Unraveling the Heterogeneous Mutational Signature of Spontaneously Developing Tumors in MLH1−/− Mice

 , ,
, ,
Abstract
1. Introduction
2. Results
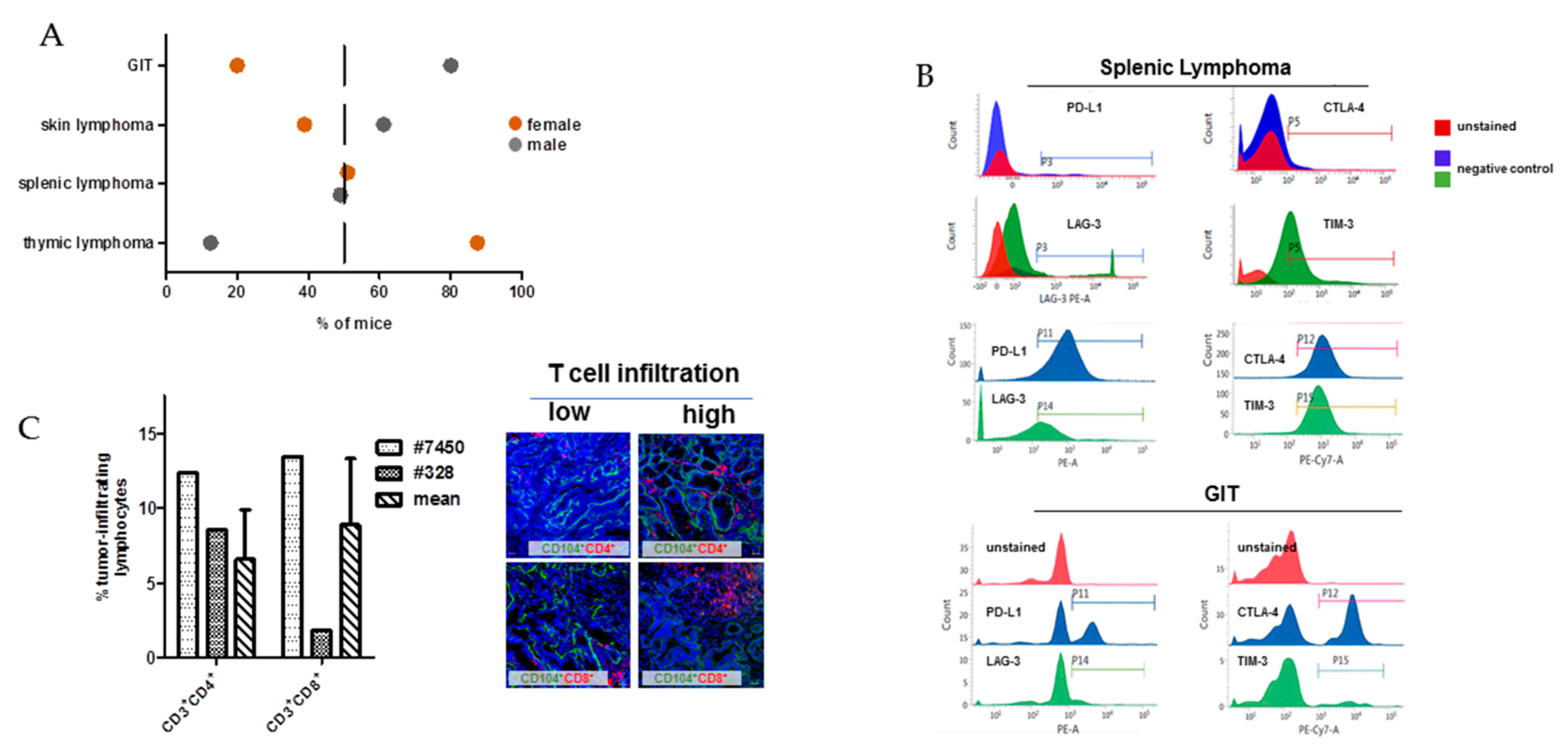
2.1. In Vivo and Ex Vivo Data
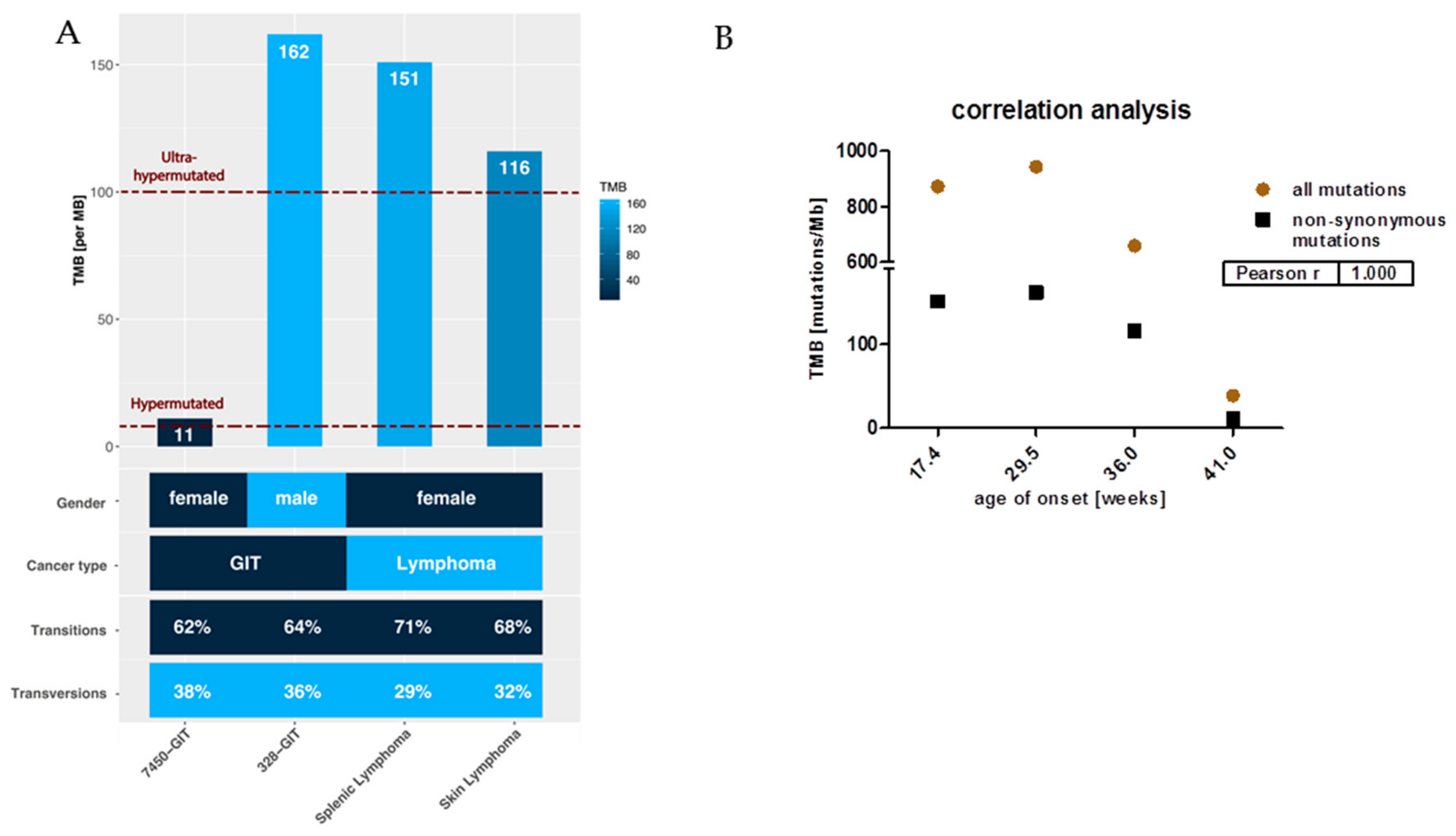
2.2. Mutational Landscape of MLH1−/− Tumors
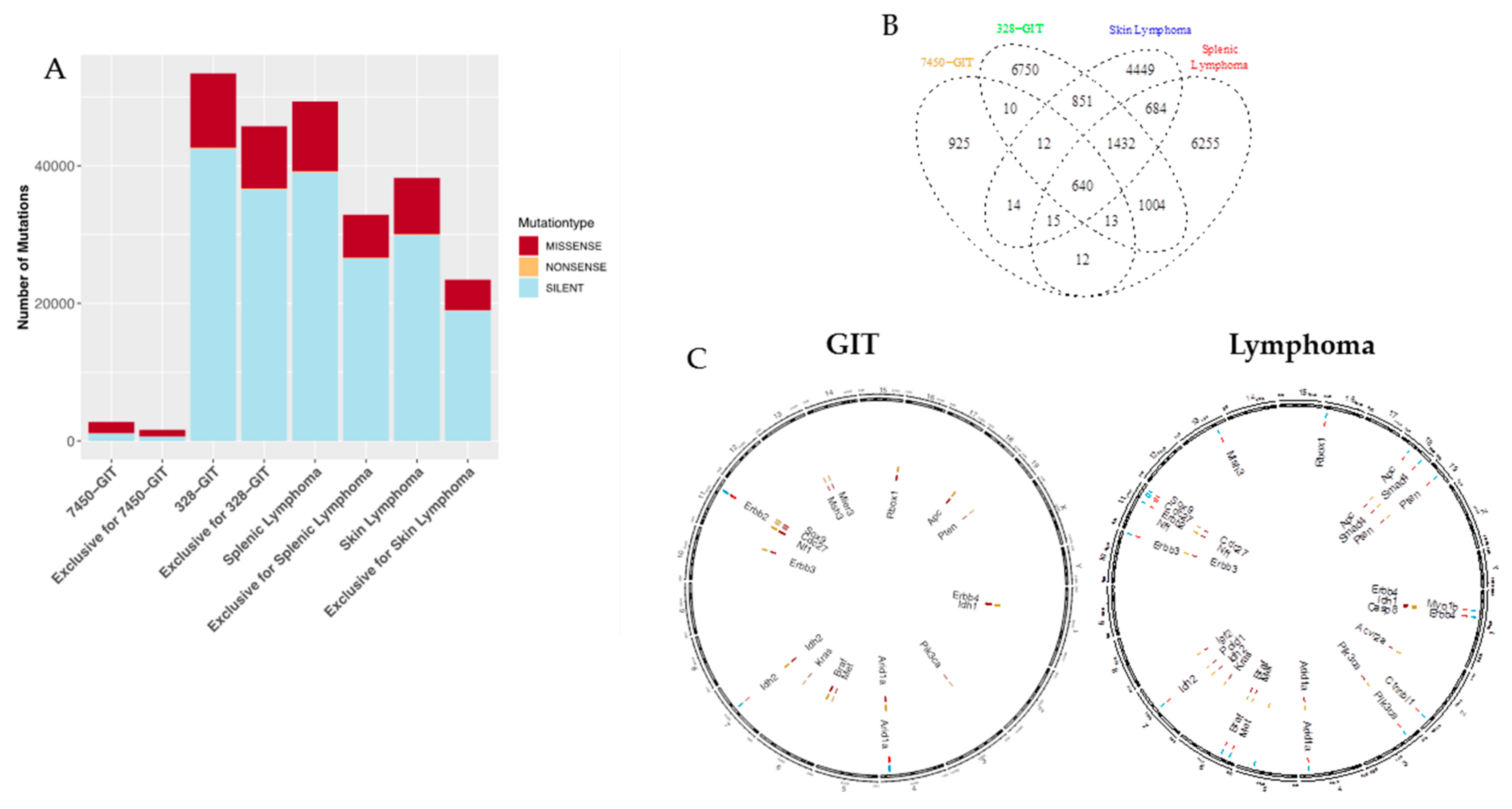
2.3. Mutation Types and Shared Mutations
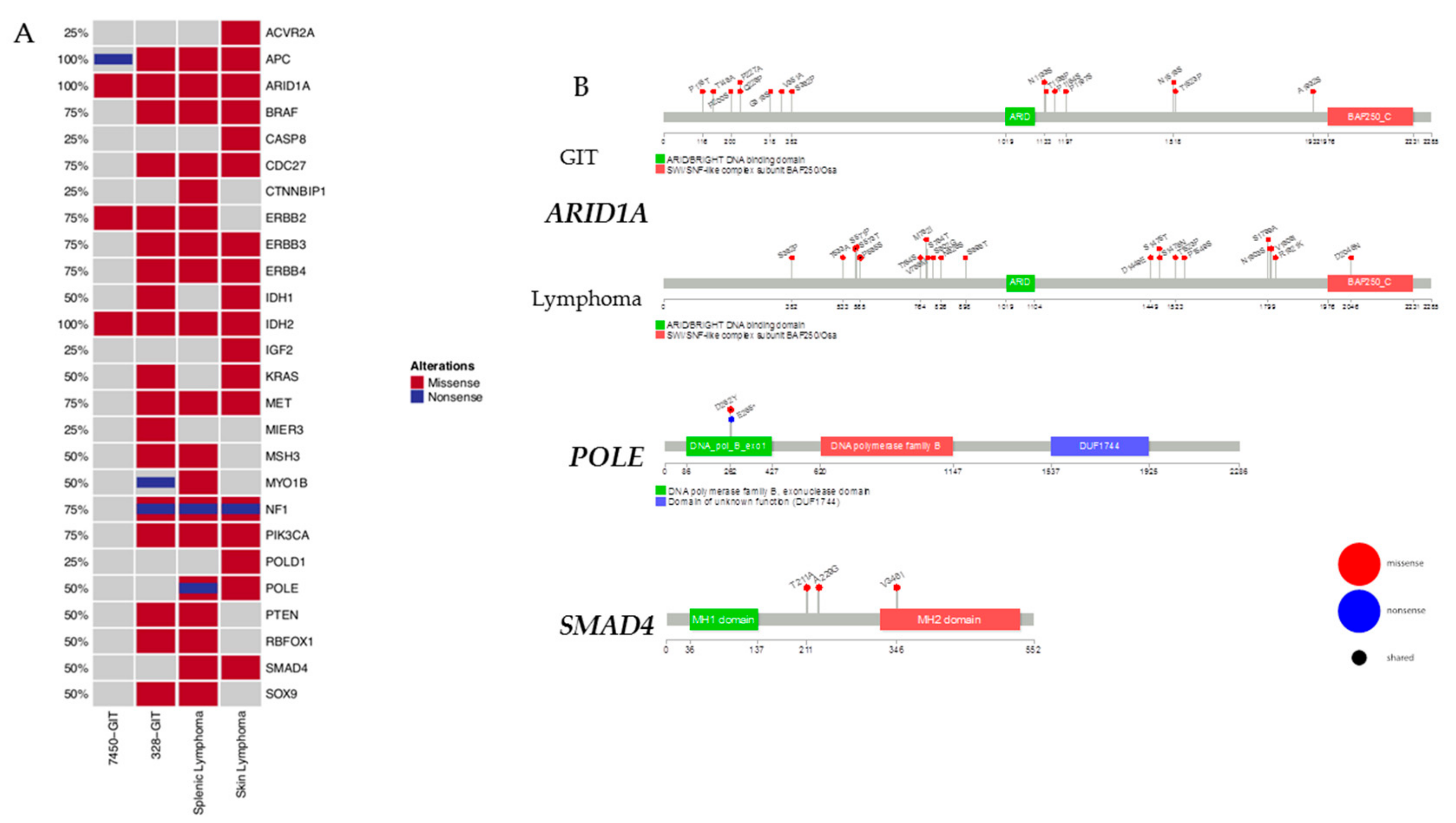
2.4. Mutational Frequencies and Types of Alterations for Selected Gene Sets Based on Prior Knowledge in Mouse and Human
2.5. Pairwise Exclusively SNV in GIT and Lymphomas
2.6. Prevalence and Hotspot Regions of GIT and Lymphomas in ARID1A, POLE, and SMAD4
2.7. Somatic Mutation Distribution and Nucleotide Changes in Chromosome X
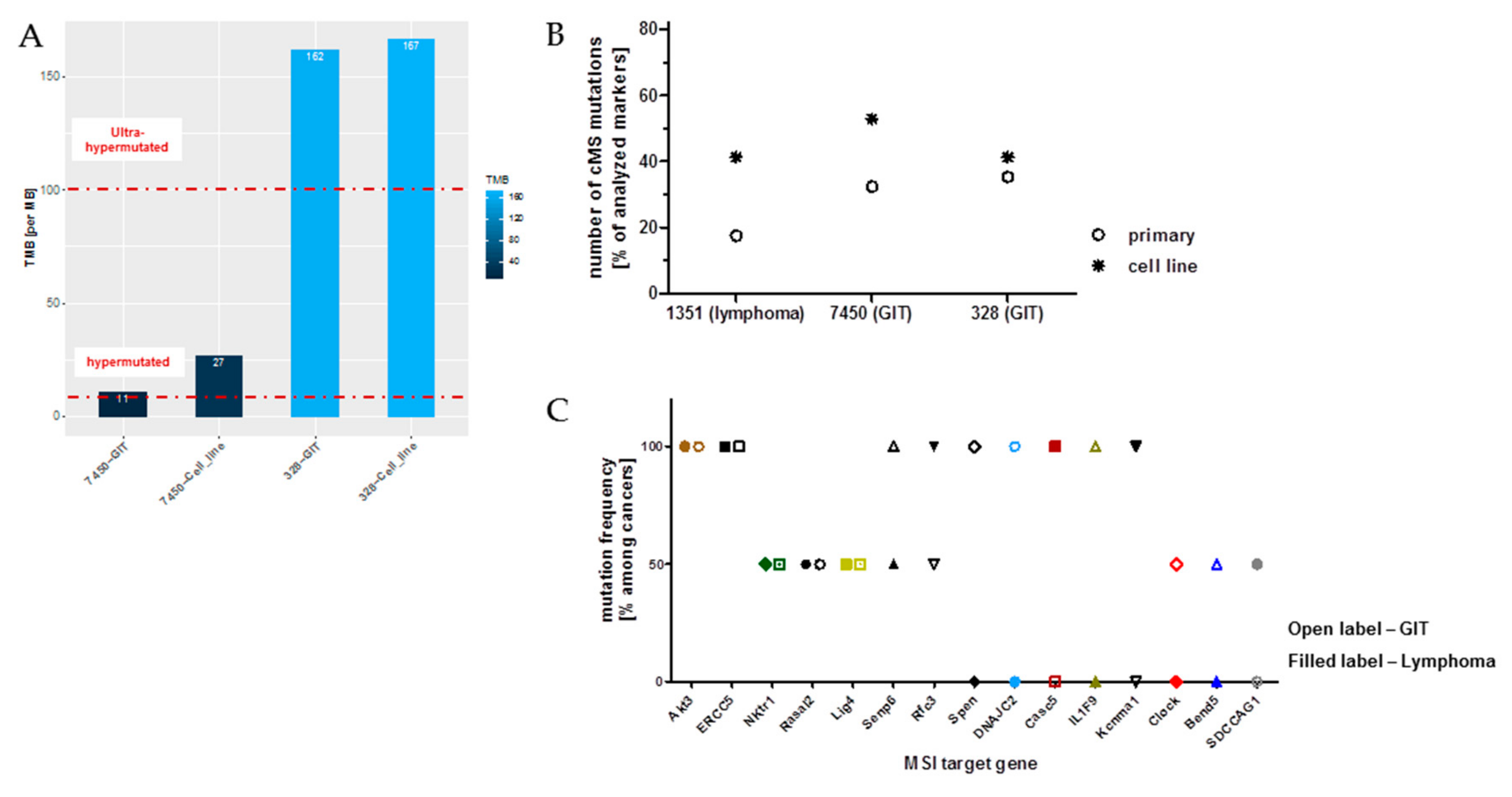
2.8. Microsatellite Instability (MSI) Pathway—Prognostic, Predictive and Therapeutic Implications
2.9. Ongoing Mutations in MLH1−/− Tumors
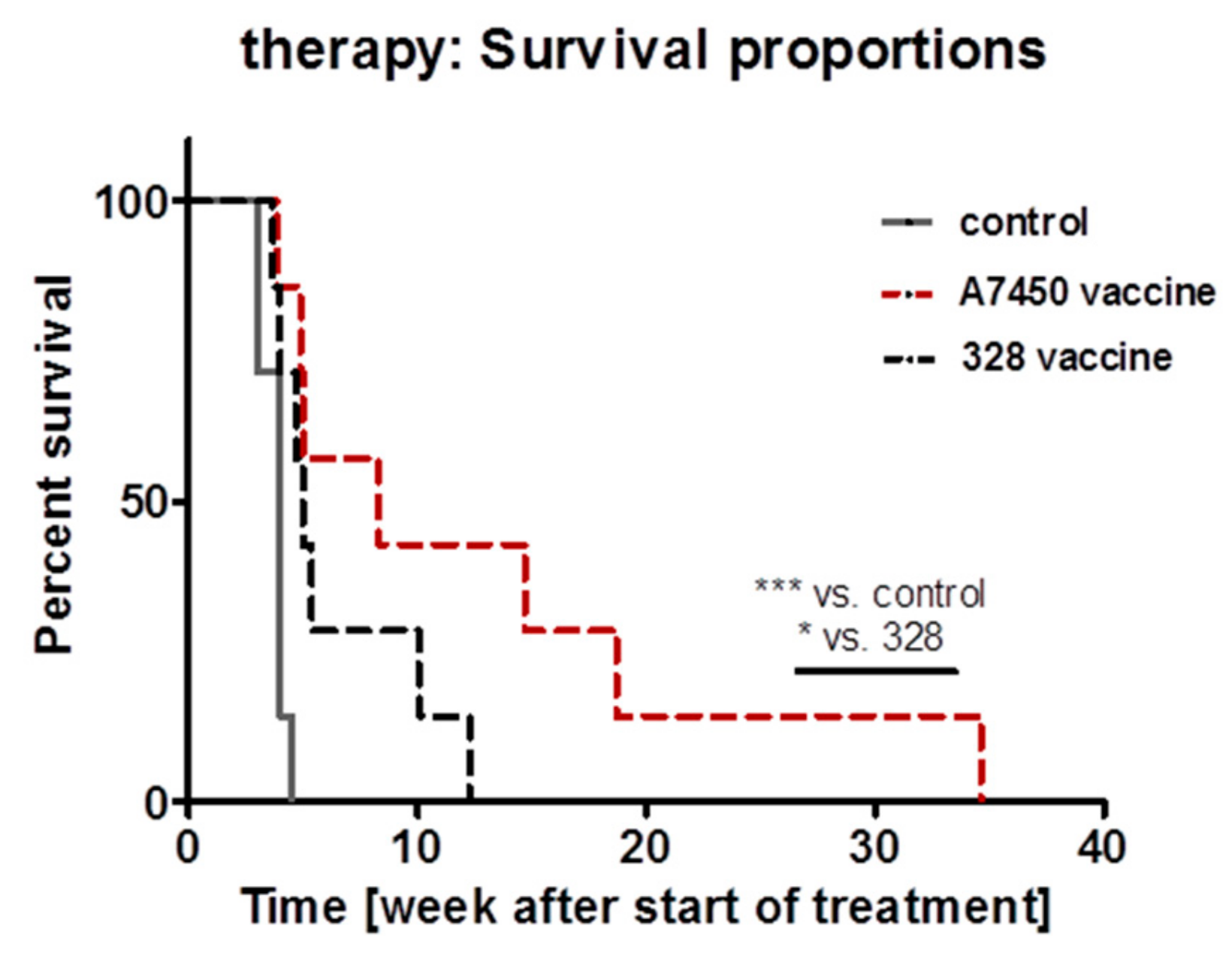
2.10. In Vivo Vaccination Approach
3. Discussion
4. Critical Limitation of the Study
5. Materials and Methods
5.1. In Vivo Mouse Model and Sample Acquisition
5.2. Whole-Exome Sequencing (WES) Analysis
5.3. Data Visualization
5.4. Hypermutation and TMB Calculation
5.5. Coding Microsatellite (cMS) Frameshift Mutation Analysis
5.6. In Vivo Vaccination with MLH1−/− Tumor Lysates
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Gupta, S.; Chatterjee, S.; Mukherjee, A.; Mutsuddi, M. Whole exome sequencing: Uncovering causal genetic variants for ocular diseases. Exp. Eye Res. 2017, 164, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Roche, M.I.; Berg, J.S. Incidental Findings with Genomic Testing: Implications for Genetic Counseling Practice. Curr. Genet. Med. Rep. 2015, 3, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Maletzki, C.; Gladbach, Y.S.; Hamed, M.; Fuellen, G.; Semmler, M.