Gene-Specific Targeting of DNA Methylation in the Mammalian Genome


Abstract
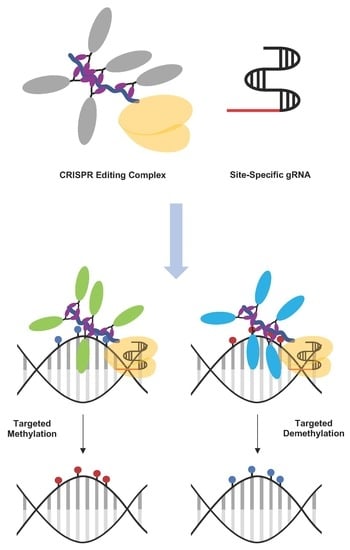
1. Introduction
2. Regulation and Maintenance Mechanisms of DNA Methylation
2.1. Methylation by De Novo and Maintenance Mechanisms
2.2. Active and Passive DNA Demethylation
3. DNA Methylation in Cancer
3.1. DNA Methylation as a Driving Force for the Functional Hallmarks of Cancer
3.2. Establishing Causality between DNA Methylation and Transcriptional Control
4. Gene-Specific Editing of DNA Methylation in the Mammalian Genome
4.1. ZNFs and TALEs
4.2. CRISPR-Based Editing Systems
4.2.1. Basic Components of CRISPR
4.2.2. Strategies for CRISPR-dCas9-Based Targeted Methylation
4.2.3. Strategies for CRISPR-dCas9-Based Targeted Demethylation
5. Application
5.1. In Vivo Applications
5.2. Potential Therapeutic Applications
6. Technical Considerations
6.1. Off-Target Effects of CRISPR-dCas9 Tools
6.2. Controls
6.3. Techniques Used for Further Analysis Post-Methylation-Editing
6.4. Expression of the dCas9 Effector and gRNA
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5caC | 5-carbocylcytosine |
| 5fC | 5-formylcytosine |
| 5hmC | 5-hydroxymethylcytosine |
| 5hmU | 5-hydroxymethyluracil |
| 5mC | 5-methylcytosine |
| 6mA | 6-methyladenine |
| AID | activation-induced deaminase |
| Ala | alanine |
| APOBEC | apolipoprotein B mRNA-editing enzyme complex |
| Asp | aspartic acid |
| ATP | adenosine triphosphate |
| BACH2 | broad complex-tramtrack-bric a brac and Cap’n’collar homology 2 |
| BCL2 | B cell lymphoma 2 |
| BER | base excision repair |
| bp | base pair |
| BRCA1 | breast cancer 1 |
| C | cytosine |
| CH3 | methyl group |
| ChIP | chromatin immunoprecipitation |
| CpG | cytosine-guanine dinucleotide |
| CRISPR | clustered regulatory interspaced short palindromic repeats |
| crRNA | CRISPR RNA |
| dCas9 | deactivated Cas9 |
| DNA | deoxyribonucleic acid |
| DNMT | DNA methyltransferase |
| DNMTi | DNA methyltransferase inhibitor |
| EBF3 | early B cell factor 3 |
| FMR1 | fragile X mental retardation 1 |
| FoxP3 | Forkhead box P3 |
| G | guanine |
| GATA4 | GATA binding protein 4 |
| Gly | glycine |
| GPR17 | G protein-coupled receptor 17 |
| gRNA | guide RNA |
| HIF-1α | hypoxia-inducible factor 1-alpha |
| His | histidine |
| HOXA | homeobox A |
| HOXD12 | homeobox D12 |
| hTERT | human telomerase reverse transcriptase |
| Igf2/H19 | insulin-like growth factor 2/H19 |
| KRAB | Krupple-associated box |
| MAGE | melanoma antigen gene |
| MGMT | O-6-methylguanine-DNA methyltransferases |
| mRNA | messenger RNA |
| MTase | methyltransferase |
| MyoD | myoblast determination protein 1 |
| p16/CDKN2A | p16INK4a/cyclin-dependent kinase inhibitor 2A |
| PAM | protospacer adjacent motif |
| PCR | polymerase chain reaction |
| RASSF1A | Ras association domain-containing protein 1 |
| RB | retinoblastoma |
| RNA | ribonucleic acid |
| RNA-seq | RNA-sequencing |
| ROS1CD | ROS1 5mC DNA glycosylase |
| RT-qPCR | quantitative reverse transcription PCR |
| SAH | S-adenosyl-homocysteine |
| SAM | S-adenosyl-L-methionine |
| Ser | serine |
| sgRNA | single-guide RNA |
| SMRT | single-molecule real-time |
| SunTag | SUperNova TAGging |
| TALE | transcription activator-like effector |
| TDG | thymine DNA glycosylase |
| TET | ten-eleven translocation |
| TIMP2 | tissue inhibitor of metalloproteinases 2 |
| tracrRNA | trans-activating CRISPR RNA |
