Replication Stress Response Links RAD52 to Protecting Common Fragile Sites

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
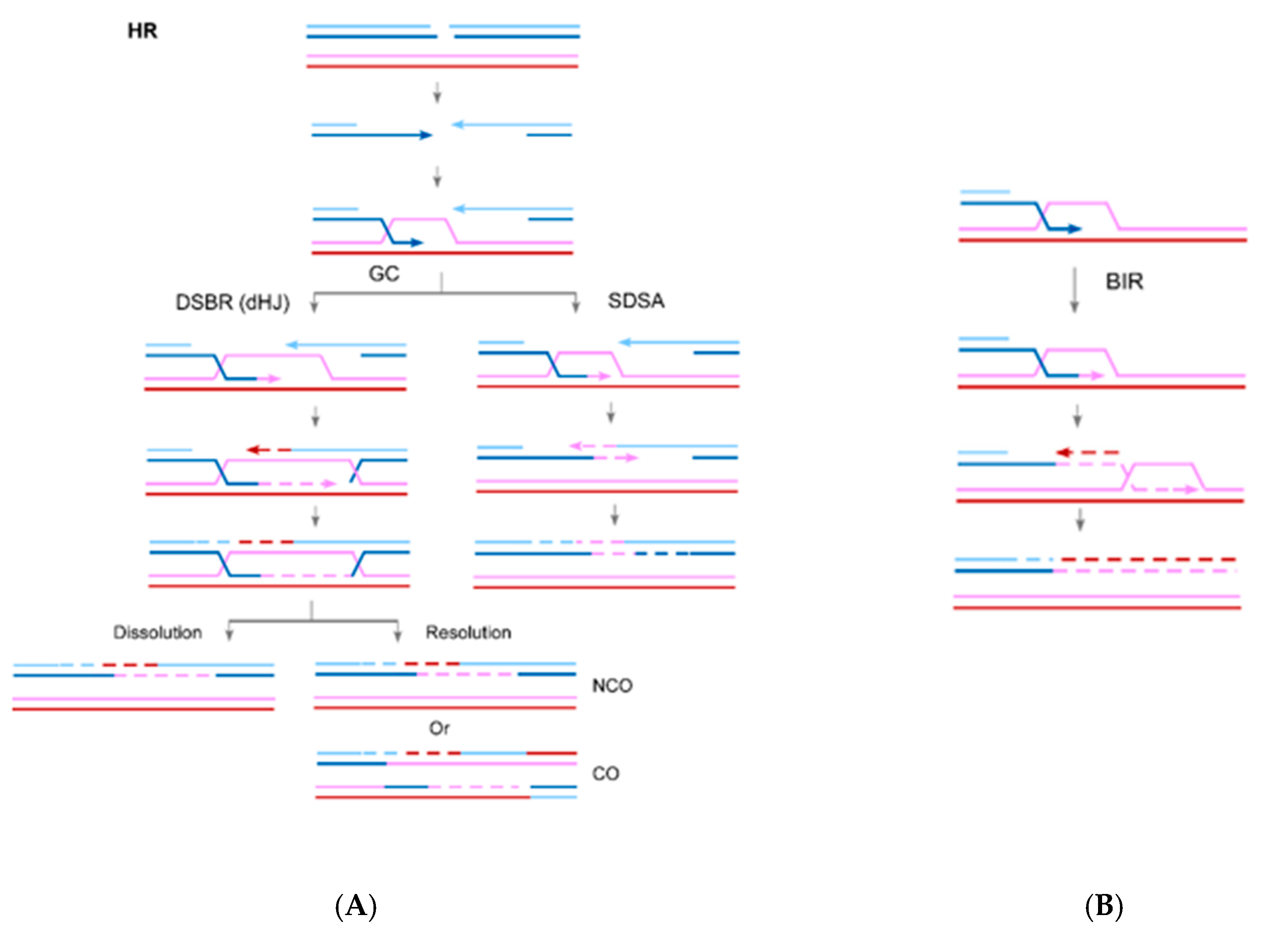
2. The Role of RAD52 in HR
3. The Role of RAD52 in Break-Induced Replication (BIR)
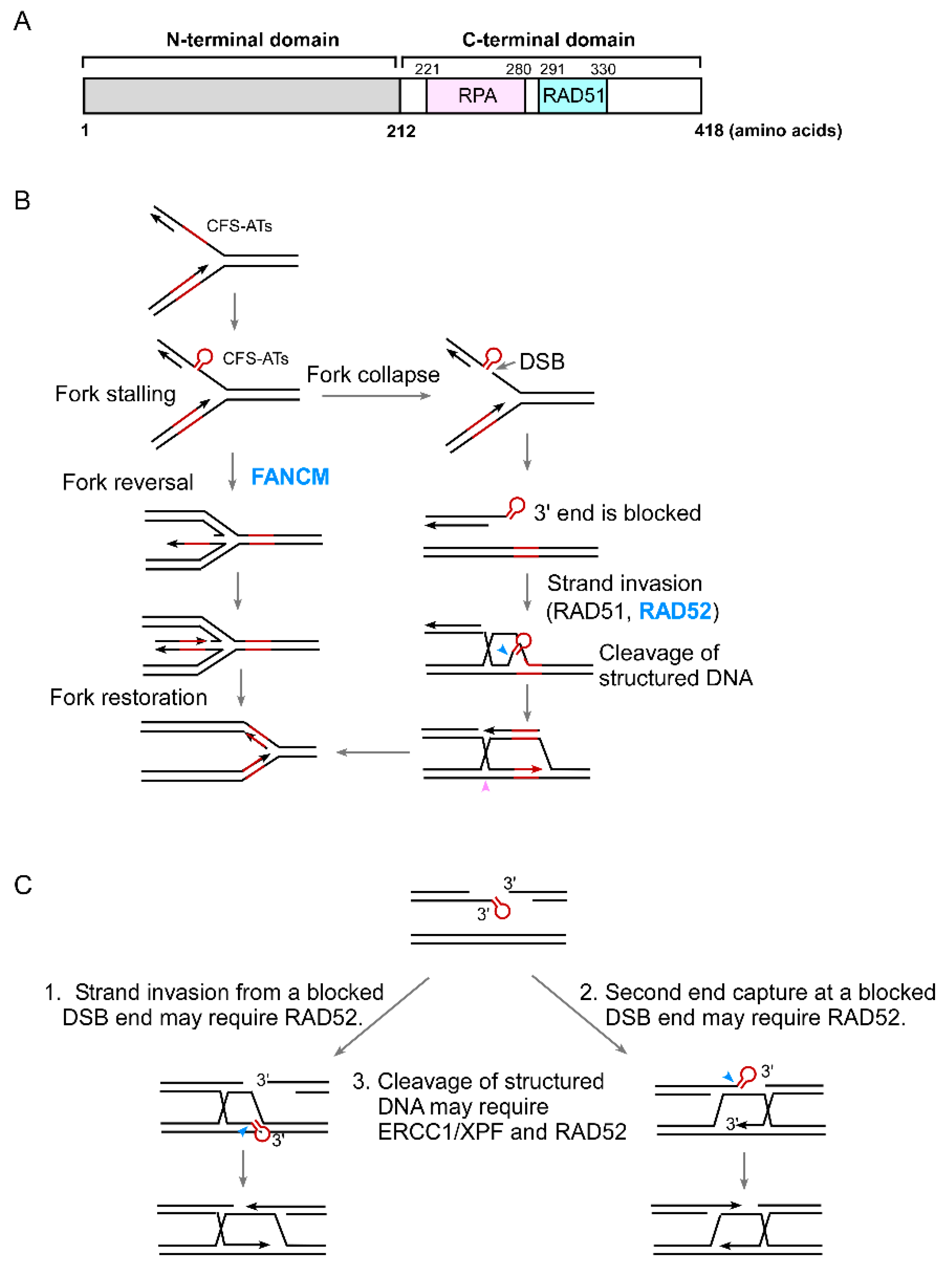
4. The Role of RAD52 in the Protection of Structure-Prone DNA Sequences
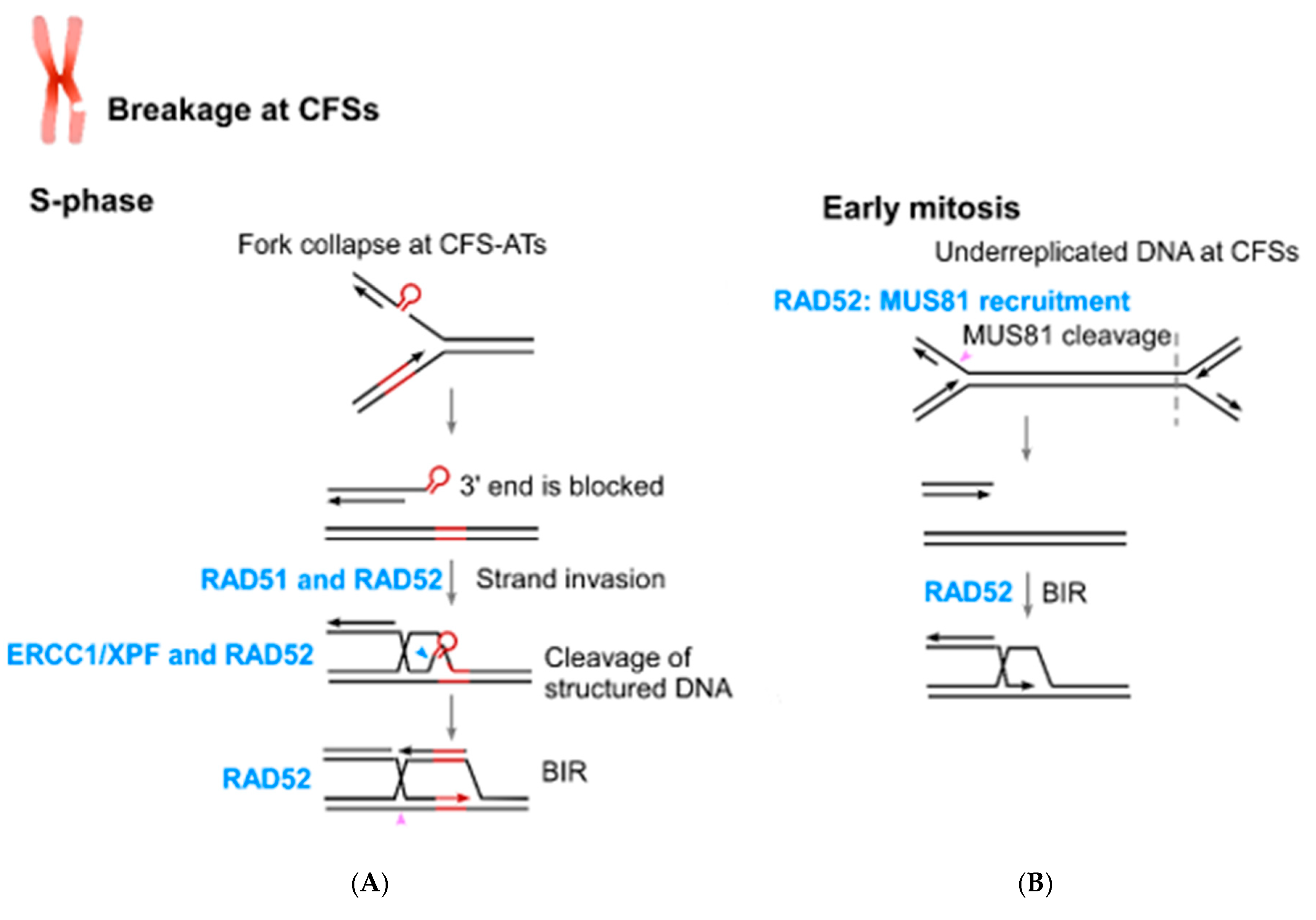
5. The Role of RAD52 in Coping with Replication Stress
6. The Connection of RAD52 with CFS Protection
7. RAD52 as a New Target for Cancer Therapy
8. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Gomez-Gonzalez, B. Genome instability: A mechanistic view of its causes and consequences. Nat. Rev. Genet. 2008, 9, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, E.M.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Jasin, M.; Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013, 5, a01274. [Google Scholar] [CrossRef]
- Paques, F.; Haber, J.E. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1999, 63, 349–404. [Google Scholar] [PubMed]
- Filippo, S.J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.R.; Lovett, S.T.; Haber, J.E. Break-induced DNA replication. Cold Spring Harb. Perspect. Biol. 2013, 5, a010397. [Google Scholar] [CrossRef] [PubMed]
- Sakofsky, J.C.; Malkova, A. Break induced replication in eukaryotes: Mechanisms, functions, and consequences. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 395–413. [Google Scholar] [CrossRef]
- Llorente, B.; Smith, C.E.; Symington, L.S. Break-induced replication: What is it and what is it for? Cell Cycle 2008, 7, 859–864. [Google Scholar] [CrossRef]
- Verma, P.; Greenberg, R.A. Noncanonical views of homology-directed DNA repair. Genes Dev. 2016, 30, 1138–1154. [Google Scholar] [CrossRef] [PubMed]
- Lydeard, J.R.; Jain, S.; Yamaguchi, M.; Haber, J.E. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 2007, 448, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Costantino, L.; Sotiriou, S.K.; Rantala, J.K.; Magin, S.; Mladenov, E.; Helleday, T.; Haber, J.E.; Iliakis, G.; Kallioniemi, O.P.; Halazonetis, T.D. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 2014, 343, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Malkova, A.; Ivanov, E.L.; Haber, J.E. Double-strand break repair in the absence of RAD51 in yeast: A possible role for break-induced DNA replication. Proc. Natl. Acad. Sci. USA 1996, 93, 7131–7136. [Google Scholar] [CrossRef] [PubMed]
