KRAS Mutations Predict Response and Outcome in Advanced Lung Adenocarcinoma Patients Receiving First-Line Bevacizumab and Platinum-Based Chemotherapy

 , ,
, ,  ,
, 
Abstract
1. Introduction
2. Results
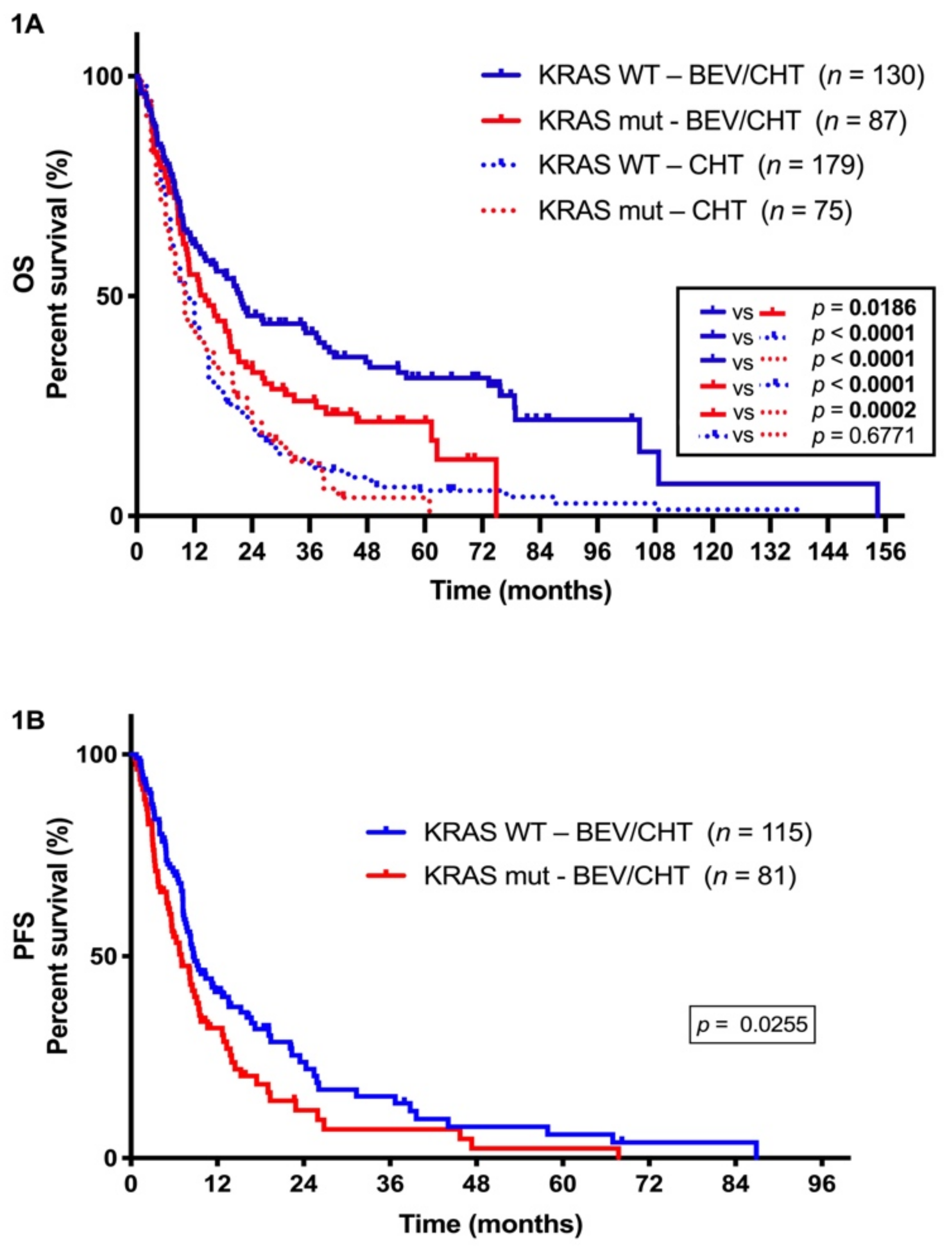
2.1. Incidence of KRAS Mutations in LADC Patients Treated with Bevacizumab and Chemotherapy
2.2. The Presence of KRAS Mutations has Clinical Utility in Predicting Disease Outcome in LADC Patients Receiving Concurrent Antiangiogenic and Chemotherapy
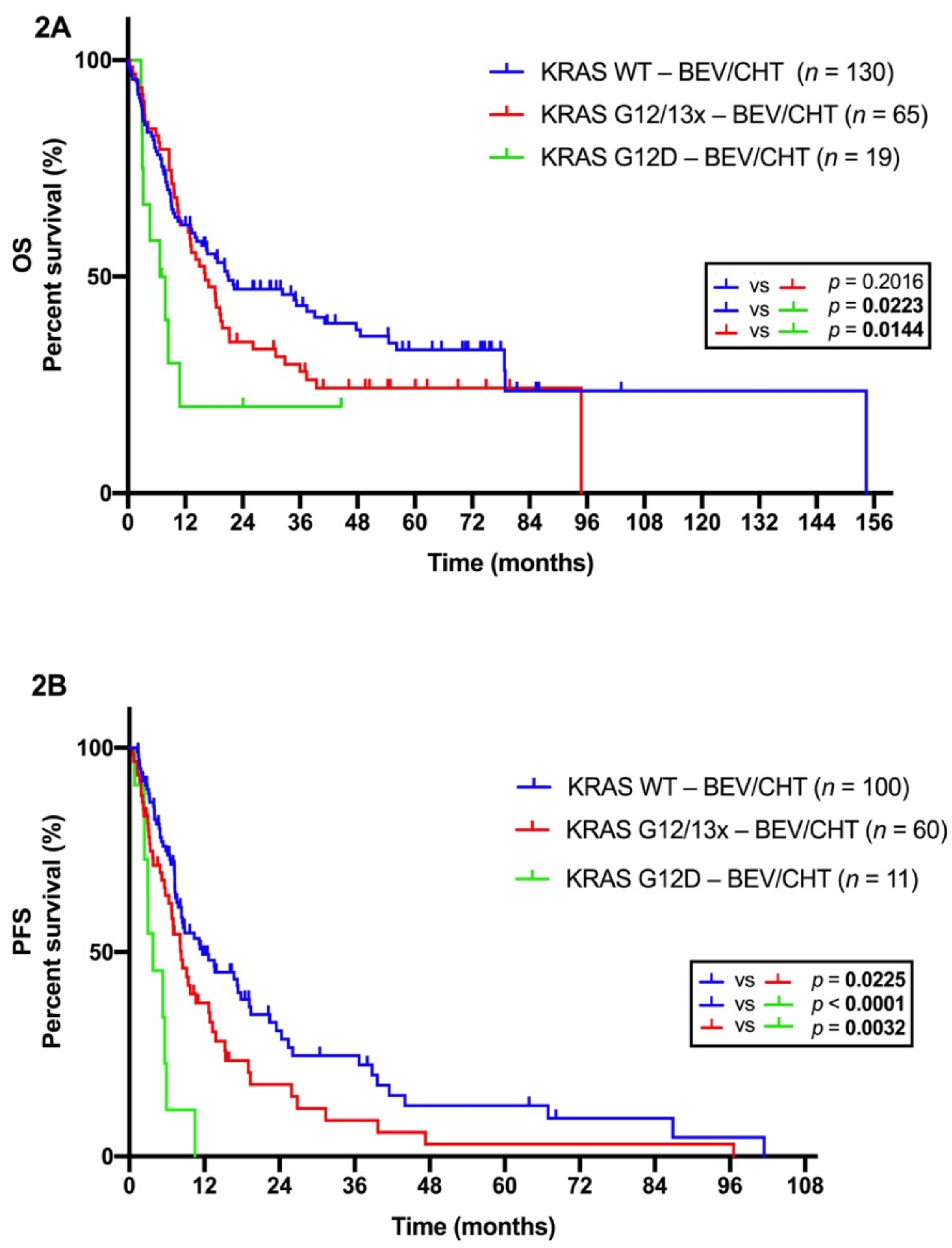
2.3. Distinct Efficacy of BEV/CHT in Advanced LADC Patients with Different Subtype-Specific KRAS Mutations
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Molecular Diagnosis
4.3. Statistical Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fernandez-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef]
- O’Bryan, J.P. Pharmacological targeting of RAS: Recent success with direct inhibitors. Pharmacol. Res. 2019, 139, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, C.R.; Jamal-Hanjani, M.; Forster, M.; Blackhall, F. KRAS: Reasons for optimism in lung cancer. Eur. J. Cancer 2018, 99, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Timar, J. The clinical relevance of KRAS gene mutation in non-small-cell lung cancer. Curr. Opin. Oncol. 2014, 26, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Rodenhuis, S.; van de Wetering, M.L.; Mooi, W.J.; Evers, S.G.; van Zandwijk, N.; Bos, J.L. Mutational activation of the K-ras oncogene. A possible pathogenetic factor in adenocarcinoma of the lung. N. Engl. J. Med. 1987, 317, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Slebos, R.J.; Kibbelaar, R.E.; Dalesio, O.; Kooistra, A.; Stam, J.; Meijer, C.J.; Wagenaar, S.S.; Vanderschueren, R.G.; van Zandwijk, N.; Mooi, W.J.; et al. K-ras oncogene activation as a prognostic marker in adenocarcinoma of the lung. N. Engl. J. Med. 1990, 323, 561–565. [Google Scholar] [CrossRef]
- Kern, J.A.; Slebos, R.J.; Top, B.; Rodenhuis, S.; Lager, D.; Robinson, R.A.; Weiner, D.; Schwartz, D.A. C-erbB-2 expression and codon 12 K-ras mutations both predict shortened survival for patients with pulmonary adenocarcinomas. J. Clin. Investig. 1994, 93, 516–520. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Steinberg, S.M.; Oie, H.K.; Mulshine, J.L.; Phelps, R.; Viallet, J.; Pass, H.; Minna, J.D.; Gazdar, A.F. Ras gene mutations in non-small cell lung cancers are associated with shortened survival irrespective of treatment intent. Cancer Res. 1991, 51, 4999–5002. [Google Scholar]
- Huncharek, M.; Muscat, J.; Geschwind, J.F. K-ras oncogene mutation as a prognostic marker in non-small cell lung cancer: A combined analysis of 881 cases. Carcinogenesis 1999, 20, 1507–1510. [Google Scholar] [CrossRef]
- Mascaux, C.; Iannino, N.; Martin, B.; Paesmans, M.; Berghmans, T.; Dusart, M.; Haller, A.; Lothaire, P.; Meert, A.P.; Noel, S.; et al. The role of RAS oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br. J. Cancer 2005, 92, 131–139. [Google Scholar] [CrossRef]