| UHRF1 | ubiquitin-like containing PHD and RING finger domains 1 |
| VEGF-2 | vascular endothelial growth factor 2 |
| VHL | Von Hippel-Lindau |
| WT1 | Wilms tumor 1 |
| ZNF | zinc finger |
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. Ca A Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef]
- Vizoso, M.; Ferreira, H.J.; Lopez-Serra, P.; Carmona, F.J.; Martínez-Cardús, A.; Girotti, M.R.; Villanueva, A.; Guil, S.; Moutinho, C.; Liz, J.; et al. Epigenetic activation of a cryptic TBC1D16 transcript enhances melanoma progression by targeting EGFR. Nat. Med. 2015, 21, 741. [Google Scholar] [CrossRef] [PubMed]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef]
- Timp, W.; Feinberg, A.P. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat. Rev. Cancer 2013, 13, 497. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Chen, D.; Chen, D. Study on shape of modern folk robes and structure recovery. J. Silk 2015, 52, 42–47. [Google Scholar]
- Chatterjee, A.; Stockwell, P.A.; Ahn, A.; Rodger, E.J.; Leichter, A.L.; Eccles, M.R. Genome-wide methylation sequencing of paired primary and metastatic cell lines identifies common DNA methylation changes and a role for EBF3 as a candidate epigenetic driver of melanoma metastasis. Oncotarget 2017, 8, 6085–6101. [Google Scholar] [CrossRef]
- Chatterjee, A.; Rodger, E.J.; Eccles, M.R. Epigenetic drivers of tumourigenesis and cancer metastasis. Semin. Cancer Biol. 2018, 51, 149–159. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef]
- Inbar-Feigenberg, M.; Choufani, S.; Butcher, D.T.; Roifman, M.; Weksberg, R. Basic concepts of epigenetics. Fertil. Steril. 2013, 99, 607–615. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2012, 38, 23. [Google Scholar] [CrossRef] [PubMed]
- Bestor, T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402. [Google Scholar] [CrossRef] [PubMed]
- Hata, K.; Okano, M.; Lei, H.; Li, E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Dev. Camb. Engl. 2002, 129, 1983–1993. [Google Scholar]
- Kaneda, M.; Okano, M.; Hata, K.; Sado, T.; Tsujimoto, N.; Li, E.; Sasaki, H. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 2004, 429, 900–903. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Xu, G.L.; Bestor, T.H.; Bourchis, D.; Hsieh, C.L.; Tommerup, N.; Bugge, M.; Hulten, M.; Qu, X.; Russo, J.J.; Viegas-Pequignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999, 402, 187–191. [Google Scholar] [CrossRef]
- Jin, Z.; Liu, Y. DNA methylation in human diseases. Genes Dis. 2018, 5, 1–8. [Google Scholar] [CrossRef]
- Cortellino, S.; Xu, J.; Sannai, M.; Moore, R.; Caretti, E.; Cigliano, A.; Le Coz, M.; Devarajan, K.; Wessels, A.; Soprano, D. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011, 146, 67–79. [Google Scholar] [CrossRef]
- Sadakierska-Chudy, A.; Kostrzewa, R.M.; Filip, M. A comprehensive view of the epigenetic landscape part I: DNA methylation, passive and active DNA demethylation pathways and histone variants. Neurotox. Res. 2015, 27, 84–97. [Google Scholar] [CrossRef]
- Hill, P.W.S.; Amouroux, R.; Hajkova, P. DNA demethylation, Tet proteins and 5-hydroxymethylcytosine in epigenetic reprogramming: An emerging complex story. Genomics 2014, 104, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Nabel, C.S.; Jia, H.; Ye, Y.; Shen, L.; Goldschmidt, H.L.; Stivers, J.T.; Zhang, Y.; Kohli, R.M. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat. Chem. Biol. 2012, 8, 751. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Rodger, E.J.; Ahn, A.; Stockwell, P.A.; Parry, M.; Motwani, J.; Gallagher, S.J.; Shklovskaya, E.; Tiffen, J.; Eccles, M.R.; et al. Marked Global DNA Hypomethylation Is Associated with Constitutive PD-L1 Expression in Melanoma. Science 2018, 4, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011, 17, 330. [Google Scholar] [CrossRef]