- Malkova, A.; Naylor, M.L.; Yamaguchi, M.; Ira, G.; Haber, J.E. RAD51-dependent break-induced replication differs in kinetics and checkpoint responses from RAD51-mediated gene conversion. Mol. Cell. Biol. 2005, 25, 933–944. [Google Scholar] [CrossRef]
- Davis, A.P.; Symington, L.S. RAD51-dependent break-induced replication in yeast. Mol. Cell. Biol. 2004, 24, 2344–2351. [Google Scholar] [CrossRef] [PubMed]
- Game, J.C.; Mortimer, R.K. A genetic study of x-ray sensitive mutants in yeast. Mutat Res. 1974, 24, 281–292. [Google Scholar] [CrossRef]
- Symington, L.S. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol. Mol. Biol. Rev. 2002, 66, 630–670. [Google Scholar] [CrossRef]
- Rijkers, T.; Van, D.O.J.; Morolli, B.; Rolink, A.G.; Baarends, W.M.; Van Sloun, P.P.; Lohman, P.H.; Pastink, A. Targeted inactivation of mouse RAD52 reduces homologous recombination but not resistance to ionizing radiation. Mol. Cell Biol. 1998, 18, 6423–6429. [Google Scholar] [CrossRef]
- Lim, D.S.; Hasty, P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol. Cell Biol. 1996, 16, 7133–7143. [Google Scholar] [CrossRef]
- Tsuzuki, T.; Fujii, Y.; Sakumi, K.; Tominaga, Y.; Nakao, K.; Sekiguchi, M.; Matsushiro, A.; Yoshimura, Y.; Morita, T. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc. Natl. Acad. Sci. USA 1996, 93, 6236–6240. [Google Scholar] [CrossRef] [PubMed]
- Lok, B.H.; Carley, A.C.; Tchang, B.; Powell, S.N. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene 2013, 32, 3552–3558. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, S.K.; Kamileri, I.; Lugli, N.; Evangelou, K.; Da-Re, C.; Huber, F.; Padayachy, L.; Tardy, S.; Nicati, N.L.; Barriot, S.; et al. Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. Mol. Cell 2016, 64, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, S.; Oaks, J.; Ren, J.; Li, L.; Wu, X. The concerted roles of FANCM and Rad52 in the protection of common fragile sites. Nat. Commun. 2018, 9, 2791. [Google Scholar] [CrossRef] [PubMed]
