Current Management of Pheochromocytoma/Paraganglioma: A Guide for the Practicing Clinician in the Era of Precision Medicine

 , ,
, , 
Abstract
1. Introduction
2. Genetics
2.1. Cluster 1: Pseudohypoxic Krebs Cycle-Related Genes and Pseudohypoxia VHL/EPAS1-Related Genes
2.2. Cluster 2: Kinase Signaling-Related Genes
2.3. Cluster 3: Wnt Signaling-Related Genes
2.4. Summary: Cluster 1, 2 and 3
Conclusion/Practical Tips
3. Diagnosis
3.1. Biochemistry
Conclusion/Practical Tips
3.2. Imaging for Diagnosis, Staging, and Follow-Up
- (1)
- Metastatic SDHB-related PCCs/PGLs ([68Ga]Ga-DOTA-SSA 99% vs. [18F]FDOPA 61% vs. [18F]FDG PET/CT 86%) [61];
- (2)
- SDHA-related PCCs/PGLs (highest lesion detection sensitivity: [68Ga]Ga-DOTA-SSA > [18F]FDG > [18F]FDOPA PET/CT) [65];
- (3)
- SDHD-related PCCs/PGLs ([68Ga]Ga-DOTA-SSA 99% vs. [18F]FDOPA 87% vs. [18F]FDG PET/CT 62%) [66];
- (4)
- Pediatric SDHx-related PGLs/PCCs ([68Ga]Ga-DOTA-SSA 94% vs. [18F]FDG PET/CT 79%) [67];
- (5)
- (6)
- (7)
- Sporadic metastatic PCCs/PGLs ([68Ga]Ga-DOTA-SSA 98% vs. [18F]FDOPA 75% vs. [18F]FDG PET/CT 49%) [71].
- (1)
- (Sporadic) PCCs ([18F]FDOPA 94% vs. [68Ga]Ga-DOTA-SSA PET/CT 81%) [69];
- (2)
- Moreover, for EPAS1 (HIF2A)-, PHD1/2-, FH-, and MAX-related PGLs, [18F]FDOPA PET/CT seems to be the imaging modality with the highest lesion-based sensitivity (for MAX-related PCCs/PGLs [18F]FDOPA 91% vs. [68Ga]Ga-DOTA-SSA 57% vs. [18F]FDG PET/CT 18%) although these findings have to be confirmed in a larger patient group with FH mutations [72,73,74,75];
- (3)
- VHL-related PGLs/PCCs belong to the pseudohypoxia group without leading to succinate accumulation and show high [18F]FDOPA uptake but variable [18F]FDG uptake which probably makes [18F]FDOPA PET/CT the most sensitive imaging modality in VHL-related PCCs/PGLs;
- (4)
- In the kinase signaling group, [18F]FDOPA also seems to be the most sensitive radiopharmaceutical due to high uptake by the tumor and low uptake in the remaining adrenal gland; however, a head-to-head comparison with [68Ga]Ga-DOTA-SSA and [18F]FDG PET/CT has only been performed for MAX-related PCCs/PGLs as yet [74].
Conclusion/Practical Tips
3.3. Biopsy Is Not Recommended
- ○
- There is another extra-adrenal malignancy in the patient’s history;
- ○
- The tumor is non-functioning (especially non-functioning PCCs/PGLs);
- ○
- Not judged as benign on imaging;
- ○
- Biopsy would change patient management.
Conclusion/Practical Tips
3.4. Immunohistochemistry: Biomarkers
4. Follow-Up
4.1. Conclusion/Practical Tips
4.2. Perspectives
5. Therapy
5.1. Targeted Endoradionuclide Therapy Using [131I]MIBG or Radiolabeled Somatostatin Analogs (PRRT)
Conclusion/Practical Tips
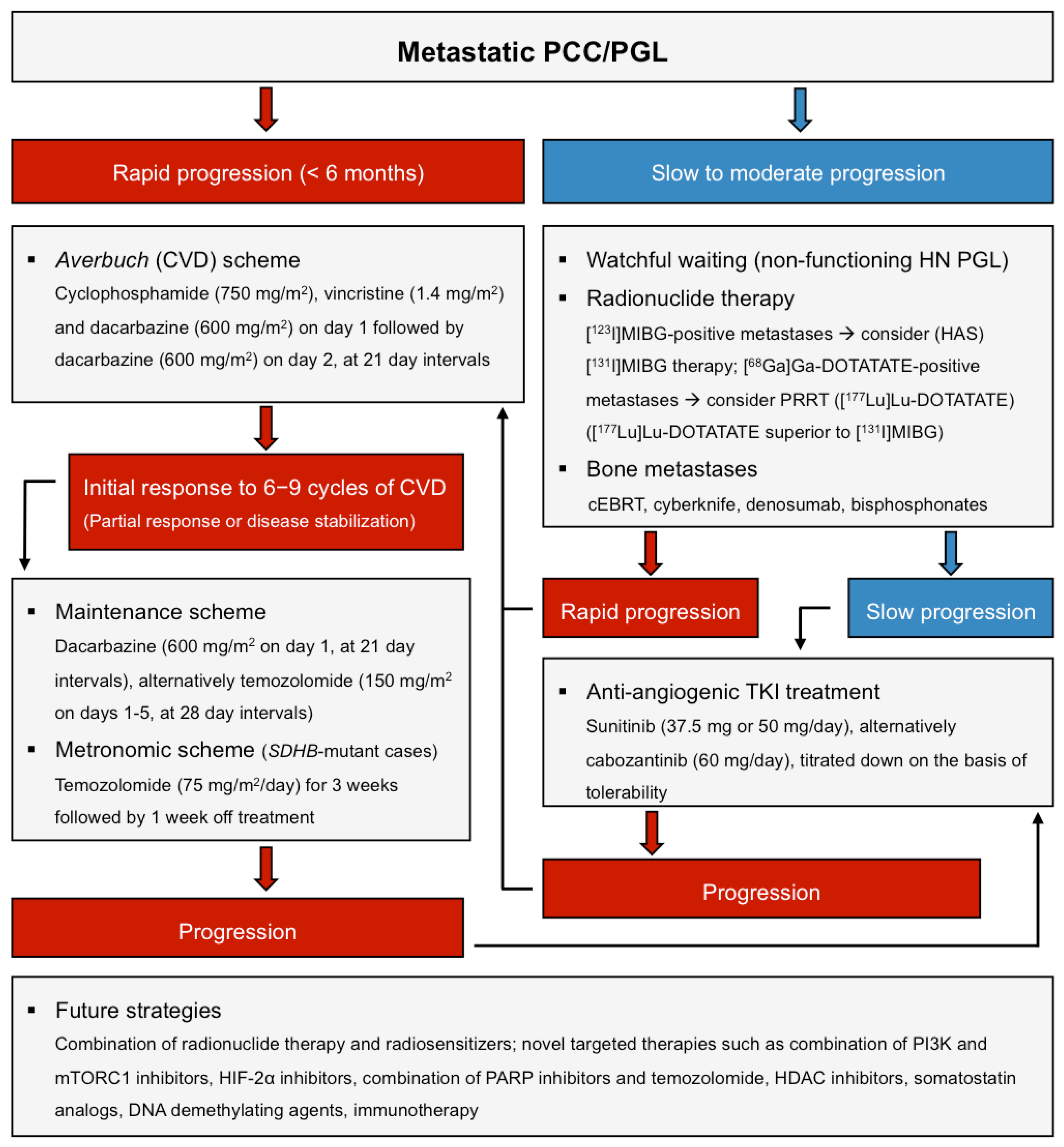
5.2. Chemotherapy
Conclusion/Practical Tips
5.3. Targeted Therapy and Immunotherapy
Conclusion/Practical Tips
6. Outlook
Funding
Conflicts of Interest
References
- Beard, C.M.; Sheps, S.G.; Kurland, L.T.; Carney, J.A.; Lie, J.T. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin. Proc. 1983, 58, 802–804. [Google Scholar] [PubMed]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Burnichon, N.; Vescovo, L.; Amar, L.; Libe, R.; De, R.A.; Venisse, A.; Jouanno, E.; Laurendeau, I.; Parfait, B.; Bertherat, J.; et al. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum. Mol. Genet. 2011, 20, 3974–3985. [Google Scholar] [CrossRef] [PubMed]
- Sutton, M.G.; Sheps, S.G.; Lie, J.T. Prevalence of clinically unsuspected pheochromocytoma. Review of a 50-year autopsy series. Mayo Clin. Proc. 1981, 56, 354–360. [Google Scholar] [CrossRef]
- Pamporaki, C.; Hamplova, B.; Peitzsch, M.; Prejbisz, A.; Beuschlein, F.; Timmers, H.; Fassnacht, M.; Klink, B.; Lodish, M.; Stratakis, C.A.; et al. Characteristics of pediatric vs adult pheochromocytomas and paragangliomas. J. Clin. Endocrinol. Metab. 2017, 102, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Prejbisz, A.; Peitzsch, M.; Pamporaki, C.; Masjkur, J.; Rogowski-Lehmann, N.; Langton, K.; Tsourdi, E.; Peczkowska, M.; Fliedner, S.; et al. Biochemical diagnosis of chromaffin cell tumors in patients at high and low risk of disease: Plasma versus urinary free or deconjugated O-methylated catecholamine metabolites. Clin. Chem. 2018, 64, 1646–1656. [Google Scholar] [CrossRef]
- Geroula, A.; Deutschbein, T.; Langton, K.; Masjkur, J.R.; Pamporaki, C.; Peitzsch, M.; Fliedner, S.; Timmers, H.J.; Bornstein, S.R.; Beuschlein, F.; et al. Pheochromocytoma and paraganglioma: Clinical feature based disease probability in relation to catecholamine biochemistry and reason for disease suspicion. Eur. J. Endocrinol. 2019. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Klink, B.; Richter, S.; Lenders, J.W.; Robledo, M. Metabologenomics of phaeochromocytoma and paraganglioma: An integrated approach for personalised biochemical and genetic testing. Clin. Biochem. Rev. 2017, 38, 69–100. [Google Scholar]
- Timmers, H.J.; Pacak, K.; Huynh, T.T.; Abu-Asab, M.; Tsokos, M.; Merino, M.J.; Baysal, B.E.; Adams, K.T.; Eisenhofer, G. Biochemically silent abdominal paragangliomas in patients with mutations in the succinate dehydrogenase subunit B gene. J. Clin. Endocrinol. Metab. 2008, 93, 4826–4832. [Google Scholar] [CrossRef]
- Remine, W.H.; Chong, G.C.; Van Heerden, J.A.; Sheps, S.G.; Harrison, E.G., Jr. Current management of pheochromocytoma. Ann. Surg. 1974, 179, 740–748. [Google Scholar] [CrossRef]
- Proye, C.A.; Vix, M.; Jansson, S.; Tisell, L.E.; Dralle, H.; Hiller, W. “The” pheochromocytoma: A benign, intra-adrenal, hypertensive, sporadic unilateral tumor. Does it exist? World J. Surg. 1994, 18, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, R.E.; O’Neill, J.A., Jr.; Holcomb, G.W., 3rd; Morgan, W.M., 3rd; Neblett, W.W., 3rd; Oates, J.A.; Brown, N.; Nadeau, J.; Smith, B.; Page, D.L.; et al. Clinical experience over 48 years with pheochromocytoma. Ann. Surg. 1999, 229, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Mannelli, M.; Ianni, L.; Cilotti, A.; Conti, A. Pheochromocytoma in Italy: A multicentric retrospective study. Eur. J. Endocrinol. 1999, 141, 619–624. [Google Scholar] [CrossRef] [PubMed]
- John, H.; Ziegler, W.H.; Hauri, D.; Jaeger, P. Pheochromocytomas: Can malignant potential be predicted? Urology 1999, 53, 679–683. [Google Scholar] [CrossRef]
- Elder, E.E.; Skog, A.L.H.; Hoog, A.; Hamberger, B. The management of benign and malignant pheochromocytoma and abdominal paraganglioma. Eur. J. Surg. Oncol. 2003, 29, 278–283. [Google Scholar] [CrossRef]