- Ziller, M.J.; Gu, H.; Müller, F.; Donaghey, J.; Tsai, L.T.Y.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013, 500, 477. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Chatterjee, A.; Macaulay, E.C.; Rodger, E.J.; Stockwell, P.A.; Parry, M.F.; Roberts, H.E.; Slatter, T.L.; Hung, N.A.; Devenish, C.J.; Morison, I.M. Placental Hypomethylation Is More Pronounced in Genomic Loci Devoid of Retroelements. G3 (Bethesda) 2016, 6, 1911–1921. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Dammann, R.; Schagdarsurengin, U.; Strunnikova, M.; Rastetter, M.; Seidel, C.; Liu, L.; Tommasi, S.; Pfeifer, G.P. Epigenetic inactivation of the Ras-association domain family 1 (RASSF1A) gene and its function in human carcinogenesis. Histol. Histopathol. 2003, 18, 665–677. [Google Scholar] [CrossRef]
- Teitz, T.; Wei, T.; Valentine, M.B.; Vanin, E.F.; Grenet, J.; Valentine, V.A.; Behm, F.G.; Look, A.T.; Lahti, J.M.; Kidd, V.J. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat. Med. 2000, 6, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Merlo, A.; Mao, L.; Lapidus, R.G.; Issa, J.P.; Davidson, N.E.; Sidransky, D.; Baylin, S.B. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995, 55, 4525–4530. [Google Scholar] [PubMed]
- Herman, J.G.; Latif, F.; Weng, Y.; Lerman, M.I.; Zbar, B.; Liu, S.; Samid, D.; Duan, D.S.; Gnarra, J.R.; Linehan, W.M. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc. Natl. Acad. Sci. USA 1994, 91, 9700–9704. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.P.; Keaney, J.F., Jr. Epigenetic control of angiogenesis via DNA methylation. Circulation 2011, 123, 2916–2918. [Google Scholar] [CrossRef]
- Greger, V.; Passarge, E.; Hopping, W.; Messmer, E.; Horsthemke, B. Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum. Genet. 1989, 83, 155–158. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Maio, M.; Covre, A.; Fratta, E.; Di Giacomo, A.M.; Taverna, P.; Natali, P.G.; Coral, S.; Sigalotti, L. Molecular Pathways: At the Crossroads of Cancer Epigenetics and Immunotherapy. Clin. Cancer Res. 2015, 21, 4040–4047. [Google Scholar] [CrossRef]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249. [Google Scholar] [CrossRef]
- Dear, A.E. Epigenetic Modulators and the New Immunotherapies. N. Engl. J. Med. 2016, 374, 684–686. [Google Scholar] [CrossRef]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2016, 164, 1073. [Google Scholar] [CrossRef]
- Wrangle, J.; Wang, W.; Koch, A.; Easwaran, H.; Mohammad, H.P.; Vendetti, F.; Vancriekinge, W.; Demeyer, T.; Du, Z.; Parsana, P.; et al. Alterations of immune response of Non-Small Cell Lung Cancer with Azacytidine. Oncotarget 2013, 4, 2067–2079. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2009, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Horn, G.; Moulton, K.; Oza, A.; Byler, S.; Kokolus, S.; Longacre, M. Cancer Development, Progression, and Therapy: An Epigenetic Overview. Int. J. Mol. Sci. 2013, 14, 21087–21113. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetic gene silencing in cancer: The DNA hypermethylome. Hum. Mol. Genet. 2007, 16, R50–R59. [Google Scholar] [CrossRef] [PubMed]