- Adzuma, K.; Ogawa, T.; Ogawa, H. Primary structure of the RAD52 gene in Saccharomyces cerevisiae. Mol. Cell Biol. 1984, 4, 2735–2744. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Denison, K.; Lobb, R.; Gatewood, J.M.; Chen, D.J. The human and mouse homologs of the yeast RAD52 gene: cDNA cloning, sequence analysis, assignment to human chromosome 12p12.2-p13, and mRNA expression in mouse tissues. Genomics 1995, 25, 199–206. [Google Scholar] [CrossRef]
- Park, M.S.; Ludwig, D.L.; Stigger, E.; Lee, S.H. Physical interaction between human RAD52 and RPA is required for homologous recombination in mammalian cells. J. Biol. Chem. 1996, 271, 18996–19000. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Cloud, K.G.; Chen, D.J.; Park, M.S. Specific interactions between the human RAD51 and RAD52 proteins. J. Biol. Chem. 1996, 271, 148–152. [Google Scholar] [CrossRef]
- Mortensen, U.H.; Bendixen, C.; Sunjevaric, I.; Rothstein, R. DNA strand annealing is promoted by the yeast Rad52 protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10729–10734. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; New, J.H.; Kowalczykowski, S.C. DNA annealing by RAD52 protein is stimulated by specific interaction with the complex of replication protein A and single-stranded DNA. Proc. Natl. Acad. Sci. USA 1998, 95, 6049–6054. [Google Scholar] [CrossRef] [PubMed]
- New, J.H.; Sugiyama, T.; Zaitseva, E.; Kowalczykowski, S.C. Rad52 protein stimulates DNA strand exchange by Rad51 and replication protein A. Nature 1998, 391, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Sung, P. Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J. Biol. Chem. 1997, 272, 28194–28197. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, A.; Ogawa, T. Stimulation by Rad52 of yeast Rad51-mediated recombination. Nature 1998, 391, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.A.; Masson, J.Y.; McIlwraith, M.J.; Stasiak, A.Z.; Stasiak, A.; Venkitaraman, A.R.; West, S.C. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol. Cell 2001, 7, 273–282. [Google Scholar] [CrossRef]
- Chen, C.C.; Feng, W.; Lim, P.X.; Kass, E.M.; Jasin, M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu. Rev. Cancer Biol. 2018, 2, 313–336. [Google Scholar] [CrossRef]