- Shepherd, F.A.; Domerg, C.; Hainaut, P.; Janne, P.A.; Pignon, J.P.; Graziano, S.; Douillard, J.Y.; Brambilla, E.; Le Chevalier, T.; Seymour, L.; et al. Pooled analysis of the prognostic and predictive effects of KRAS mutation status and KRAS mutation subtype in early-stage resected non-small-cell lung cancer in four trials of adjuvant chemotherapy. J. Clin. Oncol. 2013, 31, 2173–2181. [Google Scholar] [CrossRef] [PubMed]
- Cserepes, M.; Ostoros, G.; Lohinai, Z.; Raso, E.; Barbai, T.; Timar, J.; Rozsas, A.; Moldvay, J.; Kovalszky, I.; Fabian, K.; et al. Subtype-specific KRAS mutations in advanced lung adenocarcinoma: A retrospective study of patients treated with platinum-based chemotherapy. Eur. J. Cancer 2014, 50, 1819–1828. [Google Scholar] [CrossRef] [PubMed]
- Garassino, M.C.; Marabese, M.; Rusconi, P.; Rulli, E.; Martelli, O.; Farina, G.; Scanni, A.; Broggini, M. Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Ann. Oncol. 2011, 22, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Gentzler, R.D.; Yentz, S.E.; Patel, J.D. Bevacizumab in advanced NSCLC: Chemotherapy partners and duration of use. Curr. Treat. Options Oncol. 2013, 14, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.; Wang, J.; Lv, X.J.; Wang, Q.; Qiu, L.X.; Lin, X.Q.; Yu, L.K.; Song, Y. Prognostic value of vascular endothelial growth factor expression in patients with lung cancer: A systematic review with meta-analysis. J. Thorac. Oncol. 2009, 4, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Sandler, A.; Gray, R.; Perry, M.C.; Brahmer, J.; Schiller, J.H.; Dowlati, A.; Lilenbaum, R.; Johnson, D.H. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N. Engl. J. Med. 2006, 355, 2542–2550. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.H.; Fehrenbacher, L.; Novotny, W.F.; Herbst, R.S.; Nemunaitis, J.J.; Jablons, D.M.; Langer, C.J.; DeVore, R.F., 3rd; Gaudreault, J.; Damico, L.A.; et al. Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J. Clin. Oncol. 2004, 22, 2184–2191. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; von Pawel, J.; Zatloukal, P.; Ramlau, R.; Gorbounova, V.; Hirsh, V.; Leighl, N.; Mezger, J.; Archer, V.; Moore, N.; et al. Phase III trial of cisplatin plus gemcitabine with either placebo or bevacizumab as first-line therapy for nonsquamous non-small-cell lung cancer: AVAil. J. Clin. Oncol. 2009, 27, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Twelves, C.; Chmielowska, E.; Havel, L.; Popat, S.; Swieboda-Sadlej, A.; Sawrycki, P.; Bycott, P.; Ingrosso, A.; Kim, S.; Williams, J.A.; et al. Randomised phase II study of axitinib or bevacizumab combined with paclitaxel/carboplatin as first-line therapy for patients with advanced non-small-cell lung cancer. Ann. Oncol. 2014, 25, 132–138. [Google Scholar] [CrossRef]