- Hanada, M.; Delia, D.; Aiello, A.; Stadtmauer, E.; Reed, J.C. bcl-2 Gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood 1993, 82, 1820–1828. [Google Scholar] [PubMed]
- Nishigaki, M.; Aoyagi, K.; Danjoh, I.; Fukaya, M.; Yanagihara, K.; Sakamoto, H.; Yoshida, T.; Sasaki, H. Discovery of Aberrant Expression of R-RAS by Cancer-Linked DNA Hypomethylation in Gastric Cancer Using Microarrays. Cancer Res. 2005, 65, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.M.; Glazer, C.A.; Mithani, S.K.; Ochs, M.F.; Sun, W.; Bhan, S.; Vostrov, A.; Abdullaev, Z.; Lobanenkov, V.; Gray, A.; et al. Coordinated activation of candidate proto-oncogenes and cancer testes antigens via promoter demethylation in head and neck cancer and lung cancer. PLoS ONE 2009, 4, e4961. [Google Scholar] [CrossRef] [PubMed]
- Koslowski, M.; Luxemburger, U.; Türeci, Ö.; Sahin, U. Tumor-associated CpG demethylation augments hypoxia-induced effects by positive autoregulation of HIF-1α. Oncogene 2010, 30, 876. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D’Anna, F.; Kuchnio, A.; Ploumakis, A.; Ghesquière, B.; Van Dyck, L.; Boeckx, B.; Schoonjans, L.; et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016, 537, 63. [Google Scholar] [CrossRef]
- Chatterjee, A.; Stockwell, P.A.; Rodger, E.J.; Parry, M.F.; Eccles, M.R. scan_tcga tools for integrated epigenomic and transcriptomic analysis of tumor subgroups. Epigenomics 2016, 8, 1315–1330. [Google Scholar] [CrossRef] [PubMed]
- Guillaumet-Adkins, A.; Richter, J.; Odero, M.D.; Sandoval, J.; Agirre, X.; Catala, A.; Esteller, M.; Prósper, F.; Calasanz, M.J.; Buño, I.; et al. Hypermethylation of the alternative AWT1 promoter in hematological malignancies is a highly specific marker for acute myeloid leukemias despite high expression levels. J. Hematol. Oncol. 2014, 7, 4. [Google Scholar] [CrossRef]
- Parashar, G.; Capalash, N. Expression of the TIMP2 gene is not regulated by promoter hypermethylation in the Caski cell line. Oncol. Lett. 2012, 3, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Guilleret, I.; Yan, P.; Grange, F.; Braunschweig, R.; Bosman, F.T.; Benhattar, J. Hypermethylation of the human telomerase catalytic subunit (hTERT) gene correlates with telomerase activity. Int. J. Cancer 2002, 101, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Castelo-Branco, P.; Choufani, S.; Mack, S.; Gallagher, D.; Zhang, C.; Lipman, T.; Zhukova, N.; Walker, E.J.; Martin, D.; Merino, D.; et al. Methylation of the TERT promoter and risk stratification of childhood brain tumours: An integrative genomic and molecular study. Lancet Oncol. 2013, 14, 534–542. [Google Scholar] [CrossRef]
- Choi, J.H.; Park, S.H.; Park, J.; Park, B.G.; Cha, S.J.; Kong, K.H.; Lee, K.H.; Park, A.J. Site-specific methylation of CpG nucleotides in the hTERT promoter region can control the expression of hTERT during malignant progression of colorectal carcinoma. Biochem. Biophys. Res. Commun. 2007, 361, 615–620. [Google Scholar] [CrossRef] [PubMed]
- De Wilde, J.; Kooter, J.M.; Overmeer, R.M.; Claassen-Kramer, D.; Meijer, C.J.; Snijders, P.J.; Steenbergen, R.D. hTERT promoter activity and CpG methylation in HPV-induced carcinogenesis. BMC Cancer 2010, 10, 271. [Google Scholar] [CrossRef]
