Congestive Heart Failure After Myocardial Infarction. Circ. Res. 2018, 122, 742–751. [Google Scholar] [CrossRef]
- Moffatt, P.; Thomas, G.; Sellin, K.; Bessette, M.-C.; Lafrenière, F.; Akhouayri, O.; St-Arnaud, R.; Lanctôt, C. Osteocrin Is a Specific Ligand of the Natriuretic Peptide Clearance Receptor That Modulates Bone Growth. J. Biol. Chem. 2007, 282, 36454–36462. [Google Scholar] [CrossRef]
- Whitham, M.; Febbraio, M.A. The Ever-Expanding Myokinome: Discovery Challenges and Therapeutic Implications. Nat. Rev. Drug Discov. 2016, 15, 719–729. [Google Scholar] [CrossRef]
- Arany, Z. PGC-1 Coactivators and Skeletal Muscle Adaptations in Health and Disease. Curr. Opin. Genet. Dev. 2008, 18, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, G.B.; Olivari, D.; Re Cecconi, A.D.; Talamini, L.; Ottoboni, L.; Lecker, S.H.; Stretch, C.; Baracos, V.E.; Bathe, O.F.; Resovi, A.; et al. Activation of the SDF1/CXCR4 Pathway Retards Muscle Atrophy during Cancer Cachexia. Oncogene 2016, 35, 6212–6222. [Google Scholar] [CrossRef] [PubMed]
- Silvennoinen, M.; Ahtiainen, J.P.; Hulmi, J.J.; Pekkala, S.; Taipale, R.S.; Nindl, B.C.; Laine, T.; Häkkinen, K.; Selänne, H.; Kyröläinen, H.; et al. PGC-1 Isoforms and Their Target Genes Are Expressed Differently in Human Skeletal Muscle Following Resistance and Endurance Exercise. Physiol. Rep. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Steensberg, A.; Fischer, C.; Keller, C.; Keller, P.; Plomgaard, P.; Febbraio, M.; Saltin, B. Searching for the Exercise Factor: Is IL-6 a Candidate? J. Muscle Res. Cell Motil. 2003, 24, 113–119. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Baltgalvis, K.A.; Puppa, M.J.; Sato, S.; Baynes, J.W.; Carson, J.A. Muscle Oxidative Capacity during IL-6-Dependent Cancer Cachexia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R201–R211. [Google Scholar] [CrossRef]
- Barreto, R.; Mandili, G.; Witzmann, F.A.; Novelli, F.; Zimmers, T.A.; Bonetto, A. Cancer and Chemotherapy Contribute to Muscle Loss by Activating Common Signaling Pathways. Front. Physiol. 2016, 7, 472. [Google Scholar] [CrossRef]
- Pretto, F.; Ghilardi, C.; Moschetta, M.; Bassi, A.; Rovida, A.; Scarlato, V.; Talamini, L.; Fiordaliso, F.; Bisighini, C.; Damia, G.; et al. Sunitinib prevents cachexia and prolongs survival of mice bearing renal cancer by restraining STAT3 and MuRF-1 activation in muscle. Oncotarget 2015, 6, 3043–3054. [Google Scholar]
- Busquets, S.; Toledo, M.; Orpi, M.; Massa, D.; Porta, M.; Capdevila, E.; Padilla, N.; Frailis, V.; Lopez-Soriano, F.J.; Han, H.Q.; et al. Myostatin Blockage Using ActRIIB Antagonism in Mice Bearing the Lewis Lung Carcinoma Results in the Improvement of Muscle Wasting and Physical Performance. J. Cachexia Sarcopenia Muscle 2012, 3, 37–43. [Google Scholar] [CrossRef]
- Wang, W.; Andersson, M.; Iresjö, B.-M.; Lönnroth, C.; Lundholm, K. Effects of Ghrelin on Anorexia in Tumor-Bearing Mice with Eicosanoid-Related Cachexia. Int. J. Oncol. 2006, 28, 1393–1400. [Google Scholar] [CrossRef]
- Montalvo, R.N.; Hardee, J.P.; VanderVeen, B.N.; Carson, J.A. Resistance Exercise’s Ability to Reverse Cancer-Induced Anabolic Resistance. Exerc. Sport Sci. Rev. 2018, 46, 247–253. [Google Scholar] [CrossRef]
- Nader, G.A.; Esser, K.A. Intracellular Signaling Specificity in Skeletal Muscle in Response to Different Modes of Exercise. J. Appl. Physiol. Bethesda Md 1985 2001, 90, 1936–1942. [Google Scholar] [CrossRef]