- Patel, J.D.; Socinski, M.A.; Garon, E.B.; Reynolds, C.H.; Spigel, D.R.; Olsen, M.R.; Hermann, R.C.; Jotte, R.M.; Beck, T.; Richards, D.A.; et al. PointBreak: A randomized phase III study of pemetrexed plus carboplatin and bevacizumab followed by maintenance pemetrexed and bevacizumab versus paclitaxel plus carboplatin and bevacizumab followed by maintenance bevacizumab in patients with stage IIIB or IV nonsquamous non-small-cell lung cancer. J. Clin. Oncol. 2013, 31, 4349–4357. [Google Scholar] [CrossRef]
- Tolnay, E.; Ghimessy, Á.; Juhász, E.; Sztancsik, Z.; Losonczy, G.; Dombi, P.; Vennes, Z.; Helf, L.; Csada, E.; Sárosi, V. The efficacy and safety of bevacizumab in addition to platinum-based chemotherapy for the first-line treatment of patients with advanced nonsquamous non-small-cell lung cancer: Final results of AVALANCHE, an observational cohort study. Oncol. Lett. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; Tam, A.; Pomerantz, J.; Wong, M.; Holash, J.; Bardeesy, N.; Shen, Q.; O’Hagan, R.; Pantginis, J.; Zhou, H.; et al. Essential role for oncogenic Ras in tumour maintenance. Nature 1999, 400, 468–472. [Google Scholar] [CrossRef]
- Hurwitz, H.I.; Yi, J.; Ince, W.; Novotny, W.F.; Rosen, O. The clinical benefit of bevacizumab in metastatic colorectal cancer is independent of K-ras mutation status: analysis of a phase III study of bevacizumab with chemotherapy in previously untreated metastatic colorectal cancer. Oncologist 2009, 14, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Ince, W.L.; Jubb, A.M.; Holden, S.N.; Holmgren, E.B.; Tobin, P.; Sridhar, M.; Hurwitz, H.I.; Kabbinavar, F.; Novotny, W.F.; Hillan, K.J.; et al. Association of k-ras, b-raf, and p53 status with the treatment effect of bevacizumab. J. Natl. Cancer Inst. 2005, 97, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Bencsikova, B.; Bortlicek, Z.; Halamkova, J.; Ostrizkova, L.; Kiss, I.; Melichar, B.; Pavlik, T.; Dusek, L.; Valik, D.; Vyzula, R.; et al. Efficacy of bevacizumab and chemotherapy in the first-line treatment of metastatic colorectal cancer: Broadening KRAS-focused clinical view. BMC Gastroenterol. 2015, 15, e37. [Google Scholar] [CrossRef]
- Stremitzer, S.; Stift, J.; Gruenberger, B.; Tamandl, D.; Aschacher, T.; Wolf, B.; Wrba, F.; Gruenberger, T. KRAS status and outcome of liver resection after neoadjuvant chemotherapy including bevacizumab. Br. J. Surg. 2012, 99, 1575–1582. [Google Scholar] [CrossRef]
- Fiala, O.; Buchler, T.; Mohelnikova-Duchonova, B.; Melichar, B.; Matejka, V.M.; Holubec, L.; Kulhankova, J.; Bortlicek, Z.; Bartouskova, M.; Liska, V.; et al. G12V and G12A KRAS mutations are associated with poor outcome in patients with metastatic colorectal cancer treated with bevacizumab. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 6823–6830. [Google Scholar] [CrossRef]