- Takasawa, K.; Arai, Y.; Yamazaki-Inoue, M.; Toyoda, M.; Akutsu, H.; Umezawa, A.; Nishino, K. DNA hypermethylation enhanced telomerase reverse transcriptase expression in human-induced pluripotent stem cells. Hum. Cell 2018, 31, 78–86. [Google Scholar] [CrossRef]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef]
- Issa, J.P.; Kantarjian, H.M. Targeting DNA methylation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 3938–3946. [Google Scholar] [CrossRef]
- Christman, J.K. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef] [PubMed]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 2010, 186, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Eccles, M.R. DNA Methylation and Epigenomics: New Technologies and Emerging Concepts. Genome Biol. 2015, 16, 103. [Google Scholar] [CrossRef] [PubMed]
- Grimmer, M.R.; Stolzenburg, S.; Ford, E.; Lister, R.; Blancafort, P.; Farnham, P.J. Analysis of an artificial zinc finger epigenetic modulator: Widespread binding but limited regulation. Nucleic Acids Res. 2014, 42, 10856–10868. [Google Scholar] [CrossRef]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Enriquez, P. CRISPR-Mediated Epigenome Editing. Yale J. Biol. Med. 2016, 89, 471–486. [Google Scholar] [PubMed]
- Oka, M.; Rodic, N.; Graddy, J.; Chang, L.J.; Terada, N. CpG sites preferentially methylated by Dnmt3a in vivo. J. Biol. Chem. 2006, 281, 9901–9908. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.X.; Mann, J.R.; Hsieh, C.L.; Riggs, A.D.; Chedin, F. Physical and functional interactions between the human DNMT3L protein and members of the de novo methyltransferase family. J. Cell. Biochem. 2005, 95, 902–917. [Google Scholar] [CrossRef] [PubMed]
- Vojta, A.; Dobrinic, P.; Tadic, V.; Bockor, L.; Korac, P.; Julg, B.; Klasic, M.; Zoldos, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef] [PubMed]
- Stepper, P.; Kungulovski, G.; Jurkowska, R.Z.; Chandra, T.; Krueger, F.; Reinhardt, R.; Reik, W.; Jeltsch, A.; Jurkowski, T.P. Efficient targeted DNA methylation with chimeric dCas9-Dnmt3a-Dnmt3L methyltransferase. Nucleic Acids Res. 2017, 45, 1703–1713. [Google Scholar] [CrossRef]
- Amabile, A.; Migliara, A.; Capasso, P.; Biffi, M.; Cittaro, D.; Naldini, L.; Lombardo, A. Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell 2016, 167, 219–232.e214. [Google Scholar] [CrossRef]
- Suetake, I.; Shinozaki, F.; Miyagawa, J.; Takeshima, H.; Tajima, S. DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J. Biol. Chem. 2004, 279, 27816–27823. [Google Scholar] [CrossRef]
- Jia, D.; Jurkowska, R.Z.; Zhang, X.; Jeltsch, A.; Cheng, X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 2007, 449, 248–251. [Google Scholar] [CrossRef]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Su, J.; Lei, Y.; Brunetti, L.; Gundry, M.C.; Zhang, X.; Jeong, M.; Li, W.; Goodell, M.A. DNA epigenome editing using CRISPR-Cas SunTag-directed DNMT3A. Genome Biol. 2017, 18, 176. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Zhang, X.; Su, J.; Jeong, M.; Gundry, M.C.; Huang, Y.H.; Zhou, Y.; Li, W.; Goodell, M.A. Targeted DNA methylation in vivo using an engineered dCas9-MQ1 fusion protein. Nat. Commun. 2017, 8, 16026. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA Methylation in the Mammalian Genome. Cell 2016, 167, 233–247.e217. [Google Scholar] [CrossRef] [PubMed]
- Saunderson, E.A.; Stepper, P.; Gomm, J.J.; Hoa, L.; Morgan, A.; Allen, M.D.; Jones, J.L.; Gribben, J.G.; Jurkowski, T.P.; Ficz, G. Hit-and-run epigenetic editing prevents senescence entry in primary breast cells from healthy donors. Nat. Commun. 2017, 8, 1450. [Google Scholar] [CrossRef] [PubMed]