- Reddy, G.; Golub, E.I.; Radding, C.M. Human Rad52 protein promotes single-strand DNA annealing followed by branch migration. Mutat. Res. 1997, 377, 53–59. [Google Scholar] [CrossRef]
- Stark, J.M.; Pierce, A.J.; Oh, J.; Pastink, A.; Jasin, M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol. Cell Biol. 2004, 24, 9305–9316. [Google Scholar] [CrossRef]
- Shinohara, A.; Shinohara, M.; Ohta, T.; Matsuda, S.; Ogawa, T. Rad52 forms ring structures and co-operates with RPA in single-strand DNA annealing. Genes Cells 1998, 3, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Lao, J.P.; Oh, S.D.; Shinohara, M.; Shinohara, A.; Hunter, N. Rad52 promotes postinvasion steps of meiotic double-strand-break repair. Mol. Cell 2008, 29, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Yonetani, Y.; Hochegger, H.; Sonoda, E.; Shinya, S.; Yoshikawa, H.; Takeda, S.; Yamazoe, M. Differential and collaborative actions of Rad51 paralog proteins in cellular response to DNA damage. Nucleic. Acids Res. 2005, 33, 4544–4552. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, A.; Tachiiri, S.; Sonoda, E.; Thompson, L.H.; Dhar, P.K.; Hiraoka, M.; Takeda, S.; Zhang, Y.; Reth, M.; Takata, M. Rad52 partially substitutes for the Rad51 paralog XRCC3 in maintaining chromosomal integrity in vertebrate cells. EMBO J. 2001, 20, 5513–5520. [Google Scholar] [CrossRef] [PubMed]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.W.; Shen, H.; Liu, Y.; Hickson, I.D. Replication stress activates DNA repair synthesis in mitosis. Nature 2015, 528, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Dilley, R.L.; Verma, P.; Cho, N.W.; Winters, H.D.; Wondisford, A.R.; Greenberg, R.A. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 2016, 539, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Roumelioti, F.M.; Sotiriou, S.K.; Katsini, V.; Chiourea, M.; Halazonetis, T.D.; Gagos, S. Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO Rep. 2016, 17, 1731–1737. [Google Scholar] [CrossRef]
- Min, J.; Wright, W.E.; Shay, J.W. Alternative lengthening of telomeres can be maintained by preferential elongation of lagging strands. Nucleic Acids Res. 2017, 45, 2615–2628. [Google Scholar] [CrossRef]