- Bruera, G.; Cannita, K.; Di Giacomo, D.; Lamy, A.; Frebourg, T.; Sabourin, J.C.; Tosi, M.; Alesse, E.; Ficorella, C.; Ricevuto, E. Worse prognosis of KRAS c.35 G > A mutant metastatic colorectal cancer (MCRC) patients treated with intensive triplet chemotherapy plus bevacizumab (FIr-B/FOx). BMC Med. 2013, 11, e59. [Google Scholar] [CrossRef]
- Chaft, J.E.; Rusch, V.; Ginsberg, M.S.; Paik, P.K.; Finley, D.J.; Kris, M.G.; Price, K.A.; Azzoli, C.G.; Fury, M.G.; Riely, G.J.; et al. Phase II trial of neoadjuvant bevacizumab plus chemotherapy and adjuvant bevacizumab in patients with resectable nonsquamous non-small-cell lung cancers. J. Thorac. Oncol. 2013, 8, 1084–1090. [Google Scholar] [CrossRef]
- Brady, A.K.; McNeill, J.D.; Judy, B.; Bauml, J.; Evans, T.L.; Cohen, R.B.; Langer, C.; Vachani, A.; Aggarwal, C. Survival outcome according to KRAS mutation status in newly diagnosed patients with stage IV non-small cell lung cancer treated with platinum doublet chemotherapy. Oncotarget 2015, 6, 30287–30294. [Google Scholar] [CrossRef]
- Loupakis, F.; Falcone, A.; Masi, G.; Fioravanti, A.; Kerbel, R.S.; Del Tacca, M.; Bocci, G. Vascular endothelial growth factor levels in immunodepleted plasma of cancer patients as a possible pharmacodynamic marker for bevacizumab activity. J. Clin. Oncol. 2007, 25, 1816–1818. [Google Scholar] [CrossRef] [PubMed]
- Hegde, P.S.; Jubb, A.M.; Chen, D.; Li, N.F.; Meng, Y.G.; Bernaards, C.; Elliott, R.; Scherer, S.J.; Chen, D.S. Predictive impact of circulating vascular endothelial growth factor in four phase III trials evaluating bevacizumab. Clin. Cancer Res. 2013, 19, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Said, R.; Hong, D.S.; Warneke, C.L.; Lee, J.J.; Wheler, J.J.; Janku, F.; Naing, A.; Falchook, G.S.; Fu, S.; Piha-Paul, S.; et al. P53 mutations in advanced cancers: Clinical characteristics, outcomes, and correlation between progression-free survival and bevacizumab-containing therapy. Oncotarget 2013, 4, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Lazar, V.; Validire, P.; Hansson, J.; Lacroix, L.; Soria, J.C.; Pawitan, Y.; Kurzrock, R. VEGF-a expression correlates with TP53 mutations in non-small cell lung cancer: Implications for antiangiogenesis therapy. Cancer Res. 2015, 75, 1187–1190. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Leighl, N.B.; Tsao, M.S.; Shepherd, F.A. KRAS mutations as prognostic and predictive markers in non-small cell lung cancer. J. Thorac. Oncol. 2013, 8, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Chaft, J.E.; Dagogo-Jack, I.; Santini, F.C.; Eng, J.; Yeap, B.Y.; Izar, B.; Chin, E.; Jones, D.R.; Kris, M.G.; Shaw, A.T.; et al. Clinical outcomes of patients with resected, early-stage ALK-positive lung cancer. Lung Cancer 2018, 122, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Rapisarda, A.; Melillo, G. Overcoming disappointing results with antiangiogenic therapy by targeting hypoxia. Nat. Rev. Clin. Oncol. 2012, 9, 378–390. [Google Scholar] [CrossRef]