- Pflueger, C.; Tan, D.; Swain, T.; Nguyen, T.; Pflueger, J.; Nefzger, C.; Polo, J.M.; Ford, E.; Lister, R. A modular dCas9-SunTag DNMT3A epigenome editing system overcomes pervasive off-target activity of direct fusion dCas9-DNMT3A constructs. Genome Res. 2018, 28, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Xiong, T.; Meister, G.E.; Workman, R.E.; Kato, N.C.; Spellberg, M.J.; Turker, F.; Timp, W.; Ostermeier, M.; Novina, C.D. Targeted DNA methylation in human cells using engineered dCas9-methyltransferases. Sci. Rep. 2017, 7, 6732. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Angstman, J.F.; Richardson, M.E.; Linder, S.J.; Cascio, V.M.; Tsai, S.Q.; Ho, Q.H.; Sander, J.D.; Reyon, D.; Bernstein, B.E.; et al. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat. Biotechnol. 2013, 31, 1137. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kazemier, H.G.; de Groote, M.L.; Ruiters, M.H.J.; Xu, G.-L.; Rots, M.G. Induced DNA demethylation by targeting Ten-Eleven Translocation 2 to the human ICAM-1 promoter. Nucleic Acids Res. 2013, 42, 1563–1574. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.R.; Cui, Y.; Lubecka, K.; Stefanska, B.; Irudayaraj, J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget 2016, 7, 46545–46556. [Google Scholar] [CrossRef]
- Xu, X.; Tao, Y.; Gao, X.; Zhang, L.; Li, X.; Zou, W.; Ruan, K.; Wang, F.; Xu, G.-L.; Hu, R. A CRISPR-based approach for targeted DNA demethylation. Cell Discov. 2016, 2, 16009. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Noguchi, H.; Horii, T.; Nakabayashi, K.; Kimura, M.; Okamura, K.; Sakai, A.; Nakashima, H.; Hata, K.; Nakashima, K.; et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat. Biotechnol. 2016, 34, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Wu, H.; Krzisch, M.; Wu, X.; Graef, J.; Muffat, J.; Hnisz, D.; Li, C.H.; Yuan, B.; Xu, C.; et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 2018, 172, 979–992.e976. [Google Scholar] [CrossRef] [PubMed]
- Parrilla-Doblas, J.T.; Ariza, R.R.; Roldán-Arjona, T. Targeted DNA demethylation in human cells by fusion of a plant 5-methylcytosine DNA glycosylase to a sequence-specific DNA binding domain. Epigenetics 2017, 12, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Wang, J.; Sun, W.; Huang, W.; Cai, Z.; Zhao, G.; Wang, J. Reprogrammable CRISPR/dCas9-based recruitment of DNMT1 for site-specific DNA demethylation and gene regulation. Cell Discov. 2019, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Gregory, D.J.; Zhang, Y.; Kobzik, L.; Fedulov, A.V. Specific transcriptional enhancement of inducible nitric oxide synthase by targeted promoter demethylation. Epigenetics 2013, 8, 1205–1212. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Kanamori, M.; Someya, K.; Nakatsukasa, H.; Yoshimura, A. Stabilization of Foxp3 expression by CRISPR-dCas9-based epigenome editing in mouse primary T cells. Epigenetics Chromatin 2017, 10, 24. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, L.; Bell, P.; McMenamin, D.; He, Z.; White, J.; Yu, H.; Xu, C.; Morizono, H.; Musunuru, K.; et al. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat. Biotechnol. 2016, 34, 334. [Google Scholar] [CrossRef]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Rivera, R.M.C.; Madhavan, S.; Pan, X.; Ran, F.A.; Yan, W.X.; et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef]