-L.; Stenzel, J.; Linnebacher, M. Cellular vaccination of MLH1−/− mice—An immunotherapeutic proof of concept study. Oncoimmunology 2018, 7, e1408748. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, M.; Janikovits, J.; von Knebel Doeberitz, M.; Kloor, M. Complex pattern of immune evasion in MSI colorectal cancer. Oncoimmunology 2018, 7, e1445453. [Google Scholar] [CrossRef]
- Kloor, M.; Von Knebel Doeberitz, M. The immune biology of microsatellite-unstable cancer. Trends Cancer 2016, 2, 121–133. [Google Scholar] [CrossRef]
- Bakry, D.; Aronson, M.; Durno, C.; Rimawi, H.; Farah, R.; Alharbi, Q.K.; Alharbi, M.; Shamvil, A.; Ben-Shachar, S.; Mistry, M.; et al. Genetic and clinical determinants of constitutional mismatch repair deficiency syndrome: Report from the constitutional mismatch repair deficiency consortium. Eur. J. Cancer 2014, 50, 987–996. [Google Scholar] [CrossRef]
- Wimmer, K.; Rosenbaum, T.; Messiaen, L. Connections between constitutional mismatch repair deficiency syndrome and neurofibromatosis type 1. Clin. Genet. 2016, 91, 507–519. [Google Scholar] [CrossRef]
- Maletzki, C.; Beyrich, F.; Hühns, M.; Klar, E.; Linnebacher, M. The mutational profile and infiltration pattern of murine MLH1−/− tumors: Concurrences, disparities and cell line establishment for functional analysis. Oncotarget 2016, 7, 53583–53598. [Google Scholar] [CrossRef]
- Daino, K.; Ishikawa, A.; Suga, T.; Amasaki, Y.; Kodama, Y.; Shang, Y.; Hirano-Sakairi, S.; Nishimura, M.; Nakata, A.; Yoshida, M.; et al. Mutational landscape of T-cell lymphoma in mice lacking the DNA mismatch repair gene Mlh1: No synergism with ionizing radiation. Carcinogenesis 2019, 40, 216–224. [Google Scholar] [CrossRef]