- Araki, S.; Izumiya, Y.; Hanatani, S.; Rokutanda, T.; Usuku, H.; Akasaki, Y.; Takeo, T.; Nakagata, N.; Walsh, K.; Ogawa, H. Akt1-Mediated Skeletal Muscle Growth Attenuates Cardiac Dysfunction and Remodeling after Experimental Myocardial Infarction. Circ. Heart Fail. 2012, 5, 116–125. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Goldberg, A.L.; Etlinger, J.D.; Goldspink, D.F.; Jablecki, C. Mechanism of Work-Induced Hypertrophy of Skeletal Muscle. Med. Sci. Sports 1975, 7, 185–198. [Google Scholar] [PubMed]
- Terena, S.M.L.; Fernandes, K.P.S.; Bussadori, S.K.; Deana, A.M.; Mesquita-Ferrari, R.A. Systematic Review of the Synergist Muscle Ablation Model for Compensatory Hypertrophy. Rev. Assoc. Medica Bras. 2017, 63, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Sasso, J.P.; Eves, N.D.; Christensen, J.F.; Koelwyn, G.J.; Scott, J.; Jones, L.W. A Framework for Prescription in Exercise-Oncology Research. J. Cachexia Sarcopenia Muscle 2015, 6, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Degens, H.; Meessen, N.E.; Wirtz, P.; Binkhorst, R.A. The Development of Compensatory Hypertrophy in the Plantaris Muscle of the Rat. Ann. Anat. Anat. Anz. 1995, 177, 285–289. [Google Scholar] [CrossRef]
- Pattison, J.S.; Folk, L.C.; Madsen, R.W.; Booth, F.W. Selected Contribution: Identification of Differentially Expressed Genes between Young and Old Rat Soleus Muscle during Recovery from Immobilization-Induced Atrophy. J. Appl. Physiol. 2003, 95, 2171–2179. [Google Scholar] [CrossRef][Green Version]
- Yasui, A.; Nishizawa, H.; Okuno, Y.; Morita, K.; Kobayashi, H.; Kawai, K.; Matsuda, M.; Kishida, K.; Kihara, S.; Kamei, Y.; et al. Foxo1 Represses Expression of Musclin, a Skeletal Muscle-Derived Secretory Factor. Biochem. Biophys. Res. Commun. 2007, 364, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Sable, C.L.; Filippa, N.; Hemmings, B.; Van Obberghen, E. CAMP Stimulates Protein Kinase B in a Wortmannin-Insensitive Manner. FEBS Lett. 1997, 409, 253–257. [Google Scholar] [CrossRef]
- Yano, S.; Tokumitsu, H.; Soderling, T.R. Calcium Promotes Cell Survival through CaM-K Kinase Activation of the Protein-Kinase-B Pathway. Nature 1998, 396, 584–587. [Google Scholar] [CrossRef]
- Sakamoto, K.; Hirshman, M.F.; Aschenbach, W.G.; Goodyear, L.J. Contraction Regulation of Akt in Rat Skeletal Muscle. J. Biol. Chem. 2002, 277, 11910–11917. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Staiger, H.; Haas, C.; Machicao, F.; Haring, H.U. The PPAR Gamma Agonist Troglitazone Induces Musclin MRNA Expression in Human Myotubes. Horm. Metab. Res. 2006, 38, 614–616. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Spiegelman, B.M. Peroxisome Proliferator-Activated Receptor-Gamma Coactivator 1 Alpha (PGC-1 Alpha): Transcriptional Coactivator and Metabolic Regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Garcia, S.; Nissanka, N.; Mareco, E.A.; Rossi, S.; Peralta, S.; Diaz, F.; Rotundo, R.L.; Carvalho, R.F.; Moraes, C.T. Overexpression of PGC-1α in Aging Muscle Enhances a Subset of Young-like Molecular Patterns. Aging Cell 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.S.; Ghosh, S.; Newman, S.; Salbaum, J.M. A Map of the PGC-1α- and NT-PGC-1α-Regulated Transcriptional Network in Brown Adipose Tissue. Sci. Rep. 2018, 8, 7876. [Google Scholar] [CrossRef]