- Tabebordbar, M.; Zhu, K.; Cheng, J.K.W.; Chew, W.L.; Widrick, J.J.; Yan, W.X.; Maesner, C.; Wu, E.Y.; Xiao, R.; Ran, F.A.; et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016, 351, 407–411. [Google Scholar] [CrossRef]
- Swiech, L.; Heidenreich, M.; Banerjee, A.; Habib, N.; Li, Y.; Trombetta, J.; Sur, M.; Zhang, F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 2014, 33, 102. [Google Scholar] [CrossRef] [PubMed]
- Azad, N.; Zahnow, C.A.; Rudin, C.M.; Baylin, S.B. The future of epigenetic therapy in solid tumours--lessons from the past. Nat. Rev. Clin. Oncol. 2013, 10, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Jakovcevski, M.; Akbarian, S. Epigenetic mechanisms in neurological disease. Nat. Med. 2012, 18, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Rodger, E.J.; Chatterjee, A.; Morison, I.M. 5-hydroxymethylcytosine: A potential therapeutic target in cancer. Epigenomics 2014, 6, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Polstein, L.R.; Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Bledsoe, P.; Song, L.; Safi, A.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Genome-wide specificity of DNA binding, gene regulation, and chromatin remodeling by TALE- and CRISPR/Cas9-based transcriptional activators. Genome Res. 2015, 25, 1158–1169. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Scott, D.A.; Kriz, A.J.; Chiu, A.C.; Hsu, P.D.; Dadon, D.B.; Cheng, A.W.; Trevino, A.E.; Konermann, S.; Chen, S.; et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 2014, 32, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Lu, G.; Xie, Z.; Lou, M.; Luo, J.; Guo, L.; Zhang, Y. Genome-wide identification of CRISPR/Cas9 off-targets in human genome. Cell Res. 2014, 24, 1009–1012. [Google Scholar] [CrossRef] [PubMed]
- O’Geen, H.; Henry, I.M.; Bhakta, M.S.; Meckler, J.F.; Segal, D.J. A genome-wide analysis of Cas9 binding specificity using ChIP-seq and targeted sequence capture. Nucleic Acids Res. 2015, 43, 3389–3404. [Google Scholar] [CrossRef]
- Ehrlich, M.; Gama-Sosa, M.A.; Huang, L.-H.; Midgett, R.M.; Kuo, K.C.; McCune, R.A.; Gehrke, C. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues or cells. Nucleic Acids Res. 1982, 10, 2709–2721. [Google Scholar] [CrossRef]
- McDonald, J.I.; Celik, H.; Rois, L.E.; Fishberger, G.; Fowler, T.; Rees, R.; Kramer, A.; Martens, A.; Edwards, J.R.; Challen, G.A. Reprogrammable CRISPR/Cas9-based system for inducing site-specific DNA methylation. Biol. Open 2016, 5, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Rodger, E.J.; Morison, I.M.; Eccles, M.R.; Stockwell, P.A. Tools and Strategies for Analysis of Genome-Wide and Gene-Specific DNA Methylation Patterns. Methods Mol. Biol. 2017, 1537, 249–277. [Google Scholar] [CrossRef]
- Pulecio, J.; Verma, N.; Mejia-Ramirez, E.; Huangfu, D.; Raya, A. CRISPR/Cas9-Based Engineering of the Epigenome. Cell Stem Cell 2017, 21, 431–447. [Google Scholar] [CrossRef] [PubMed]
- Flusberg, B.A.; Webster, D.R.; Lee, J.H.; Travers, K.J.; Olivares, E.C.; Clark, T.A.; Korlach, J.; Turner, S.W. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 2010, 7, 461. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Thakore, P.I.; D’Ippolito, A.M.; Song, L.; Safi, A.; Shivakumar, N.K.; Kabadi, A.M.; Reddy, T.E.; Crawford, G.E.; Gersbach, C.A. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 2015, 12, 1143. [Google Scholar] [CrossRef] [PubMed]