- Campbell, B.B.; Light, N.; Fabrizio, D.; Zatzman, M.; Fuligni, F.; de Borja, R.; Davidson, S.; Edwards, M.; Elvin, J.A.; Hodel, K.P.; et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell 2017, 171, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017, 171, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Maletzki, C.; Wiegele, L.; Nassar, I.; Stenzel, J.; Junghanss, C. Chemo-immunotherapy improves long-term survival in a preclinical model of MMR-D-related cancer. J. Immunother. Cancer 2019, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Ayhan, A.; Mao, T.-L.; Seckin, T.; Wu, C.-H.; Guan, B.; Ogawa, H.; Futagami, M.; Mizukami, H.; Yokoyama, Y.; Kurman, R.J.; et al. Loss of ARID1A expression is an early molecular event in tumor progression from ovarian endometriotic cyst to clear cell and endometrioid carcinoma. Int. J. Gynecol. Cancer 2012, 22, 1310–1315. [Google Scholar] [CrossRef]
- Fakhri, B.; Lim, K.H. Molecular landscape and sub-classification of gastrointestinal cancers: A review of literature. J. Gastrointest. Oncol. 2017, 8, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Zenonos, K.; Kyprianou, K. RAS signaling pathways, mutations and their role in colorectal cancer. World J. Gastrointest. Oncol. 2013, 5, 97–101. [Google Scholar] [CrossRef]
- Hayashi, T.; Desmeules, P.; Smith, R.S.; Drilon, A.; Somwar, R.; Ladanyi, M. RASA1 and NF1 are preferentially co-mutated and define a distinct genetic subset of smoking-associated non-small cell lung carcinomas sensitive to MEK inhibition. Clin. Cancer Res. 2018, 24, 1436–1447. [Google Scholar] [CrossRef] [PubMed]