- Amar, L.; Servais, A.; Gimenez-Roqueplo, A.P.; Zinzindohoue, F.; Chatellier, G.; Plouin, P.F. Year of diagnosis, features at presentation, and risk of recurrence in patients with pheochromocytoma or secreting paraganglioma. J. Clin. Endocrinol. Metab. 2005, 90, 2110–2116. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Lenders, J.W.; Siegert, G.; Bornstein, S.R.; Friberg, P.; Milosevic, D.; Mannelli, M.; Linehan, W.M.; Adams, K.; Timmers, H.J.; et al. Plasma methoxytyramine: A novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J. Cancer 2012, 48, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, O.; Young, W.F., Jr.; Gruber, L.; Smestad, J.; Yan, Q.; Ponce, O.J.; Prokop, L.; Murad, M.H.; Bancos, I. Outcomes of patients with metastatic phaeochromocytoma and paraganglioma: A systematic review and meta-analysis. Clin. Endocrinol. 2017, 87, 440–450. [Google Scholar] [CrossRef]
- Lam, A.K. Update on adrenal tumours in 2017 World Health Organization (WHO) of endocrine tumours. Endocr. Pathol. 2017, 28, 213–227. [Google Scholar] [CrossRef]
- Asa, S.L.; Ezzat, S.; Mete, O. The diagnosis and clinical significance of paragangliomas in unusual locations. J. Clin. Med. 2018, 7, 280. [Google Scholar] [CrossRef]
- Koh, P.S.; Koong, J.K.; Westerhout, C.J.; Yoong, B.K. Education and imaging. Hepatobiliary and pancreatic: A huge liver paraganglioma. J. Gastroenterol. Hepatol. 2013, 28, 1075. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Ding, Z.Y.; Zhang, B.; Chen, L.; Li, G.X.; Wu, J.J.; Zhang, B.; Chen, X.P.; Zhu, P. Primary functioning hepatic paraganglioma mimicking hepatocellular carcinoma: A case report and literature review. Medicine 2018, 97, e0293. [Google Scholar] [CrossRef] [PubMed]
- Gucer, H.; Mete, O. Endobronchial gangliocytic paraganglioma: Not all keratin-positive endobronchial neuroendocrine neoplasms are pulmonary carcinoids. Endocr. Pathol. 2014, 25, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Liang, Q.L.; Jiang, L.; Liu, Q.L.; Ou, W.T.; Li, D.H.; Zhang, H.J.; Yuan, G.L. Primary pulmonary paraganglioma: A case report and review of literature. Medicine 2015, 94, e1271. [Google Scholar] [CrossRef]
- Fiorentino, G.; Annunziata, A.; de Rosa, N. Primary paraganglioma of the lung: A case report. J. Med. Case Rep. 2015, 9, 166. [Google Scholar] [CrossRef]
- Thompson, L.D. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: A clinicopathologic and immunophenotypic study of 100 cases. Am J. Surg. Pathol. 2002, 26, 551–566. [Google Scholar] [CrossRef]
- Kimura, N.; Takayanagi, R.; Takizawa, N.; Itagaki, E.; Katabami, T.; Kakoi, N.; Rakugi, H.; Ikeda, Y.; Tanabe, A.; Nigawara, T.; et al. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr. Relat. Cancer 2014, 21, 405–414. [Google Scholar] [CrossRef]
- Kimura, N.; Takekoshi, K.; Naruse, M. Risk stratification on pheochromocytoma and paraganglioma from laboratory and clinical medicine. J. Clin. Med. 2018, 7, 242. [Google Scholar] [CrossRef]
- Stenman, A.; Zedenius, J.; Juhlin, C.C. The value of histological algorithms to predict the malignancy potential of pheochromocytomas and abdominal paragangliomas—A meta-analysis and systematic review of the literature. Cancers 2019, 11, 225. [Google Scholar] [CrossRef]
- Stenman, A.; Zedenius, J.; Juhlin, C.C. Retrospective application of the pathologic tumor-node-metastasis classification system for pheochromocytoma and abdominal paraganglioma in a well characterized cohort with long-term follow-up. Surgery 2019. [Google Scholar] [CrossRef]
- Roman-Gonzalez, A.; Jimenez, C. Malignant pheochromocytoma-paraganglioma: Pathogenesis, TNM staging, and current clinical trials. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Crona, J.; Taieb, D.; Pacak, K. New perspectives on pheochromocytoma and paraganglioma: Toward a molecular classification. Endocr. Rev. 2017, 38, 489–515. [Google Scholar] [CrossRef] [PubMed]
- Jochmanova, I.; Pacak, K. Genomic landscape of pheochromocytoma and paraganglioma. Trends Cancer 2018, 4, 6–9. [Google Scholar] [CrossRef]
- Nölting, S.; Grossman, A.; Pacak, K. Metastatic Phaeochromocytoma: Spinning towards more promising treatment options. Exp. Clin. Endocrinol. Diabetes 2018. [Google Scholar] [CrossRef] [PubMed]
- Jochmanova, I.; Yang, C.; Zhuang, Z.; Pacak, K. Hypoxia-inducible factor signaling in pheochromocytoma: Turning the rudder in the right direction. J. Natl. Cancer Inst. 2013, 105, 1270–1283. [Google Scholar] [CrossRef] [PubMed]
- Tella, S.H.; Taieb, D.; Pacak, K. HIF-2alpha: Achilles’ heel of pseudohypoxic subtype paraganglioma and other related conditions. Eur. J. Cancer 2017, 86, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Timmers, H.J.; Kozupa, A.; Eisenhofer, G.; Raygada, M.; Adams, K.T.; Solis, D.; Lenders, J.W.; Pacak, K. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. J. Clin. Endocrinol. Metab. 2007, 92, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; de Cubas, A.A.; Garcia-Martin, R.; Richter, S.; Peitzsch, M.; Menschikowski, M.; Lenders, J.W.; Timmers, H.J.; Mannelli, M.; Opocher, G.; et al. Opposing effects of HIF1α and HIF2α on chromaffin cell phenotypic features and tumor cell proliferation: Insights from MYC-associated factor X. Int. J. Cancer 2014, 135, 2054–2064. [Google Scholar] [CrossRef] [PubMed]
- Plouin, P.F.; Amar, L.; Dekkers, O.M.; Fassnacht, M.; Gimenez-Roqueplo, A.P.; Lenders, J.W.; Lussey-Lepoutre, C.; Steichen, O.; Guideline Working Group. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur. J. Endocrinol. 2016, 174, G1–G10. [Google Scholar] [CrossRef] [PubMed]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: The American Thyroid Association guidelines task force on thyroid nodules and differentiated thyroid cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef]
- Hageman, J.C.; Pegues, D.A.; Jepson, C.; Bell, R.L.; Guinan, M.; Ward, K.W.; Cohen, M.D.; Hindler, J.A.; Tenover, F.C.; McAllister, S.K.; et al. Vancomycin-intermediate Staphylococcus aureus in a home health-care patient. Emerg. Infect. Dis. 2001, 7, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.W.; Duh, Q.Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.P.; Grebe, S.K.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr.; Endocrine, S. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef] [PubMed]
- Darr, R.; Pamporaki, C.; Peitzsch, M.; Miehle, K.; Prejbisz, A.; Peczkowska, M.; Weismann, D.; Beuschlein, F.; Sinnott, R.; Bornstein, S.R.; et al. Biochemical diagnosis of phaeochromocytoma using plasma-free normetanephrine, metanephrine and methoxytyramine: Importance of supine sampling under fasting conditions. Clin. Endocrinol. 2014, 80, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Weismann, D.; Peitzsch, M.; Raida, A.; Prejbisz, A.; Gosk, M.; Riester, A.; Willenberg, H.S.; Klemm, R.; Manz, G.; Deutschbein, T.; et al. Measurements of plasma metanephrines by immunoassay vs liquid chromatography with tandem mass spectrometry for diagnosis of pheochromocytoma. Eur. J. Endocrinol. 2015, 172, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.; Leung, A.A.; Sadrzadeh, H.; Pamporaki, C.; Pacak, K.; Deutschbein, T.; Fliedner, S.; Kline, G.A. A high rate of modestly elevated plasma normetanephrine in a population referred for suspected PPGL when measured in a seated position. Eur. J. Endocrinol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Olson, S.W.; Yoon, S.; Baker, T.; Prince, L.K.; Oliver, D.; Abbott, K.C. Longitudinal plasma metanephrines preceding pheochromocytoma diagnosis: A retrospective case-control serum repository study. Eur. J. Endocrinol. 2016, 174, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Huynh, T.T.; Pacak, K.; Brouwers, F.M.; Walther, M.M.; Linehan, W.M.; Munson, P.J.; Mannelli, M.; Goldstein, D.S.; Elkahloun, A.G. Distinct gene expression profiles in norepinephrine-and epinephrine-producing hereditary and sporadic pheochromocytomas: Activation of hypoxia-driven angiogenic pathways in von Hippel-Lindau syndrome. Endocr. Relat. Cancer 2004, 11, 897–911. [Google Scholar] [CrossRef]