- Kroll, C.; Rathert, P. Stable Expression of Epigenome Editors via Viral Delivery and Genomic Integration. Methods Mol. Biol. 2018, 1767, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.-Y.; Liu, D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2014, 33, 73. [Google Scholar] [CrossRef]





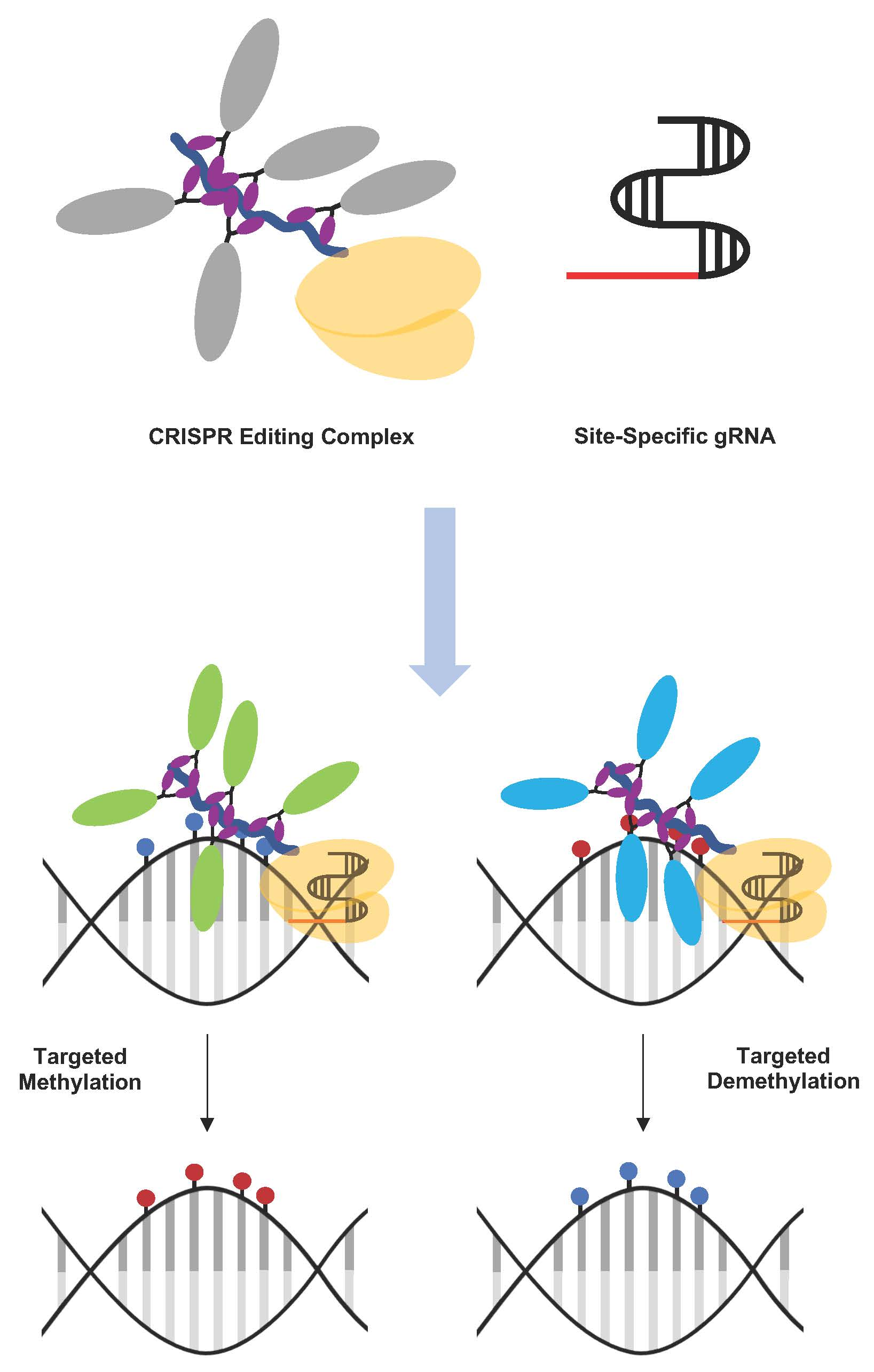{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hallmark | Gene | Gene Function |
|---|---|---|
| Self-sufficiency in growth signals | RASSF1A | Regulation of Ras pathway [31] |
| Evading apoptosis | Caspase-8 | Initiation of apoptosis [32] |
| Insensitivity to antigrowth signals | p16/CDKN2A | Cyclin-kinase inhibitor [33] |
| Tissue invasion and metastasis | VHL (Von Hippel-Lindau) | Suppression of metastasis [34] |
| Sustained angiogenesis | VEGF-2 | Crucial for angiogenesis [35] |
| Limitless replicative potential | RB (Retinoblastoma) | Cell cycle regulation [36] |
| dCas9 Tool | Feature |
|---|---|
| dCas9-DNMT3A | Targeted CpG methylation-altered CTCG looping and local gene expression [85] |
| dCas9-DNMT3ACD | Targeted CpG methylation of the promoter silences gene expression; high off-target DNA methylation is observed using unspecified sgRNAs [77] |
| dCas9-DNMT3ACD-DNMT3L | Multimerization of DNMT3A-DNMT3L complexes on the promoter to induce long term hypermethylation and gene silencing [78,86] |
| dCas9-DNMT3ACD, DNMT3L, KRAB | Triple-engineered transcriptional repressors (ETRs): Using a combination of Cas9-DNMT3A, dCas9-DNMT3L and dCas9-KRAB to promote long-term silencing of endogenous genes [79] |
| dCas9-SunTag-DNMT3A | SunTag recruits multiple copies of antibody-fused DNMT3A to increase CpG methylation [83] |
| dCas9-MQ1 | In vivo application in mice by zygote microinjection [84] |
| dCas9-SunTag-DNMT3ACD | Modular SunTag shows reduction of off-target events [87] |
| dCas9-Split M.SssI | Catalytic domain is split for higher specificity [88] |
| dCas9 Tool | Feature |
|---|---|
| dCas9-TET1CD | Targeted demethylation of the BRCA promoter activates gene expression [91] |
| dCas9-TET1CD, MS2-TET1CD | Modified sgRNA (sgRNA2.0) were constructed using bacteriophage MS2 RNA elements [92] |
| dCas-TET1CD | Demethylation of CGG repeats induced an active chromatin conformation [94] |
| dCas9-SunTag-TET1CD | The linker length of original SunTag was changed to 22 amino acids, improving targeted demethylation efficiency [93] |
| Gal4-ROS1CD | Direct removal of 5mC is induced by ROS1CD glycosylase, without hydroxymethylation [95] |
| dCas9-R2 | A short RNA sequence with stem-loop structure is fused to the sgRNA scaffold and binds DNMT1, inhibiting DNMT1 action to prevent DNA methylation [96] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urbano, A.; Smith, J.; Weeks, R.J.; Chatterjee, A. Gene-Specific Targeting of DNA Methylation in the Mammalian Genome. Cancers 2019, 11, 1515. https://doi.org/10.3390/cancers11101515
Urbano A, Smith J, Weeks RJ, Chatterjee A. Gene-Specific Targeting of DNA Methylation in the Mammalian Genome. Cancers. 2019; 11(10):1515. https://doi.org/10.3390/cancers11101515
Chicago/Turabian StyleUrbano, Arthur, Jim Smith, Robert J. Weeks, and Aniruddha Chatterjee. 2019. "Gene-Specific Targeting of DNA Methylation in the Mammalian Genome" Cancers 11, no. 10: 1515. https://doi.org/10.3390/cancers11101515
APA StyleUrbano, A., Smith, J., Weeks, R. J., & Chatterjee, A. (2019). Gene-Specific Targeting of DNA Methylation in the Mammalian Genome. Cancers, 11(10), 1515. https://doi.org/10.3390/cancers11101515