- Sampson, E.M.; Haque, Z.K.; Ku, M.C.; Tevosian, S.G.; Albanese, C.; Pestell, R.G.; Paulson, K.E.; Yee, A.S. Negative regulation of the Wnt-β-catenin pathway by the transcriptional repressor HBP1. EMBO J. 2001, 20, 4500–4511. [Google Scholar] [CrossRef]
- Kim, J.-W.; Lee, H.S.; Nam, K.H.; Ahn, S.; Kim, J.W.; Ahn, S.-H.; Park, D.J.; Kim, H.-H.; Lee, K.-W. PIK3CA mutations are associated with increased tumor aggressiveness and Akt activation in gastric cancer. Oncotarget 2017, 8, 90948–90958. [Google Scholar] [CrossRef]
- Abubaker, J.; Bavi, P.P.; Al-Harbi, S.; Siraj, A.K.; Al-Dayel, F.; Uddin, S.; Al-Kuraya, K. PIK3CA mutations are mutually exclusive with PTEN loss in diffuse large B-cell lymphoma. Leukemia 2007, 21, 2368–2370. [Google Scholar] [CrossRef][Green Version]
- Wang, J.; Yu, P.; Lin, T.; Konigsberg, W.; Steitz, T. Crystal structures of an NH2-terminal fragment of T4 DNA polymerase and its complexes with single-stranded DNA and with divalent metal ions. Biochemistry 1996, 35, 8110–8119. [Google Scholar] [CrossRef] [PubMed]
- Yingling, J.M.; Das, P.; Savage, C.; Zhang, M.; Padgett, R.W.; Wang, X.F. Mammalian dwarfins are phosphorylated in response to transforming growth factor beta and are implicated in control of cell growth. Proc. Natl. Acad. Sci. USA 1996, 93, 8940–8944. [Google Scholar] [CrossRef] [PubMed]
- Slenter, D.N.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Mélius, J.; Cirillo, E.; Coort, S.L.; Digles, D.; et al. WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018, 46, D661–D667. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Yu, C.; Hong, H.; Zhang, S.; Zong, Y.; Ma, J.; Lu, A.; Sun, J.; Zheng, M. Identification of key genes and pathways involved in microsatellite instability in colorectal cancer. Mol. Med. Rep. 2019, 19, 2065–2076. [Google Scholar] [CrossRef] [PubMed]