- Giuliano, S.; Pages, G. Mechanisms of resistance to anti-angiogenesis therapies. Biochimie 2013, 95, 1110–1119. [Google Scholar] [CrossRef]
- Smith, N.R.; Wedge, S.R.; Pommier, A.; Barry, S.T. Mechanisms that influence tumour response to VEGF-pathway inhibitors. Biochem. Soc. Trans. 2014, 42, 1601–1607. [Google Scholar] [CrossRef]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: current status and future directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- Dome, B.; Timar, J.; Ladanyi, A.; Paku, S.; Renyi-Vamos, F.; Klepetko, W.; Lang, G.; Dome, P.; Bogos, K.; Tovari, J. Circulating endothelial cells, bone marrow-derived endothelial progenitor cells and proangiogenic hematopoietic cells in cancer: From biology to therapy. Crit. Rev. Oncol. Hematol. 2009, 69, 108–124. [Google Scholar] [CrossRef] [PubMed]
- Torok, S.; Rezeli, M.; Kelemen, O.; Vegvari, A.; Watanabe, K.; Sugihara, Y.; Tisza, A.; Marton, T.; Kovacs, I.; Tovari, J.; et al. Limited tumor tissue drug penetration contributes to primary resistance against angiogenesis inhibitors. Theranostics 2017, 7, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Dome, B.; Hendrix, M.J.; Paku, S.; Tovari, J.; Timar, J. Alternative vascularization mechanisms in cancer: Pathology and therapeutic implications. Am. J. Pathol. 2007, 170, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Donnem, T.; Reynolds, A.R.; Kuczynski, E.A.; Gatter, K.; Vermeulen, P.B.; Kerbel, R.S.; Harris, A.L.; Pezzella, F. Non-angiogenic tumours and their influence on cancer biology. Nat. Rev. Cancer 2018, 18, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Frentzas, S.; Simoneau, E.; Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Kostaras, E.; Nathan, M.; Wotherspoon, A.; Gao, Z.H.; Shi, Y.; et al. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat. Med. 2016, 22, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Bilecz, A.; Daley, F.; Kostaras, E.; Nathan, M.R.; Wan, E.; Frentzas, S.; Schweiger, T.; et al. Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models. J. Pathol. 2017, 241, 362–374. [Google Scholar] [CrossRef]
- Warth, A.; Beasley, M.B.; Mino-Kenudson, M. Breaking new ground: The evolving concept of spread through air spaces (STAS). J. Thorac. Oncol. 2017, 12, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Onozato, M.L.; Kovach, A.E.; Yeap, B.Y.; Morales-Oyarvide, V.; Klepeis, V.E.; Tammireddy, S.; Heist, R.S.; Mark, E.J.; Dias-Santagata, D.; Iafrate, A.J.; et al. Tumor islands in resected early-stage lung adenocarcinomas are associated with unique clinicopathologic and molecular characteristics and worse prognosis. Am. J. Surg. Pathol. 2013, 37, 287–294. [Google Scholar] [CrossRef]
- Yagi, Y.; Aly, R.; Tabata, K.; Rekhtman, N.; Eguchi, T.; Montecalvo, J.; Manova, K.; Adusumilli, P.; Hameed, M.; Travis, W. Three-dimensional immunofluorescence analysis of dynamic vessel co-option of spread through air spaces (STAS) in lung cancer. J. Thorac. Oncol. 2018, 13, e327. [Google Scholar] [CrossRef]