- Sue, M.; Martucci, V.; Frey, F.; Lenders, J.M.; Timmers, H.J.; Peczkowska, M.; Prejbisz, A.; Swantje, B.; Bornstein, S.R.; Arlt, W.; et al. Lack of utility of SDHB mutation testing in adrenergic metastatic phaeochromocytoma. Eur. J. Endocrinol. 2015, 172, 89–95. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Lenders, J.W.; Timmers, H.; Mannelli, M.; Grebe, S.K.; Hofbauer, L.C.; Bornstein, S.R.; Tiebel, O.; Adams, K.; Bratslavsky, G.; et al. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin. Chem. 2011, 57, 411–420. [Google Scholar] [CrossRef]
- Feldman, J.M.; Blalock, J.A.; Zern, R.T.; Shelburne, J.D.; Gaede, J.T.; Farrell, R.E.; Wells, S.A., Jr. Deficiency of dopamine-β-hydroxylase: A new mechanism for normotensive pheochromocytomas. Am. J. Clin. Pathol. 1979, 72, 175–185. [Google Scholar] [CrossRef]
- Zuber, S.; Wesley, R.; Prodanov, T.; Eisenhofer, G.; Pacak, K.; Kantorovich, V. Clinical utility of chromogranin A in SDHx-related paragangliomas. Eur. J. Clin. Investig. 2014, 44, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, R.J.; Parmer, R.J.; Takiyyuddin, M.A.; O’Connor, D.T. Chromogranin A storage and secretion: Sensitivity and specificity for the diagnosis of pheochromocytoma. Medicine 1991, 70, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Bilek, R.; Vlcek, P.; Safarik, L.; Michalsky, D.; Novak, K.; Duskova, J.; Vaclavikova, E.; Widimsky, J., Jr.; Zelinka, T. Chromogranin A in the laboratory diagnosis of pheochromocytoma and paraganglioma. Cancers 2019, 11, 586. [Google Scholar] [CrossRef] [PubMed]
- Pacak, K.; Ilias, I.; Adams, K.T.; Eisenhofer, G. Biochemical diagnosis, localization and management of pheochromocytoma: Focus on multiple endocrine neoplasia type 2 in relation to other hereditary syndromes and sporadic forms of the tumour. J. Intern. Med. 2005, 257, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Pacak, K.; Huynh, T.T.; Qin, N.; Bratslavsky, G.; Linehan, W.M.; Mannelli, M.; Friberg, P.; Grebe, S.K.; Timmers, H.J.; et al. Catecholamine metabolomic and secretory phenotypes in phaeochromocytoma. Endocr. Relat. Cancer 2011, 18, 97–111. [Google Scholar] [CrossRef]
- Weise, M.; Merke, D.P.; Pacak, K.; Walther, M.M.; Eisenhofer, G. Utility of plasma free metanephrines for detecting childhood pheochromocytoma. J. Clin. Endocrinol. Metab. 2002, 87, 1955–1960. [Google Scholar] [CrossRef]
- Lenders, J.W.; Pacak, K.; Walther, M.M.; Linehan, W.M.; Mannelli, M.; Friberg, P.; Keiser, H.R.; Goldstein, D.S.; Eisenhofer, G. Biochemical diagnosis of pheochromocytoma: Which test is best? JAMA 2002, 287, 1427–1434. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Lenders, J.W.; Linehan, W.M.; Walther, M.M.; Goldstein, D.S.; Keiser, H.R. Plasma normetanephrine and metanephrine for detecting pheochromocytoma in von Hippel-Lindau disease and multiple endocrine neoplasia type 2. N. Engl. J. Med. 1999, 340, 1872–1879. [Google Scholar] [CrossRef]
- Jalil, N.D.; Pattou, F.N.; Combemale, F.; Chapuis, Y.; Henry, J.F.; Peix, J.L.; Proye, C.A. Effectiveness and limits of preoperative imaging studies for the localisation of pheochromocytomas and paragangliomas: A review of 282 cases. French Association of Surgery (AFC), and The French Association of Endocrine Surgeons (AFCE). Eur. J. Surg. 1998, 164, 23–28. [Google Scholar] [CrossRef]
- Ganguly, A.; Henry, D.P.; Yune, H.Y.; Pratt, J.H.; Grim, C.E.; Donohue, J.P.; Weinberger, M.H. Diagnosis and localization of pheochromocytoma: Detection by measurement of urinary norepinephrine excretion during sleep, plasma norepinephrine concentration and computerized axial tomography (CT-scan). Am. J. Med. 1979, 67, 21–26. [Google Scholar] [CrossRef]
- Janssen, I.; Blanchet, E.M.; Adams, K.; Chen, C.C.; Millo, C.M.; Herscovitch, P.; Taieb, D.; Kebebew, E.; Lehnert, H.; Fojo, A.T.; et al. Superiority of [68Ga]-DOTATATE PET/CT to other functional imaging modalities in the localization of SDHB-associated metastatic pheochromocytoma and paraganglioma. Clin. Cancer Res. 2015, 21, 3888–3895. [Google Scholar] [CrossRef] [PubMed]
- Timmers, H.J.; Chen, C.C.; Carrasquillo, J.A.; Whatley, M.; Ling, A.; Eisenhofer, G.; King, K.S.; Rao, J.U.; Wesley, R.A.; Adams, K.T.; et al. Staging and functional characterization of pheochromocytoma and paraganglioma by 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography. J. Natl. Cancer Inst. 2012, 104, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Suh, C.H.; Woo, S.; Kim, Y.J.; Lee, J.J. Performance of 68Ga-DOTA–Conjugated somatostatin receptor–targeting peptide PET in detection of pheochromocytoma and paraganglioma: A systematic review and metaanalysis. J. Nucl. Med. 2019, 60, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Taieb, D.; Hicks, R.J.; Hindie, E.; Guillet, B.A.; Avram, A.; Ghedini, P.; Timmers, H.J.; Scott, A.T.; Elojeimy, S.; Rubello, D.; et al. European association of nuclear medicine practice guideline/society of nuclear medicine and molecular imaging procedure standard 2019 for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2112–2137. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.; de Luna, K.; Balili, C.A.; Millo, C.; Paraiso, C.A.; Ling, A.; Gonzales, M.K.; Viana, B.; Alrezk, R.; Adams, K.T.; et al. Clinical, diagnostic, and treatment characteristics of SDHA-related metastatic pheochromocytoma and paraganglioma. Front. Oncol. 2019, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.; Ling, A.; Millo, C.; Chen, C.; Gupta, G.; Viana, B.; Gonzales, M.; Adams, K.; Herscovitch, P.; Lin, F.; et al. Superiority of 68Ga-DOTATATE PET/CT to other functional and anatomic imaging modalities in the detection of SDHD-related pheochromocytoma and paraganglioma—A comparative prospective study. J. Nucl. Med. 2018, 59 (Suppl. 1), 46. [Google Scholar]