- Zhang, J.M.; Yadav, T.; Ouyang, J.; Lan, L.; Zou, L. Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Rep. 2019, 26, 955–968. [Google Scholar] [CrossRef]
- Lukusa, T.; Fryns, J.P. Human chromosome fragility. Biochim. Biophys. Acta 2008, 1779, 3–16. [Google Scholar] [CrossRef]
- Wang, H.; Li, Y.; Truong, L.N.; Shi, L.Z.; Hwang, P.Y.; He, J.; Do, J.; Cho, M.J.; Li, H.; Negrete, A.; et al. CtIP maintains stability at common fragile sites and inverted repeats by end resection-independent endonuclease activity. Mol. Cell 2014, 54, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Ozeri-Galai, E.; Lebofsky, R.; Rahat, A.; Bester, A.C.; Bensimon, A.; Kerem, B. Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol. Cell 2011, 43, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Freudenreich, C.H. An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol. Cell 2007, 27, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Mazina, O.M.; Keskin, H.; Hanamshet, K.; Storici, F.; Mazin, A.V. Rad52 Inverse Strand Exchange Drives RNA-Templated DNA Double-Strand Break Repair. Mol. Cell 2017, 67, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.J.; Kwon, Y.; Sung, P.; Greene, E.C. Human RAD52 interactions with replication protein A and the RAD51 presynaptic complex. J. Biol. Chem. 2017, 292, 11702–11713. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Heyer, W.D. Who’s who in human recombination: BRCA2 and RAD52. Proc. Natl. Acad. Sci. USA 2011, 108, 441–442. [Google Scholar] [CrossRef] [PubMed]
- Fishman-Lobell, J.; Haber, J.E. Removal of nonhomologous DNA ends in double-strand break recombination: The role of the yeast ultraviolet repair gene RAD1. Science 1992, 258, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Bardwell, A.J.; Bardwell, L.; Tomkinson, A.E.; Friedberg, E.C. Specific cleavage of model recombination and repair intermediates by the yeast Rad1-Rad10 DNA endonuclease. Science 1994, 265, 2082–2085. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, E.L.; Haber, J.E. RAD1 and RAD10, but not other excision repair genes, are required for double-strand break-induced recombination in Saccharomyces cerevisiae. Mol. Cell Biol. 1995, 15, 2245–2251. [Google Scholar] [CrossRef]