- Rak, J.; Filmus, J.; Finkenzeller, G.; Grugel, S.; Marme, D.; Kerbel, R.S. Oncogenes as inducers of tumor angiogenesis. Cancer Metastasis Rev. 1995, 14, 263–277. [Google Scholar] [CrossRef]
- Rak, J.; Mitsuhashi, Y.; Bayko, L.; Filmus, J.; Shirasawa, S.; Sasazuki, T.; Kerbel, R.S. Mutant ras oncogenes upregulate VEGF/VPF expression: Implications for induction and inhibition of tumor angiogenesis. Cancer Res. 1995, 55, 4575–4580. [Google Scholar] [PubMed]
- Matsuo, Y.; Campbell, P.M.; Brekken, R.A.; Sung, B.; Ouellette, M.M.; Fleming, J.B.; Aggarwal, B.B.; Der, C.J.; Guha, S. K-Ras promotes angiogenesis mediated by immortalized human pancreatic epithelial cells through mitogen-activated protein kinase signaling pathways. Mol. Cancer Res. 2009, 7, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Price, T.J.; Hardingham, J.E.; Lee, C.K.; Weickhardt, A.; Townsend, A.R.; Wrin, J.W.; Chua, A.; Shivasami, A.; Cummins, M.M.; Murone, C.; et al. Impact of KRAS and BRAF gene mutation status on outcomes from the phase III AGITG MAX trial of capecitabine alone or in combination with bevacizumab and mitomycin in advanced colorectal cancer. J. Clin. Oncol. 2011, 29, 2675–2682. [Google Scholar] [CrossRef] [PubMed]
- Masi, G.; Loupakis, F.; Salvatore, L.; Fornaro, L.; Cremolini, C.; Cupini, S.; Ciarlo, A.; Del Monte, F.; Cortesi, E.; Amoroso, D.; et al. Bevacizumab with FOLFOXIRI (irinotecan, oxaliplatin, fluorouracil, and folinate) as first-line treatment for metastatic colorectal cancer: A phase 2 trial. Lancet. Oncol. 2010, 11, 845–852. [Google Scholar] [CrossRef]
- Bruera, G.; Cannita, K.; Tessitore, A.; Russo, A.; Alesse, E.; Ficorella, C.; Ricevuto, E. The prevalent KRAS exon 2 c.35 G>A mutation in metastatic colorectal cancer patients: A biomarker of worse prognosis and potential benefit of bevacizumab-containing intensive regimens? Crit. Rev. Oncol. Hematol. 2015, 93, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, M.; Ihle, M.A.; Hein, R.; Merkelbach-Bruse, S.; Scheel, A.H.; Siemanowski, J.; Bragelmann, J.; Kron, A.; Abedpour, N.; Ueckeroth, F.; et al. K-ras mutation subtypes in NSCLC and associated co-occuring mutations in other oncogenic pathways. J. Thorac. Oncol. 2019, 14, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Figueras, A.; Arbos, M.A.; Quiles, M.T.; Vinals, F.; Germa, J.R.; Capella, G. The impact of KRAS mutations on VEGF-A production and tumour vascular network. BMC Cancer 2013, 13, e125. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Renaud, S.; Guerrera, F.; Seitlinger, J.; Reeb, J.; Voegeli, A.C.; Legrain, M.; Mennecier, B.; Santelmo, N.; Falcoz, P.E.; Quoix, E.; et al. KRAS-specific amino acid substitutions are associated with different responses to chemotherapy in advanced non-small-cell lung cancer. Clin. Lung Cancer 2018, 19, 919–931. [Google Scholar] [CrossRef]