- Jha, A.; Ling, A.; Millo, C.; Gupta, G.; Viana, B.; Lin, F.I.; Herscovitch, P.; Adams, K.T.; Taieb, D.; Metwalli, A.R.; et al. Superiority of 68Ga-DOTATATE over 18F-FDG and anatomic imaging in the detection of succinate dehydrogenase mutation (SDHx)-related pheochromocytoma and paraganglioma in the pediatric population. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Janssen, I.; Chen, C.C.; Taieb, D.; Patronas, N.J.; Millo, C.M.; Adams, K.T.; Nambuba, J.; Herscovitch, P.; Sadowski, S.M.; Fojo, A.T.; et al. 68Ga-DOTATATE PET/CT in the localization of head and neck paragangliomas compared with other functional imaging modalities and CT/MRI. J. Nucl. Med. 2016, 57, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Archier, A.; Varoquaux, A.; Garrigue, P.; Montava, M.; Guerin, C.; Gabriel, S.; Beschmout, E.; Morange, I.; Fakhry, N.; Castinetti, F.; et al. Prospective comparison of 68Ga-DOTATATE and 18F-FDOPA PET/CT in patients with various pheochromocytomas and paragangliomas with emphasis on sporadic cases. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Kroiss, A.; Putzer, D.; Frech, A.; Decristoforo, C.; Uprimny, C.; Gasser, R.W.; Shulkin, B.L.; Url, C.; Widmann, G.; Prommegger, R.; et al. A retrospective comparison between 68Ga-DOTA-TOC PET/CT and 18F-DOPA PET/CT in patients with extra-adrenal paraganglioma. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Janssen, I.; Chen, C.C.; Millo, C.M.; Ling, A.; Taieb, D.; Lin, F.I.; Adams, K.T.; Wolf, K.I.; Herscovitch, P.; Fojo, A.T.; et al. PET/CT comparing 68Ga-DOTATATE and other radiopharmaceuticals and in comparison with CT/MRI for the localization of sporadic metastatic pheochromocytoma and paraganglioma. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1784–1791. [Google Scholar] [CrossRef] [PubMed]
- Darr, R.; Nambuba, J.; Del Rivero, J.; Janssen, I.; Merino, M.; Todorovic, M.; Balint, B.; Jochmanova, I.; Prchal, J.T.; Lechan, R.M.; et al. Novel insights into the polycythemia-paraganglioma-somatostatinoma syndrome. Endocr. Relat. Cancer 2016, 23, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Janssen, I.; Chen, C.C.; Zhuang, Z.; Millo, C.M.; Wolf, K.I.; Ling, A.; Lin, F.I.; Adams, K.T.; Herscovitch, P.; Feelders, R.A.; et al. Functional imaging signature of patients presenting with polycythemia/paraganglioma syndromes. J. Nucl. Med. 2017, 58, 1236–1242. [Google Scholar] [CrossRef]
- Taieb, D.; Jha, A.; Guerin, C.; Pang, Y.; Adams, K.T.; Chen, C.C.; Romanet, P.; Roche, P.; Essamet, W.; Ling, A.; et al. 18F-FDOPA PET/CT imaging of MAX-related pheochromocytoma. J. Clin. Endocrinol. Metab. 2018, 103, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- Nambuba, J.; Därr, R.; Janssen, I.; Bullova, P.; Adams, K.T.; Millo, C.; Bourdeau, I.; Kassai, A.; Yang, C.; Kebebew, E. Functional imaging experience in a germline fumarate hydratase mutation–positive patient with pheochromocytoma and paraganglioma. AACE Clin. Case Rep. 2015, 2, e176–e181. [Google Scholar] [CrossRef]
- Gild, M.L.; Naik, N.; Hoang, J.; Hsiao, E.; McGrath, R.T.; Sywak, M.; Sidhu, S.; Delbridge, L.W.; Robinson, B.G.; Schembri, G.; et al. Role of DOTATATE-PET/CT in preoperative assessment of phaeochromocytoma and paragangliomas. Clin. Endocrinol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Fassnacht, M.; Arlt, W.; Bancos, I.; Dralle, H.; Newell-Price, J.; Sahdev, A.; Tabarin, A.; Terzolo, M.; Tsagarakis, S.; Dekkers, O.M. Management of adrenal incidentalomas: European society of endocrinology clinical practice guideline in collaboration with the European network for the study of adrenal tumors. Eur. J. Endocrinol. 2016, 175, G1–G34. [Google Scholar] [CrossRef] [PubMed]
- Vanderveen, K.A.; Thompson, S.M.; Callstrom, M.R.; Young, W.F., Jr.; Grant, C.S.; Farley, D.R.; Richards, M.L.; Thompson, G.B. Biopsy of pheochromocytomas and paragangliomas: Potential for disaster. Surgery 2009, 146, 1158–1166. [Google Scholar] [CrossRef]
- Dwight, T.; Flynn, A.; Amarasinghe, K.; Benn, D.E.; Lupat, R.; Li, J.; Cameron, D.L.; Hogg, A.; Balachander, S.; Candiloro, I.L.M.; et al. TERT structural rearrangements in metastatic pheochromocytomas. Endocr. Relat. Cancer 2018, 25, 1–9. [Google Scholar] [CrossRef]
- Suh, Y.J.; Choe, J.Y.; Park, H.J. Malignancy in pheochromocytoma or paraganglioma: Integrative analysis of 176 cases in TCGA. Endocr. Pathol. 2017, 28, 159–164. [Google Scholar] [CrossRef]
- Stenman, A.; Svahn, F.; Hojjat-Farsangi, M.; Zedenius, J.; Soderkvist, P.; Gimm, O.; Larsson, C.; Juhlin, C.C. Molecular profiling of pheochromocytoma and abdominal paraganglioma stratified by the PASS algorithm reveals chromogranin B as associated with histologic prediction of malignant behavior. Am. J. Surg. Pathol. 2019, 43, 409–421. [Google Scholar] [CrossRef]
- Wang, W.; Zhong, X.; Ye, L.; Qi, Y.; Su, T.; Wei, Q.; Xie, J.; Jiang, L.; Jiang, Y.; Zhou, W.; et al. ERBB-2 overexpression as a risk factor for malignant phaeochromocytomas and paraganglinomas. Clin. Endocrinol. 2016, 84, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Udager, A.M.; Magers, M.J.; Goerke, D.M.; Vinco, M.L.; Siddiqui, J.; Cao, X.; Lucas, D.R.; Myers, J.L.; Chinnaiyan, A.M.; McHugh, J.B.; et al. The utility of SDHB and FH immunohistochemistry in patients evaluated for hereditary paraganglioma-pheochromocytoma syndromes. Hum. Pathol. 2018, 71, 47–54. [Google Scholar] [CrossRef] [PubMed]
- O’Riordain, D.S.; Young, W.F., Jr.; Grant, C.S.; Carney, J.A.; van Heerden, J.A. Clinical spectrum and outcome of functional extraadrenal paraganglioma. World J. Surg. 1996, 20, 916–921. [Google Scholar] [CrossRef] [PubMed]
- King, K.S.; Prodanov, T.; Kantorovich, V.; Fojo, T.; Hewitt, J.K.; Zacharin, M.; Wesley, R.; Lodish, M.; Raygada, M.; Gimenez-Roqueplo, A.P.; et al. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: Significant link to SDHB mutations. J. Clin. Oncol. 2011, 29, 4137–4142. [Google Scholar] [CrossRef] [PubMed]
- Grossman, A.; Pacak, K.; Sawka, A.; Lenders, J.W.; Harlander, D.; Peaston, R.T.; Reznek, R.; Sisson, J.; Eisenhofer, G. Biochemical diagnosis and localization of pheochromocytoma: Can we reach a consensus? Ann. N. Y. Acad. Sci. 2006, 1073, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Korevaar, T.I.; Grossman, A.B. Pheochromocytomas and paragangliomas: Assessment of malignant potential. Endocrine 2011, 40, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Bausch, B.; Wellner, U.; Bausch, D.; Schiavi, F.; Barontini, M.; Sanso, G.; Walz, M.K.; Peczkowska, M.; Weryha, G.; Dall’igna, P.; et al. Long-term prognosis of patients with pediatric pheochromocytoma. Endocr. Relat. Cancer 2014, 21, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Favier, J.; Amar, L.; Gimenez-Roqueplo, A.P. Paraganglioma and phaeochromocytoma: From genetics to personalized medicine. Nat. Rev. Endocrinol. 2015, 11, 101–111. [Google Scholar] [CrossRef]
- Wells, S.A., Jr.; Asa, S.L.; Dralle, H.; Elisei, R.; Evans, D.B.; Gagel, R.F.; Lee, N.; Machens, A.; Moley, J.F.; Pacini, F.; et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015, 25, 567–610. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.M.; Rhodes, L.; Blanco, I.; Chung, W.K.; Eng, C.; Maher, E.R.; Richard, S.; Giles, R.H. Von Hippel-Lindau disease: Genetics and role of genetic counseling in a multiple neoplasia syndrome. J. Clin. Oncol. 2016, 34, 2172–2181. [Google Scholar] [CrossRef] [PubMed]