- Moller, P.; Seppala, T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Evans, D.G.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.; et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: First report from the prospective Lynch syndrome database. Gut 2017, 66, 464–472. [Google Scholar] [CrossRef]
- Binder, H.; Hopp, L.; Schweiger, M.R.; Hoffmann, S.; Juhling, F.; Kerick, M.; Timmermann, B.; Siebert, S.; Grimm, C.; Nersisyan, L.; et al. Genomic and transcriptomic heterogeneity of colorectal tumours arising in Lynch syndrome. J. Pathol. 2017, 243, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Marshall, J.; Salem, M.; Grothey, A.; Goldberg, R.; Xiu, J.; Korn, W.; Shields, A.; Philip, P.; Lenz, H. O-025 Association between tumor mutation burden (TMB) and MLH1, PMS2, MSH2, and MSH6 alterations in 395 microsatellite instability-high (MSI-High) gastrointestinal (GI) tumors. Ann. Oncol. 2018, 29, mdy149. [Google Scholar] [CrossRef]
- Li, M.; Hruban, R.H.; Velculescu, V.E.; Bigner, D.; Goggins, M.; Kinzler, K.W.; Schmidt, M.K.; Zhang, X.; Iacobuzio-Donahue, C.A.; Maitra, A.; et al. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum. Mutat. 2011, 33, 100–103. [Google Scholar]
- Allo, G.; Bernardini, M.Q.; Wu, R.-C.; Shih, I.-M.; Kalloger, S.; Pollett, A.; Gilks, C.B.; Clarke, B.A. ARID1A loss correlates with mismatch repair deficiency and intact p53 expression in high-grade endometrial carcinomas. Mod. Pathol. 2014, 27, 255–261. [Google Scholar] [CrossRef]