- Ricciuti, B.; Brambilla, M.; Cortellini, A.; De Giglio, A.; Ficorella, C.; Sidoni, A.; Bellezza, G.; Crino, L.; Ludovini, V.; Baglivo, S.; et al. Clinical outcomes to pemetrexed-based versus non-pemetrexed-based platinum doublets in patients with KRAS-mutant advanced non-squamous non-small cell lung cancer. Clin. Transl. Oncol. 2019. [Google Scholar] [CrossRef]
- Lohinai, Z.; Klikovits, T.; Moldvay, J.; Ostoros, G.; Raso, E.; Timar, J.; Fabian, K.; Kovalszky, I.; Kenessey, I.; Aigner, C.; et al. KRAS-mutation incidence and prognostic value are metastatic site-specific in lung adenocarcinoma: Poor prognosis in patients with KRAS mutation and bone metastasis. Sci. Rep. 2017, 7, e39721. [Google Scholar] [CrossRef] [PubMed]
- Lohinai, Z.; Hoda, M.A.; Fabian, K.; Ostoros, G.; Raso, E.; Barbai, T.; Timar, J.; Kovalszky, I.; Cserepes, M.; Rozsas, A.; et al. Distinct epidemiology and clinical consequence of classic versus rare EGFR mutations in lung adenocarcinoma. J. Thorac. Oncol. 2015, 10, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; von Pawel, J.; Zatloukal, P.; Ramlau, R.; Gorbounova, V.; Hirsh, V.; Leighl, N.; Mezger, J.; Archer, V.; Moore, N.; et al. Overall survival with cisplatin-gemcitabine and bevacizumab or placebo as first-line therapy for nonsquamous non-small-cell lung cancer: Results from a randomised phase III trial (AVAiL). Ann. Oncol. 2010, 21, 1804–1809. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, R.; Fujimoto, D.; Kato, R.; Otoshi, T.; Kawamura, T.; Tamai, K.; Matsumoto, T.; Nagata, K.; Otsuka, K.; Nakagawa, A.; et al. The safety and efficacy of paclitaxel and carboplatin with or without bevacizumab for treating patients with advanced nonsquamous non-small cell lung cancer with interstitial lung disease. Cancer Chemother. Pharmacol. 2014, 74, 1159–1166. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| No. of Patients (%) | KRAS Status | p-Value a | |||
|---|---|---|---|---|---|
| Wild Type (%) | Mutant (%) | ||||
| A. BEV/CHT | |||||
| All patients | 247 | 152 (61.5%) | 95 (38.5%) | ||
| Age (years) b | Median: | 62 | 58 | 0.09 | |
| SD *: | 9.2 | 8.2 | |||
| Range: | 53 | 44 | |||
| Smoking c | |||||
| Never-smoker | 30 (12%) | 24 | 6 | 0.008 | |
| Ever-smoker | 167 (68%) | 93 | 74 | ||
| No data (n = 50) | |||||
| Gender c | |||||
| Female | 106 (43%) | 52 | 54 | 0.002 | |
| Male | 141 (57%) | 100 | 41 | ||
| ECOG c | |||||
| 0 | 139 (56%) | 87 | 52 | 0.056 | |
| 1 | 108 (44%) | 65 | 43 | ||
| Stage c | |||||
| III | 55 (22%) | 38 | 17 | 0.16 | |
| IV | 192 (78%) | 114 | 78 | ||
| Survival d | |||||
| Median PFS (months) | 8.63 | 7.03 | 0.0255 | ||
| Median OS (months) | 21.57 | 14.23 | 0.0186 | ||
| B. CHT | |||||
| All patients | 254 | 179 (70.5%) | 75 (29.5%) | ||
| Age (years) b | Median: | 63 | 61 | 0.297 | |
| SD *: | 7.8 | 8.7 | |||