- Rednam, S.P.; Erez, A.; Druker, H.; Janeway, K.A.; Kamihara, J.; Kohlmann, W.K.; Nathanson, K.L.; States, L.J.; Tomlinson, G.E.; Villani, A.; et al. Von Hippel-Lindau and hereditary pheochromocytoma/paraganglioma syndromes: Clinical features, genetics, and surveillance recommendations in childhood. Clin. Cancer Res. 2017, 23, e68–e75. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, D.H.; Aylsworth, A.; Carey, J.C.; Korf, B.; Marks, J.; Pyeritz, R.E.; Rubenstein, A.; Viskochil, D. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997, 278, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Brandi, M.L.; Gagel, R.F.; Angeli, A.; Bilezikian, J.P.; Beck-Peccoz, P.; Bordi, C.; Conte-Devolx, B.; Falchetti, A.; Gheri, R.G.; Libroia, A.; et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J. Clin. Endocrinol. Metab. 2001, 86, 5658–5671. [Google Scholar] [CrossRef] [PubMed]
- Buffet, A.; Ben Aim, L.; Leboulleux, S.; Drui, D.; Vezzosi, D.; Libe, R.; Ajzenberg, C.; Bernardeschi, D.; Cariou, B.; Chabolle, F.; et al. Positive impact of genetic test on the management and outcome of patients with paraganglioma and/or pheochromocytoma. J. Clin. Endocrinol. Metab. 2019, 104, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Walz, M.K.; Alesina, P.F.; Wenger, F.A.; Deligiannis, A.; Szuczik, E.; Petersenn, S.; Ommer, A.; Groeben, H.; Peitgen, K.; Janssen, O.E.; et al. Posterior retroperitoneoscopic adrenalectomy—Results of 560 procedures in 520 patients. Surgery 2006, 140, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Castinetti, F.; Qi, X.P.; Walz, M.K.; Maia, A.L.; Sanso, G.; Peczkowska, M.; Hasse-Lazar, K.; Links, T.P.; Dvorakova, S.; Toledo, R.A.; et al. Outcomes of adrenal-sparing surgery or total adrenalectomy in phaeochromocytoma associated with multiple endocrine neoplasia type 2: An international retrospective population-based study. Lancet Oncol. 2014, 15, 648–655. [Google Scholar] [CrossRef]
- Moore, M.G.; Netterville, J.L.; Mendenhall, W.M.; Isaacson, B.; Nussenbaum, B. Head and neck paragangliomas: An update on evaluation and management. Otolaryngol. Head Neck Surg. 2016, 154, 597–605. [Google Scholar] [CrossRef]
- Capatina, C.; Ntali, G.; Karavitaki, N.; Grossman, A.B. The management of head-and-neck paragangliomas. Endocr. Relat. Cancer 2013, 20, R291–R305. [Google Scholar] [CrossRef]
- Marchetti, M.; Pinzi, V.; Tramacere, I.; Bianchi, L.C.; Ghielmetti, F.; Fariselli, L. Radiosurgery for paragangliomas of the head and neck: Another step for the validation of a treatment paradigm. World Neurosurg. 2017, 98, 281–287. [Google Scholar] [CrossRef]
- Dupin, C.; Lang, P.; Dessard-Diana, B.; Simon, J.M.; Cuenca, X.; Mazeron, J.J.; Feuvret, L. Treatment of head and neck paragangliomas with external beam radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2014, 89, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.; Atanacio, A.S.; Prodanov, T.; Turkbey, B.I.; Adams, K.; Martucci, V.; Camphausen, K.; Fojo, A.T.; Pacak, K.; Kaushal, A. External beam radiation therapy in treatment of malignant pheochromocytoma and paraganglioma. Front. Oncol. 2014, 4, 166. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.P.H.; Tsoy, U.; Bancos, I.; Amodru, V.; Walz, M.K.; Tirosh, A.; Kaur, R.J.; McKenzie, T.; Qi, X.; Bandgar, T.; et al. Comparison of pheochromocytoma-specific morbidity and mortality among adults with bilateral pheochromocytomas undergoing total adrenalectomy vs cortical-sparing adrenalectomy. JAMA Netw. Open 2019, 2, e198898. [Google Scholar] [CrossRef] [PubMed]
- Roman-Gonzalez, A.; Zhou, S.; Ayala-Ramirez, M.; Shen, C.; Waguespack, S.G.; Habra, M.A.; Karam, J.A.; Perrier, N.; Wood, C.G.; Jimenez, C. Impact of surgical resection of the primary tumor on overall survival in patients with metastatic pheochromocytoma or sympathetic paraganglioma. Ann. Surg. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Ramirez, M.; Feng, L.; Habra, M.A.; Rich, T.; Dickson, P.V.; Perrier, N.; Phan, A.; Waguespack, S.; Patel, S.; Jimenez, C. Clinical benefits of systemic chemotherapy for patients with metastatic pheochromocytomas or sympathetic extra-adrenal paragangliomas: Insights from the largest single-institutional experience. Cancer 2012, 118, 2804–2812. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, O.; Young, W.F., Jr.; Iniguez-Ariza, N.M.; Kittah, N.E.; Gruber, L.; Bancos, C.; Tamhane, S.; Bancos, I. Malignant pheochromocytoma and paraganglioma: 272 patients over 55 years. J. Clin. Endocrinol. Metab. 2017, 102, 3296–3305. [Google Scholar] [CrossRef] [PubMed]
- Strajina, V.; Dy, B.M.; Farley, D.R.; Richards, M.L.; McKenzie, T.J.; Bible, K.C.; Que, F.G.; Nagorney, D.M.; Young, W.F.; Thompson, G.B. Surgical treatment of malignant pheochromocytoma and paraganglioma: Retrospective case series. Ann. Surg. Oncol. 2017, 24, 1546–1550. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Wu, D.; Yue, J. Surgical resection of multiple liver metastasis of functional malignant pheochromocytoma: A case report and literature review. J. Cancer Res. Ther. 2013, 9, S183–S185. [Google Scholar] [CrossRef] [PubMed]
- Arnas-Leon, C.; Sanchez, V.; Santana Suarez, A.D.; Quintana Arroyo, S.; Acosta, C.; Martinez Martin, F.J. Complete remission in metastatic pheochromocytoma treated with extensive surgery. Cureus 2016, 8, e447. [Google Scholar] [CrossRef] [PubMed]
- Breen, W.; Bancos, I.; Young, W.F., Jr.; Bible, K.C.; Laack, N.N.; Foote, R.L.; Hallemeier, C.L. External beam radiation therapy for advanced/unresectable malignant paraganglioma and pheochromocytoma. Adv. Radiat. Oncol. 2018, 3, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Gravel, G.; Leboulleux, S.; Tselikas, L.; Fassio, F.; Berraf, M.; Berdelou, A.; Ba, B.; Hescot, S.; Hadoux, J.; Schlumberger, M.; et al. Prevention of serious skeletal-related events by interventional radiology techniques in patients with malignant paraganglioma and pheochromocytoma. Endocrine 2018, 59, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Kohlenberg, J.; Welch, B.; Hamidi, O.; Callstrom, M.; Morris, J.; Sprung, J.; Bancos, I.; Young, W., Jr. Efficacy and safety of ablative therapy in the treatment of patients with metastatic pheochromocytoma and paraganglioma. Cancers 2019, 11, 195. [Google Scholar] [CrossRef] [PubMed]
- Plouin, P.F.; Duclos, J.M.; Soppelsa, F.; Boublil, G.; Chatellier, G. Factors associated with perioperative morbidity and mortality in patients with pheochromocytoma: Analysis of 165 operations at a single center. J. Clin. Endocrinol. Metab. 2001, 86, 1480–1486. [Google Scholar] [CrossRef] [PubMed]
- Pacak, K. Preoperative management of the pheochromocytoma patient. J. Clin. Endocrinol. Metab. 2007, 92, 4069–4079. [Google Scholar] [CrossRef]
- van der Zee, P.A.; de Boer, A. Pheochromocytoma: A review on preoperative treatment with phenoxybenzamine or doxazosin. Neth. J. Med. 2014, 72, 190–201. [Google Scholar] [PubMed]