- Hänninen, U.A.; Gylfe, A.E.; Renkonen-Sinisalo, L.; Böhm, J.; Taipale, M.; Järvinen, H.; Kondelin, J.; Katainen, R.; Mecklin, J.-P.; Tanskanen, T.; et al. Exome sequencing reveals frequent inactivating mutations in ARID1A, ARID1B, ARID2 and ARID4A in microsatellite unstable colorectal cancer. Int. J. Cancer 2014, 135, 611–623. [Google Scholar]
- Bosse, T.; ter Haar, N.T.; Seeber, L.M.; v Diest, P.J.; Hes, F.J.; Vasen, H.F.A.; Nout, R.A.; Creutzberg, C.L.; Morreau, H.; Smit, V.T. Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations, TP53 and microsatellite instability in endometrial cancer. Mod. Pathol. 2013, 26, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Caumanns, J.J.; Wisman, G.B.A.; Berns, K.; van der Zee, A.G.J.; de Jong, S. ARID1A mutant ovarian clear cell carcinoma: A clear target for synthetic lethal strategies. Biochim. Biophys. Acta-Rev. Cancer 2018, 1870, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2016, 348, 124–129. [Google Scholar] [CrossRef]
- Shlien, A.; Campbell, B.B.; De Borja, R.; Alexandrov, L.B.; Merico, D.; Wedge, D.; Van Loo, P.; Tarpey, P.S.; Coupland, P.; Behjati, S.; et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat. Genet. 2015, 47, 257–262. [Google Scholar] [CrossRef]
- Ripperger, T.; Schlegelberger, B. Acute lymphoblastic leukemia and lymphoma in the context of constitutional mismatch repair deficiency syndrome. Eur. J. Med. Genet. 2016, 59, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Cuceu, C.; Colicchio, B.; Jeandidier, E.; Junker, S.; Plassa, F.; Shim, G.; Mika, J.; Frenzel, M.; Jawhari, M.A.L.; Hempel, W.M.; et al. Independent mechanisms lead to genomic instability in Hodgkin lymphoma: Microsatellite or chromosomal instability. Cancers 2018, 10, 233. [Google Scholar] [CrossRef]