| Range: | 46 | 46 | |||
| Smoking c | |||||
| Never-smoker | 21 (8%) | 15 | 6 | 0.435 | |
| Ever-smoker | 188 (74%) | 135 | 53 | ||
| No data (n = 45) | |||||
| Gender c | |||||
| Female | 118 (46.5%) | 79 | 39 | 0.27 | |
| Male | 136 (53.5%) | 100 | 36 | ||
| ECOG | |||||
| 0 | 128 (50.5%) | 94 | 34 | 0.335 | |
| 1 | 126 (49.5%) | 85 | 41 | ||
| Stage | |||||
| III | 66 (26%) | 44 | 22 | 0.351 | |
| IV | 188 (74%) | 135 | 53 | ||
| Survival d,e | |||||
| Median OS (months) | 11 | 10 | 0.6771 | ||
| Clinicopathological Variables | PFS | OS |
|---|---|---|
| Age (continuous) | ||
| HR | 0.628 | 0.978 |
| 95% CI | 0.966–1.021) | (0.955–1.003) |
| p | 0.628 | 0.081 |
| Gender (female vs. male) | ||
| HR | 0.248 | 0.390 |
| 95% CI | (0.125–0.494) | (0.203–0.751) |
| p | 0.001 | 0.005 |
| Smoking (never- vs. ever-smokers) | ||
| HR | 0.944 | 0.968 |
| 95% CI | (0.548–1.626) | (0.562–1.669) |
| p | 0.835 | 0.907 |
| ECOG PS (0 vs. 1) | ||
| HR | 0.765 | 0.772 |
| 95% CI | (0.518–1.129) | (0.523–1.140) |
| p | 0.177 | 0.193 |
| Stage (III vs. IV) | ||
| HR | 0.879 | 0.603 |
| 95% CI | (0.531–1.455) | (0.365–0.996) |
| p | 0.617 | 0.048 |
| KRAS status (WT vs. mutant) | ||
| HR | 0.597 | 0.645 |
| 95% CI | (0.402–0.887) | (0.458–0.908) |
| p | 0.011 | 0.012 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghimessy, Á.K.; Gellert, Á.; Schlegl, E.; Hegedus, B.; Raso, E.; Barbai, T.; Timar, J.; Ostoros, G.; Megyesfalvi, Z.; Gieszer, B.; et al. KRAS Mutations Predict Response and Outcome in Advanced Lung Adenocarcinoma Patients Receiving First-Line Bevacizumab and Platinum-Based Chemotherapy. Cancers 2019, 11, 1514. https://doi.org/10.3390/cancers11101514
Ghimessy ÁK, Gellert Á, Schlegl E, Hegedus B, Raso E, Barbai T, Timar J, Ostoros G, Megyesfalvi Z, Gieszer B, et al. KRAS Mutations Predict Response and Outcome in Advanced Lung Adenocarcinoma Patients Receiving First-Line Bevacizumab and Platinum-Based Chemotherapy. Cancers. 2019; 11(10):1514. https://doi.org/10.3390/cancers11101514
Chicago/Turabian StyleGhimessy, Áron Kristof, Áron Gellert, Erzsebet Schlegl, Balazs Hegedus, Erzsebet Raso, Tamas Barbai, Jozsef Timar, Gyula Ostoros, Zsolt Megyesfalvi, Balazs Gieszer, and et al. 2019. "KRAS Mutations Predict Response and Outcome in Advanced Lung Adenocarcinoma Patients Receiving First-Line Bevacizumab and Platinum-Based Chemotherapy" Cancers 11, no. 10: 1514. https://doi.org/10.3390/cancers11101514
APA StyleGhimessy, Á. K., Gellert, Á., Schlegl, E., Hegedus, B., Raso, E., Barbai, T., Timar, J., Ostoros, G., Megyesfalvi, Z., Gieszer, B., Moldvay, J., Renyi-Vamos, F., Lohinai, Z., Hoda, M. A., Klikovits, T., Klepetko, W., Laszlo, V., & Dome, B. (2019). KRAS Mutations Predict Response and Outcome in Advanced Lung Adenocarcinoma Patients Receiving First-Line Bevacizumab and Platinum-Based Chemotherapy. Cancers, 11(10), 1514. https://doi.org/10.3390/cancers11101514