- Mak, I.Y.F.; Hayes, A.R.; Khoo, B.; Grossman, A. Peptide receptor radionuclide therapy as a novel treatment for metastatic and invasive phaeochromocytoma and paraganglioma. Neuroendocrinology 2019. [Google Scholar] [CrossRef] [PubMed]
- Pryma, D.A.; Chin, B.B.; Noto, R.B.; Dillon, J.S.; Perkins, S.; Solnes, L.; Kostakoglu, L.; Serafini, A.N.; Pampaloni, M.H.; Jensen, J.; et al. Efficacy and safety of high-specific-activity 131I-MIBG therapy in patients with advanced pheochromocytoma or paraganglioma. J. Nucl. Med. 2019, 60, 623–630. [Google Scholar] [CrossRef]
- van Hulsteijn, L.T.; Niemeijer, N.D.; Dekkers, O.M.; Corssmit, E.P. 131I-MIBG therapy for malignant paraganglioma and phaeochromocytoma: Systematic review and meta-analysis. Clin. Endocrinol. 2014, 80, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Castellani, M.R.; Seghezzi, S.; Chiesa, C.; Aliberti, G.L.; Maccauro, M.; Seregni, E.; Orunesu, E.; Luksch, R.; Bombardieri, E. 131I-MIBG treatment of pheochromocytoma: Low versus intermediate activity regimens of therapy. Q. J. Nucl. Med. Mol. Imaging 2010, 54, 100–113. [Google Scholar]
- Sze, W.C.; Grossman, A.B.; Goddard, I.; Amendra, D.; Shieh, S.C.; Plowman, P.N.; Drake, W.M.; Akker, S.A.; Druce, M.R. Sequelae and survivorship in patients treated with 131I-MIBG therapy. Br. J. Cancer 2013, 109, 565–572. [Google Scholar] [CrossRef]
- Ziegler, C.G.; Brown, J.W.; Schally, A.V.; Erler, A.; Gebauer, L.; Treszl, A.; Young, L.; Fishman, L.M.; Engel, J.B.; Willenberg, H.S.; et al. Expression of neuropeptide hormone receptors in human adrenal tumors and cell lines: Antiproliferative effects of peptide analogues. Proc. Natl. Acad. Sci. USA 2009, 106, 15879–15884. [Google Scholar] [CrossRef] [PubMed]
- van Essen, M.; Krenning, E.P.; de Jong, M.; Valkema, R.; Kwekkeboom, D.J. Peptide receptor radionuclide therapy with radiolabelled somatostatin analogues in patients with somatostatin receptor positive tumours. Acta Oncol. 2007, 46, 723–734. [Google Scholar] [CrossRef] [PubMed]
- van Essen, M.; Krenning, E.P.; Kooij, P.P.; Bakker, W.H.; Feelders, R.A.; de Herder, W.W.; Wolbers, J.G.; Kwekkeboom, D.J. Effects of therapy with [177Lu-DOTA0, Tyr3] octreotate in patients with paraganglioma, meningioma, small cell lung carcinoma, and melanoma. J. Nucl. Med. 2006, 47, 1599–1606. [Google Scholar] [PubMed]
- Zovato, S.; Kumanova, A.; Dematte, S.; Sansovini, M.; Bodei, L.; Di Sarra, D.; Casagranda, E.; Severi, S.; Ambrosetti, A.; Schiavi, F.; et al. Peptide receptor radionuclide therapy (PRRT) with 177Lu-DOTATATE in individuals with neck or mediastinal paraganglioma (PGL). Horm. Metab. Res. 2012, 44, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Forrer, F.; Riedweg, I.; Maecke, H.R.; Mueller-Brand, J. Radiolabeled DOTATOC in patients with advanced paraganglioma and pheochromocytoma. Q. J. Nucl. Med. Mol. Imaging 2008, 52, 334–340. [Google Scholar]
- Kong, G.; Grozinsky-Glasberg, S.; Hofman, M.S.; Callahan, J.; Meirovitz, A.; Maimon, O.; Pattison, D.A.; Gross, D.J.; Hicks, R.J. Efficacy of peptide receptor radionuclide therapy for functional metastatic paraganglioma and pheochromocytoma. J. Clin. Endocrinol. Metab. 2017, 102, 3278–3287. [Google Scholar] [CrossRef] [PubMed]
- Nastos, K.; Cheung, V.T.F.; Toumpanakis, C.; Navalkissoor, S.; Quigley, A.M.; Caplin, M.; Khoo, B. Peptide receptor radionuclide treatment and 131I-MIBG in the management of patients with metastatic/progressive phaeochromocytomas and paragangliomas. J. Surg. Oncol. 2017, 115, 425–434. [Google Scholar] [CrossRef]
- Pinato, D.J.; Black, J.R.; Ramaswami, R.; Tan, T.M.; Adjogatse, D.; Sharma, R. Peptide receptor radionuclide therapy for metastatic paragangliomas. Med. Oncol. 2016, 33, 47. [Google Scholar] [CrossRef] [PubMed]
- Imhof, A.; Brunner, P.; Marincek, N.; Briel, M.; Schindler, C.; Rasch, H.; Macke, H.R.; Rochlitz, C.; Muller-Brand, J.; Walter, M.A. Response, survival, and long-term toxicity after therapy with the radiolabeled somatostatin analogue [90Y-DOTA]-TOC in metastasized neuroendocrine cancers. J. Clin. Oncol. 2011, 29, 2416–2423. [Google Scholar] [CrossRef]
- Puranik, A.D.; Kulkarni, H.R.; Singh, A.; Baum, R.P. Peptide receptor radionuclide therapy with 90Y/177Lu-labelled peptides for inoperable head and neck paragangliomas (glomus tumours). Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1223–1230. [Google Scholar] [CrossRef]
- Yadav, M.P.; Ballal, S.; Bal, C. Concomitant 177Lu-DOTATATE and capecitabine therapy in malignant paragangliomas. EJNMMI Res. 2019, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Estevao, R.; Duarte, H.; Lopes, F.; Fernandes, J.; Monteiro, E. Peptide receptor radionuclide therapy in head and neck paragangliomas—Report of 14 cases. Rev. Laryngol. Otol. Rhinol. 2015, 136, 155–158. [Google Scholar]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 trial of 177Lu-DOTATATE for midgut neuroendocrine tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Navalkissoor, S.; Grossman, A. Targeted alpha particle therapy for neuroendocrine tumours: The next generation of peptide receptor radionuclide therapy. Neuroendocrinology 2019, 108, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Brabander, T.; van der Zwan, W.A.; Teunissen, J.J.M.; Kam, B.L.R.; Feelders, R.A.; de Herder, W.W.; van Eijck, C.H.J.; Franssen, G.J.H.; Krenning, E.P.; Kwekkeboom, D.J. Long-term efficacy, survival, and safety of [177Lu-DOTA0,Tyr3]octreotate in patients with gastroenteropancreatic and bronchial neuroendocrine tumors. Clin. Cancer Res. 2017, 23, 4617–4624. [Google Scholar] [CrossRef] [PubMed]
- Bodei, L.; Cremonesi, M.; Kidd, M.; Grana, C.M.; Severi, S.; Modlin, I.M.; Paganelli, G. Peptide receptor radionuclide therapy for advanced neuroendocrine tumors. Thorac. Surg. Clin. 2014, 24, 333–349. [Google Scholar] [CrossRef]
- Bodei, L.; Cremonesi, M.; Paganelli, G. Yttrium-based therapy for neuroendocrine tumors. PET Clin. 2014, 9, 71–82. [Google Scholar] [CrossRef]
- Bergsma, H.; van Lom, K.; Raaijmakers, M.; Konijnenberg, M.; Kam, B.; Teunissen, J.J.M.; de Herder, W.W.; Krenning, E.P.; Kwekkeboom, D.J. Persistent hematologic dysfunction after peptide receptor radionuclide therapy with 177Lu-DOTATATE: Incidence, course, and predicting factors in patients with gastroenteropancreatic neuroendocrine tumors. J. Nucl. Med. 2018, 59, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Schuchardt, C.; Kulkarni, H.R.; Prasad, V.; Zachert, C.; Muller, D.; Baum, R.P. The Bad Berka dose protocol: Comparative results of dosimetry in peptide receptor radionuclide therapy using 177Lu-DOTATATE, 177Lu-DOTANOC, and 177Lu-DOTATOC. In Theranostics, Gallium-68, and Other Radionuclides; Springer: Berlin/Heidelberg, Germany, 2013; Volume 194, pp. 519–536. [Google Scholar]