- Rigter, L.S.; Snaebjornsson, P.; Rosenberg, E.H.; Atmodimedjo, P.N.; Aleman, B.M.; Ten Hoeve, J.; Geurts-Giele, W.R.; van Ravesteyn, T.W.; Hoeksel, J.; Meijer, G.A.; et al. Double somatic mutations in mismatch repair genes are frequent in colorectal cancer after Hodgkin’s lymphoma treatment. Gut 2018, 67, 447–455. [Google Scholar] [CrossRef]
- Rossi, D.; Bruscaggin, A.; La Cava, P.; Galimberti, S.; Ciabatti, E.; Luminari, S.; Rigacci, L.; Tucci, A.; Pulsoni, A.; Bertoldero, G.; et al. The genotype of MLH1 identifies a subgroup of follicular lymphoma patients who do not benefit from doxorubicin: FIL-FOLL study. Haematologica 2015, 100, 517–524. [Google Scholar] [CrossRef]
- Jiang, H.; Lei, R.; Ding, S.W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M.; Zhou, S.; et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2; Springer: New York, NY, USA, 2009; ISBN 978-0-387-98140-6. [Google Scholar]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]
- Miller, C.A.; Qiao, Y.; DiSera, T.; D’Astous, B.; Marth, G.T. Bam.iobio: A web-based, real-time, sequence alignment file inspector. Nat. Methods 2014, 11, 1189. [Google Scholar] [CrossRef]
- Jay, J.J.; Brouwer, C. Lollipops in the Clinic: Information Dense Mutation Plots for Precision Medicine. PLoS ONE 2016, 11, e0160519. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MLH1−/− Number | Sex | Sample Type/Origin | Time of Onset [Weeks] |
|---|---|---|---|
| 328 | ♂ | GIT/duodenum | 29.5 |
| 7450 | ♀ | GIT/duodenum | 41.0 |
| 1351 | ♀ | Lymphoma/spleen | 17.4 |
| 1444 | ♀ | Lymphoma/skin | 36.0 |
| Gene | 7450-GIT | 328-GIT | Splenic Lymphoma | Skin Lymphoma | Survival | ||
|---|---|---|---|---|---|---|---|
| ARID1A | SILENT/MISSENSE | MISSENSE | MISSENSE | MISSENSE | OS | DFS | DSS |
| BCL2 | NONE | SILENT/MISSENSE | SILENT | SILENT/MISSENSE | |||