- Guo, Q.; Hu, H.; Liu, X.; Yang, D.; Yin, Y.; Zhang, B.; He, H.; Oh, Y.; Wu, Q.; Liu, C.; et al. C/EBPβ Mediates Palmitate-Induced Musclin Expression via the Regulation of PERK/ATF4 Pathways in Myotubes. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E1081–E1092. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.R.; da Cunha, T.F.; da Paixao, N.A.; Brum, P.C. Aerobic Exercise Training as Therapy for Cardiac and Cancer Cachexia. Life Sci. 2015, 125, 9–14. [Google Scholar] [CrossRef]
- Ranjbar, K.; Ballarò, R.; Bover, Q.; Pin, F.; Beltrà, M.; Penna, F.; Costelli, P. Combined Exercise Training Positively Affects Muscle Wasting in Tumor-Bearing Mice. Med. Sci. Sports Exerc. 2019. [Google Scholar] [CrossRef]
- Judge, S.M.; Wu, C.-L.; Beharry, A.W.; Roberts, B.M.; Ferreira, L.F.; Kandarian, S.C.; Judge, A.R. Genome-Wide Identification of FoxO-Dependent Gene Networks in Skeletal Muscle during C26 Cancer Cachexia. BMC Cancer 2014, 14, 997. [Google Scholar] [CrossRef]
- Ishikawa, K.; Hara, T.; Kato, K.; Shimomura, T.; Omori, K. Involvement of Natriuretic Peptide System in C2C12 Myocytes. Mol. Cell. Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ruas, J.L.; Estall, J.L.; Rasbach, K.A.; Choi, J.H.; Ye, L.; Boström, P.; Tyra, H.M.; Crawford, R.W.; Campbell, K.P.; et al. The Unfolded Protein Response Mediates Adaptation to Exercise in Skeletal Muscle through a PGC-1α/ATF6α Complex. Cell Metab. 2011, 13, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Pasetto, L.; Olivari, D.; Nardo, G.; Trolese, M.C.; Bendotti, C.; Piccirillo, R.; Bonetto, V. Micro-Computed Tomography for Non-Invasive Evaluation of Muscle Atrophy in Mouse Models of Disease. PLoS ONE 2018, 13, e0198089. [Google Scholar] [CrossRef] [PubMed]
- Cala, M.P.; Agulló-Ortuño, M.T.; Prieto-García, E.; González-Riano, C.; Parrilla-Rubio, L.; Barbas, C.; Díaz-García, C.V.; García, A.; Pernaut, C.; Adeva, J.; et al. Multiplatform Plasma Fingerprinting in Cancer Cachexia: A Pilot Observational and Translational Study. J. Cachexia Sarcopenia Muscle 2018, 9, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Engeli, S.; Birkenfeld, A.L.; Badin, P.-M.; Bourlier, V.; Louche, K.; Viguerie, N.; Thalamas, C.; Montastier, E.; Larrouy, D.; Harant, I.; et al. Natriuretic Peptides Enhance the Oxidative Capacity of Human Skeletal Muscle. J. Clin. Invest. 2012, 122, 4675–4679. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, K.; Itoh, H.; Tsujimoto, H.; Tamura, N.; Fukunaga, Y.; Sone, M.; Yamahara, K.; Taura, D.; Inuzuka, M.; Sonoyama, T.; et al. Natriuretic Peptides/CGMP/CGMP-Dependent Protein Kinase Cascades Promote Muscle Mitochondrial Biogenesis and Prevent Obesity. Diabetes 2009, 58, 2880–2892. [Google Scholar] [CrossRef] [PubMed]
- Murthy, K.S.; Teng, B.Q.; Zhou, H.; Jin, J.G.; Grider, J.R.; Makhlouf, G.M. G(i-1)/G(i-2)-Dependent Signaling by Single-Transmembrane Natriuretic Peptide Clearance Receptor. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G974–G980. [Google Scholar] [CrossRef] [PubMed]
- Minetti, G.C.; Feige, J.N.; Rosenstiel, A.; Bombard, F.; Meier, V.; Werner, A.; Bassilana, F.; Sailer, A.W.; Kahle, P.; Lambert, C.; et al. Galphai2 Signaling Promotes Skeletal Muscle Hypertrophy, Myoblast Differentiation, and Muscle Regeneration. Sci. Signal. 2011, 4, ra80. [Google Scholar] [CrossRef]