- Del Prete, M.; Buteau, F.A.; Arsenault, F.; Saighi, N.; Bouchard, L.O.; Beaulieu, A.; Beauregard, J.M. Personalized 177Lu-octreotate peptide receptor radionuclide therapy of neuroendocrine tumours: Initial results from the P-PRRT trial. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 728–742. [Google Scholar] [CrossRef]
- Taieb, D.; Garrigue, P.; Bardies, M.; Abdullah, A.E.; Pacak, K. Application and dosimetric requirements for gallium-68–labeled somatostatin analogues in targeted radionuclide therapy for gastroenteropancreatic neuroendocrine tumors. PET Clin. 2015, 10, 477–486. [Google Scholar] [CrossRef]
- Huizing, D.M.V.; de Wit-van der Veen, B.J.; Verheij, M.; Stokkel, M.P.M. Dosimetry methods and clinical applications in peptide receptor radionuclide therapy for neuroendocrine tumours: A literature review. EJNMMI Res. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rinke, A.; Muller, H.H.; Schade-Brittinger, C.; Klose, K.J.; Barth, P.; Wied, M.; Mayer, C.; Aminossadati, B.; Pape, U.F.; Blaker, M.; et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the PROMID Study Group. J. Clin. Oncol. 2009, 27, 4656–4663. [Google Scholar] [CrossRef] [PubMed]
- Caplin, M.E.; Pavel, M.; Cwikla, J.B.; Phan, A.T.; Raderer, M.; Sedlackova, E.; Cadiot, G.; Wolin, E.M.; Capdevila, J.; Wall, L.; et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N. Engl. J. Med. 2014, 371, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Niemeijer, N.D.; Alblas, G.; van Hulsteijn, L.T.; Dekkers, O.M.; Corssmit, E.P. Chemotherapy with cyclophosphamide, vincristine and dacarbazine for malignant paraganglioma and pheochromocytoma: Systematic review and meta-analysis. Clin. Endocrinol. 2014, 81, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Averbuch, S.D.; Steakley, C.S.; Young, R.C.; Gelmann, E.P.; Goldstein, D.S.; Stull, R.; Keiser, H.R. Malignant pheochromocytoma: Effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. Ann. Inter. Med. 1988, 109, 267–273. [Google Scholar] [CrossRef]
- Huang, H.; Abraham, J.; Hung, E.; Averbuch, S.; Merino, M.; Steinberg, S.M.; Pacak, K.; Fojo, T. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: Recommendation from a 22-year follow-up of 18 patients. Cancer 2008, 113, 2020–2028. [Google Scholar] [CrossRef] [PubMed]
- Jawed, I.; Velarde, M.; Darr, R.; Wolf, K.I.; Adams, K.; Venkatesan, A.M.; Balasubramaniam, S.; Poruchynsky, M.S.; Reynolds, J.C.; Pacak, K.; et al. Continued tumor reduction of metastatic pheochromocytoma/paraganglioma harboring succinate dehydrogenase subunit B mutations with cyclical chemotherapy. Cell. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hadoux, J.; Favier, J.; Scoazec, J.Y.; Leboulleux, S.; Al Ghuzlan, A.; Caramella, C.; Deandreis, D.; Borget, I.; Loriot, C.; Chougnet, C.; et al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int. J. Cancer 2014, 135, 2711–2720. [Google Scholar] [CrossRef] [PubMed]
- Tena, I.; Gupta, G.; Tajahuerce, M.; Benavent, M.; Cifrian, M.; Falcon, A.; Fonfria, M.; Del Olmo, M.; Reboll, R.; Conde, A.; et al. Successful second-line metronomic temozolomide in metastatic paraganglioma: Case reports and review of the literature. Clin. Med. Insights Oncol. 2018, 12. [Google Scholar] [CrossRef]
- Pegg, A.E.; Dolan, M.E.; Moschel, R.C. Structure, function, and inhibition of O6-alkylguanine-DNA alkyltransferase. In Progress in Nucleic Acid Research and Molecular Biology; Academic Press: Cambridge, MA, USA, 1995; Volume 51, pp. 167–223. [Google Scholar]
- Hegi, M.E.; Liu, L.; Herman, J.G.; Stupp, R.; Wick, W.; Weller, M.; Mehta, M.P.; Gilbert, M.R. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J. Clin. Oncol. 2008, 26, 4189–4199. [Google Scholar] [CrossRef]
- Bignami, M.; O’Driscoll, M.; Aquilina, G.; Karran, P. Unmasking a killer: DNA O6-methylguanine and the cytotoxicity of methylating agents. Mutat. Res. 2000, 462, 71–82. [Google Scholar] [CrossRef]
- Tay, C.G.; Lee, V.W.M.; Ong, L.C.; Goh, K.J.; Ariffin, H.; Fong, C.Y. Vincristine-induced peripheral neuropathy in survivors of childhood acute lymphoblastic leukaemia. Pediatric Blood Cancer 2017, 64, e26471. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Ramirez, M.; Chougnet, C.N.; Habra, M.A.; Palmer, J.L.; Leboulleux, S.; Cabanillas, M.E.; Caramella, C.; Anderson, P.; Al Ghuzlan, A.; Waguespack, S.G.; et al. Treatment with sunitinib for patients with progressive metastatic pheochromocytomas and sympathetic paragangliomas. J. Clin. Endocrinol. Metab. 2012, 97, 4040–4050. [Google Scholar] [CrossRef] [PubMed]
- O’Kane, G.M.; Ezzat, S.; Joshua, A.M.; Bourdeau, I.; Leibowitz-Amit, R.; Olney, H.J.; Krzyzanowska, M.; Reuther, D.; Chin, S.; Wang, L.; et al. A phase 2 trial of sunitinib in patients with progressive paraganglioma or pheochromocytoma: The SNIPP trial. Br. J. Cancer 2019, 120, 1113–1119. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Halabi, S.; Sanford, B.L.; Hahn, O.; Michaelson, M.D.; Walsh, M.K.; Feldman, D.R.; Olencki, T.; Picus, J.; Small, E.J.; et al. Cabozantinib versus sunitinib as initial targeted therapy for patients with metastatic renal cell carcinoma of poor or intermediate risk: The Alliance A031203 CABOSUN trial. J. Clin. Oncol. 2017, 35, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Hessel, C.; Halabi, S.; Sanford, B.; Michaelson, M.D.; Hahn, O.; Walsh, M.; Olencki, T.; Picus, J.; Small, E.J.; et al. Cabozantinib versus sunitinib as initial therapy for metastatic renal cell carcinoma of intermediate or poor risk (Alliance A031203 CABOSUN randomised trial): Progression-free survival by independent review and overall survival update. Eur. J. Cancer 2018, 94, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Fankhauser, M.; Bechmann, N.; Lauseker, M.; Goncalves, J.; Favier, J.; Klink, B.; William, D.; Gieldon, L.; Maurer, J.; Spottl, G.; et al. Synergistic highly potent targeted drug combinations in different pheochromocytoma models including human tumor cultures. Endocrinology 2019. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, P.; Tatsui, C.; Jessop, A.; Thosani, S.; Jimenez, C. Treatment for malignant pheochromocytomas and paragangliomas: 5 years of progress. Curr. Oncol. Rep. 2017, 19, 83. [Google Scholar] [CrossRef]
- Jasim, S.; Suman, V.J.; Jimenez, C.; Harris, P.; Sideras, K.; Burton, J.K.; Worden, F.P.; Auchus, R.J.; Bible, K.C. Phase II trial of pazopanib in advanced/progressive malignant pheochromocytoma and paraganglioma. Endocrine 2017, 57, 220–225. [Google Scholar] [CrossRef]
- Oh, D.Y.; Kim, T.W.; Park, Y.S.; Shin, S.J.; Shin, S.H.; Song, E.K.; Lee, H.J.; Lee, K.W.; Bang, Y.J. Phase 2 study of everolimus monotherapy in patients with nonfunctioning neuroendocrine tumors or pheochromocytomas/paragangliomas. Cancer 2012, 118, 6162–6170. [Google Scholar] [CrossRef]