| PIK3CA | SILENT | SILENT/MISSENSE | SILENT/MISSENSE | SILENT/MISSENSE | DFS | ||
| SMAD4 | SILENT | SILENT | SILENT | SILENT/MISSENSE | OS | DSS | |
| Gene | Splenic Lymphoma | Skin Lymphoma | Survival | ||
|---|---|---|---|---|---|
| AKT2 | SILENT/MISSENSE | SILENT/MISSENSE | OS | DFS | DSS |
| CASP4 | NA | NA | |||
| CASP9 | MISSENSE | NA | |||
| CERS1 | NA | NA | DFS | ||
| ECHS1 | SILENT | NA | |||
| EGR1 | SILENT | SILENT | OS | DFS | DSS |
| INADL | NA | NA | |||
| MAPK1 | SILENT | SILENT | |||
| RAC1 | SILENT | SILENT | DSS | ||
| RHOA | SILENT | SILENT | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gladbach, Y.S.; Wiegele, L.; Hamed, M.; Merkenschläger, A.-M.; Fuellen, G.; Junghanss, C.; Maletzki, C. Unraveling the Heterogeneous Mutational Signature of Spontaneously Developing Tumors in MLH1−/− Mice. Cancers 2019, 11, 1485. https://doi.org/10.3390/cancers11101485
Gladbach YS, Wiegele L, Hamed M, Merkenschläger A-M, Fuellen G, Junghanss C, Maletzki C. Unraveling the Heterogeneous Mutational Signature of Spontaneously Developing Tumors in MLH1−/− Mice. Cancers. 2019; 11(10):1485. https://doi.org/10.3390/cancers11101485
Chicago/Turabian StyleGladbach, Yvonne Saara, Leonie Wiegele, Mohamed Hamed, Anna-Marie Merkenschläger, Georg Fuellen, Christian Junghanss, and Claudia Maletzki. 2019. "Unraveling the Heterogeneous Mutational Signature of Spontaneously Developing Tumors in MLH1−/− Mice" Cancers 11, no. 10: 1485. https://doi.org/10.3390/cancers11101485
APA StyleGladbach, Y. S., Wiegele, L., Hamed, M., Merkenschläger, A.-M., Fuellen, G., Junghanss, C., & Maletzki, C. (2019). Unraveling the Heterogeneous Mutational Signature of Spontaneously Developing Tumors in MLH1−/− Mice. Cancers, 11(10), 1485. https://doi.org/10.3390/cancers11101485