- Wiegant, J.; van Hall, T.; van der Burg, M.; Colombo, M.; Tanke, H.J.; Offringa, R.; Rosenberg, C. Application of Multicolor Fluorescence in Situ Hybridization Analysis for Detection of Cross-Contamination and in Vitro Progression in Commonly Used Murine Tumor Cell Lines. Cancer Genet. Cytogenet. 2002, 139, 126–132. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Grosu, P.; Townsend, J.P.; Hartl, D.L.; Cavalieri, D. Pathway Processor: A Tool for Integrating Whole-Genome Expression Results into Metabolic Networks. Genome Res. 2002, 12, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenführer, J.; Lengauer, T. Improved Scoring of Functional Groups from Gene Expression Data by Decorrelating GO Graph Structure. Bioinforma. Oxf. Engl. 2006, 22, 1600–1607. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, R.; Goldberg, A.L. The P97/VCP ATPase Is Critical in Muscle Atrophy and the Accelerated Degradation of Muscle Proteins. EMBO J. 2012, 31, 3334–3350. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, S.D.; Suneja, M.; Ebert, S.M.; Bongers, K.S.; Fox, D.K.; Malmberg, S.E.; Alipour, F.; Shields, R.K.; Adams, C.M. MRNA Expression Signatures of Human Skeletal Muscle Atrophy Identify a Natural Compound That Increases Muscle Mass. Cell Metab. 2011, 13, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate Normalization of Real-Time Quantitative RT-PCR Data by Geometric Averaging of Multiple Internal Control Genes. Genome Biol. 2002, 3, research0034. [Google Scholar] [CrossRef]
- Pedersen, L.; Idorn, M.; Olofsson, G.H.; Lauenborg, B.; Nookaew, I.; Hansen, R.H.; Johannesen, H.H.; Becker, J.C.; Pedersen, K.S.; Dethlefsen, C.; et al. Voluntary Running Suppresses Tumor Growth through Epinephrine- and IL-6-Dependent NK Cell Mobilization and Redistribution. Cell Metab. 2016, 23, 554–562. [Google Scholar] [CrossRef]
- Catoire, M.; Mensink, M.; Kalkhoven, E.; Schrauwen, P.; Kersten, S. Identification of Human Exercise-Induced Myokines Using Secretome Analysis. Physiol. Genom. 2014, 46, 256–267. [Google Scholar] [CrossRef]
- Neufer, P.D.; Bamman, M.M.; Muoio, D.M.; Bouchard, C.; Cooper, D.M.; Goodpaster, B.H.; Booth, F.W.; Kohrt, W.M.; Gerszten, R.E.; Mattson, M.P.; et al. Understanding the Cellular and Molecular Mechanisms of Physical Activity-Induced Health Benefits. Cell Metab. 2015, 22, 4–11. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Re Cecconi, A.D.; Forti, M.; Chiappa, M.; Zhu, Z.; Zingman, L.V.; Cervo, L.; Beltrame, L.; Marchini, S.; Piccirillo, R. Musclin, A Myokine Induced by Aerobic Exercise, Retards Muscle Atrophy During Cancer Cachexia in Mice. Cancers 2019, 11, 1541. https://doi.org/10.3390/cancers11101541
Re Cecconi AD, Forti M, Chiappa M, Zhu Z, Zingman LV, Cervo L, Beltrame L, Marchini S, Piccirillo R. Musclin, A Myokine Induced by Aerobic Exercise, Retards Muscle Atrophy During Cancer Cachexia in Mice. Cancers. 2019; 11(10):1541. https://doi.org/10.3390/cancers11101541
Chicago/Turabian StyleRe Cecconi, Andrea D., Mara Forti, Michela Chiappa, Zhiyong Zhu, Leonid V. Zingman, Luigi Cervo, Luca Beltrame, Sergio Marchini, and Rosanna Piccirillo. 2019. "Musclin, A Myokine Induced by Aerobic Exercise, Retards Muscle Atrophy During Cancer Cachexia in Mice" Cancers 11, no. 10: 1541. https://doi.org/10.3390/cancers11101541
APA StyleRe Cecconi, A. D., Forti, M., Chiappa, M., Zhu, Z., Zingman, L. V., Cervo, L., Beltrame, L., Marchini, S., & Piccirillo, R. (2019). Musclin, A Myokine Induced by Aerobic Exercise, Retards Muscle Atrophy During Cancer Cachexia in Mice. Cancers, 11(10), 1541. https://doi.org/10.3390/cancers11101541