- Druce, M.R.; Kaltsas, G.A.; Fraenkel, M.; Gross, D.J.; Grossman, A.B. Novel and evolving therapies in the treatment of malignant phaeochromocytoma: Experience with the mTOR inhibitor everolimus (RAD001). Horm. Metab. Res. 2009, 41, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Nölting, S.; Garcia, E.; Alusi, G.; Giubellino, A.; Pacak, K.; Korbonits, M.; Grossman, A.B. Combined blockade of signalling pathways shows marked anti-tumour potential in phaeochromocytoma cell lines. J. Mol. Endocrinol. 2012, 49, 79–96. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nölting, S.; Giubellino, A.; Tayem, Y.; Young, K.; Lauseker, M.; Bullova, P.; Schovanek, J.; Anver, M.; Fliedner, S.; Korbonits, M.; et al. Combination of 13-Cis retinoic acid and lovastatin: Marked anti-tumor potential in vivo in a pheochromocytoma allograft model in female athymic nude mice. Endocrinology 2014. [Google Scholar] [CrossRef] [PubMed]
- Nölting, S.; Maurer, J.; Spottl, G.; Aristizabal Prada, E.T.; Reuther, C.; Young, K.; Korbonits, M.; Goke, B.; Grossman, A.; Auernhammer, C.J. Additive anti-tumor effects of lovastatin and everolimus in vitro through simultaneous inhibition of signaling pathways. PLoS ONE 2015, 10, e0143830. [Google Scholar] [CrossRef] [PubMed]
- Giubellino, A.; Bullova, P.; Nolting, S.; Turkova, H.; Powers, J.F.; Liu, Q.; Guichard, S.; Tischler, A.S.; Grossman, A.B.; Pacak, K. Combined inhibition of mTORC1 and mTORC2 signaling pathways is a promising therapeutic option in inhibiting pheochromocytoma tumor growth: In vitro and in vivo studies in female athymic nude mice. Endocrinology 2013, 154, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Liu, Y.; Pacak, K.; Yang, C. Pheochromocytomas and paragangliomas: From genetic diversity to targeted therapies. Cancers 2019, 11, 436. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, S.M.; Sitkovsky, M. A2A adenosine receptor antagonists to weaken the hypoxia-HIF-1alpha driven immunosuppression and improve immunotherapies of cancer. Curr. Opin. Pharmacol. 2016, 29, 90–96. [Google Scholar] [CrossRef]
- Labiano, S.; Palazon, A.; Bolanos, E.; Azpilikueta, A.; Sanchez-Paulete, A.R.; Morales-Kastresana, A.; Quetglas, J.I.; Perez-Gracia, J.L.; Gurpide, A.; Rodriguez-Ruiz, M.; et al. Hypoxia-induced soluble CD137 in malignant cells blocks CD137L-costimulation as an immune escape mechanism. Oncoimmunology 2016, 5, e1062967. [Google Scholar] [CrossRef]
- Chouaib, S.; Noman, M.Z.; Kosmatopoulos, K.; Curran, M.A. Hypoxic stress: Obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 2017, 36, 439–445. [Google Scholar] [CrossRef]


{kind=link}
| Genes | Cluster 1 (Pseudohypoxic Krebs Cycle-Related): SDHx (SDHA, B, C, D, F2), FH, MDH2 (10–15% of PCC/PGL) | Cluster 1 (Pseudohypoxia VHL/EPAS1-Related: VHL, EPAS1/2 (HIF2A), PHD1/2 (15–20% of PCC/PGL) | Cluster 2 (Kinase Signaling-Related): RET, NF1, MAX, TMEM127, HRAS (50–60% of PCC/PGL) | Cluster 3 (Wnt Signaling-Related): CSDE1, MAML3 5–10% of PCC/PGL) |
|---|---|---|---|---|
| Percentage of germline mutations [2] | Germline 100% | Germline 25% | Germline 20% | Germline 0% |
| Signaling pathways | Pseudohypoxia, Krebs cycle-related, HIF-2α stabilization and signaling | Pseudohypoxia, VHL/EPAS1-related signaling | Kinase signaling: PI3K/AKT, RAS/RAF/ERK, mTORC1/p70S6K | Wnt signaling |
| Biochemistry | Normetanephrine, 3-methoxytyramine | Normetanephrine | Normetanephrine and metanephrine or metanephrine alone | Normetanephrine, metanephrine |
| Imaging | [68Ga]Ga-DOTA-SSA PET/CT (possibly except for FH) | [18F]FDOPA PET/CT (possibly also for FH) | [18F]FDOPA PET/CT in most | Unknown |
| Tumor location | Mostly extra-adrenal | Adrenal, extra-adrenal | Adrenal | Unknown |
| Metastatic risk | High-Intermediate | Intermediate-Low | Low | Intermediate |
| Age of presentation | Early (under 20–30 year-old) | Early, some often during childhood | Late but some can present early | Unknown |
| Radionuclide Imaging Method for Diagnosis, Staging, and Follow-Up | (Sporadic) PGL, Multifocal/Metastatic Disease, SDHx | (Sporadic) PCC (Except SDHx): VHL, EPAS1 (HIF2A), FH, PHD1/2, Kinase Signaling-Associated (NF1, RET, MAX) |
|---|---|---|
| First choice | [68Ga]Ga-DOTA-SSA PET/CT reveals the predictive power for efficacy of PRRT | [18F]FDOPA PET/CT |
| Second choice | [18F]FDG PET/CT for SDHx-related PGL (except for SDHD-related HN PGL) | [68Ga]Ga-DOTA-SSA PET/CT (except for EPAS1 (HIF2A), PHD1/2) |
| Second choice | [18F]FDOPA PET/CT for SDHD-related HN PGL | [18F]FDG PET/CT for EPAS1/2 (HIF2A), PHD1/2 |
| Follow-Up | High Risk Group: Completely Resected Metastatic PCC/PGL, Completely Resected Non-Metastatic Sympathetic PGL, SDHA, SDHB, SDHD (Except for HN PGL), EPAS1 (HIF2A), PHD1/2 | Intermediate Risk Group: Completely Resected HN PGL, Completely Resected High-Risk PCC, SDHAF2, SDHC, VHL Type 2, FH (Data Is Limited), TMEM127, MAX, RET (High Risk Allele), Recurrence, Multiplicity | Low Risk Group: Completely Resected Low-Risk PCC, VHL Type 1, NF1, RET (Low to Moderate Risk Allele) |
|---|---|---|---|
| Clinical/metanephrines | 6–12 months | 12 months | 12–24 months |
| Imaging (preferably MRI *, less frequently CT *, but an alternate approach is allowed and by some recommended) | 12–24 months (shorter 12-months interval may be based on metastases, the initial size of a tumor, secretory pattern, and young age), consider CT, especially for lung involvement | 24–36 months (36 months applies for small sporadic metanephrine secreting tumors) | Optional at screening |
| Special cases | Completely resected metastatic PGL/PCC, SDHA/B PCC/PGL, sympathetic PGL: May consider radionuclide imaging 3–4 months after surgery if abnormal biochemistry or non-functioning PCC/PGL Completely resected metastatic PGL/metastatic PCC (independent of biochemistry): MRI 6 months, 12 months after surgery, then MRI yearly, consider specific radionuclide imaging every 24–36 months |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nölting, S.; Ullrich, M.; Pietzsch, J.; Ziegler, C.G.; Eisenhofer, G.; Grossman, A.; Pacak, K. Current Management of Pheochromocytoma/Paraganglioma: A Guide for the Practicing Clinician in the Era of Precision Medicine. Cancers 2019, 11, 1505. https://doi.org/10.3390/cancers11101505
Nölting S, Ullrich M, Pietzsch J, Ziegler CG, Eisenhofer G, Grossman A, Pacak K. Current Management of Pheochromocytoma/Paraganglioma: A Guide for the Practicing Clinician in the Era of Precision Medicine. Cancers. 2019; 11(10):1505. https://doi.org/10.3390/cancers11101505
Chicago/Turabian StyleNölting, Svenja, Martin Ullrich, Jens Pietzsch, Christian G. Ziegler, Graeme Eisenhofer, Ashley Grossman, and Karel Pacak. 2019. "Current Management of Pheochromocytoma/Paraganglioma: A Guide for the Practicing Clinician in the Era of Precision Medicine" Cancers 11, no. 10: 1505. https://doi.org/10.3390/cancers11101505
APA StyleNölting, S., Ullrich, M., Pietzsch, J., Ziegler, C. G., Eisenhofer, G., Grossman, A., & Pacak, K. (2019). Current Management of Pheochromocytoma/Paraganglioma: A Guide for the Practicing Clinician in the Era of Precision Medicine. Cancers, 11(10), 1505. https://doi.org/10.3390/cancers11101505