- Al-Minawi, A.Z.; Saleh-Gohari, N.; Helleday, T. The ERCC1/XPF endonuclease is required for efficient single-strand annealing and gene conversion in mammalian cells. Nucleic Acids Res. 2008, 36, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef] [PubMed]
- Adair, G.M.; Rolig, R.L.; Moore-Faver, D.; Zabelshansky, M.; Wilson, J.H.; Nairn, R.S. Role of ERCC1 in removal of long non-homologous tails during targeted homologous recombination. EMBO J. 2000, 19, 5552–5561. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lu, H.; Wang, Z.; Hu, Q.; Wang, H.; Xiang, R.; Chiba, T.; Wu, X. ERCC1/XPF Is Important for Repair of DNA Double-Strand Breaks Containing Secondary Structures. iScience 2019, 16, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Motycka, T.A.; Bessho, T.; Post, S.M.; Sung, P.; Tomkinson, A.E. Physical and functional interaction between the XPF/ERCC1 endonuclease and hRad52. J. Biol. Chem. 2004, 279, 13634–13639. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Sugawara, N.; Lydeard, J.; Vaze, M.; Tanguy, L.G.N.; Haber, J.E. A recombination execution checkpoint regulates the choice of homologous recombination pathway during DNA double-strand break repair. Genes Dev. 2009, 23, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Beach, A.; Haber, J.E. Homology Requirements and Competition between Gene Conversion and Break-Induced Replication during Double-Strand Break Repair. Mol. Cell 2017, 65, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Sridharan, S.; van Wietmarschen, N.; Maman, Y.; Callen, E.; Stanlie, A.; Wu, W.; Wu, X.; Day, A.; Wong, N.; et al. Dual Roles of Poly(dA:dT) Tracts in Replication Initiation and Fork Collapse. Cell 2018, 174, 1127–1142. [Google Scholar] [CrossRef]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef]
- Malacaria, E.; Pugliese, G.M.; Honda, M.; Marabitti, V.; Aiello, F.A.; Spies, M.; Franchitto, A.; Pichierri, P. Rad52 prevents excessive replication fork reversal and protects from nascent strand degradation. Nat. Commun. 2019, 10, 1412. [Google Scholar] [CrossRef]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Vassiliou, L.V.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Tsantoulis, P.K.; Kotsinas, A.; Sfikakis, P.P.; Evangelou, K.; Sideridou, M.; Levy, B.; Mo, L.; Kittas, C.; Wu, X.R.; Papavassiliou, A.G.; et al. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene 2008, 27, 3256–3264. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.N.; Opresko, P.L.; Meng, X.; Lee, M.Y.; Eckert, K.A. DNA structure and the Werner protein modulate human DNA polymerase delta-dependent replication dynamics within the common fragile site FRA16D. Nucleic Acids Res. 2010, 38, 1149–1162. [Google Scholar] [CrossRef] [PubMed]
- Zlotorynski, E.; Rahat, A.; Skaug, J.; Ben-Porat, N.; Ozeri, E.; Hershberg, R.; Levi, A.; Scherer, S.W.; Margalit, H.; Kerem, B. Molecular basis for expression of common and rare fragile sites. Mol. Cell Biol. 2003, 23, 7143–7151. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Lucas, I.; Young, D.J.; Davis, E.M.; Karrison, T.; Rest, J.S.; Le Beau, M.M. Common fragile sites are characterized by histone hypoacetylation. Hum. Mol. Genet. 2009, 18, 4501–4512. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sarni, D.; Kerem, B. The complex nature of fragile site plasticity and its importance in cancer. Curr. Opin. Cell Biol. 2016, 40, 131–136. [Google Scholar] [CrossRef]
- Naim, V.; Wilhelm, T.; Debatisse, M.; Rosselli, F. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 2013, 15, 1008–1015. [Google Scholar] [CrossRef]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre, M.; Nuciforo, P.G.; Bensimon, A. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Sola, D.; Ying, C.Y.; Grandori, C.; Ruggiero, L.; Chen, B.; Li, M.; Galloway, D.A.; Gu, W.; Gautier, J.; Dalla-Favera, R. Non-transcriptional control of DNA replication by c-Myc. Nature 2007, 448, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.; Fenster, B.E.; Ito, H.; Takeda, K.; Bae, N.S.; Hirai, T.; Yu, Z.X.; Ferrans, V.J.; Howard, B.H.; Finkel, T. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J. Biol. Chem. 1999, 274, 7936–7940. [Google Scholar] [CrossRef] [PubMed]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Neidhardt, G.; Hauke, J.; Ramser, J.; Gross, E.; Gehrig, A.; Muller, C.R.; Kahlert, A.K.; Hackmann, K.; Honisch, E.; Niederacher, D.; et al. Association Between Loss-of-Function Mutations Within the FANCM Gene and Early-Onset Familial Breast Cancer. JAMA Oncol. 2017, 3, 1245–1248. [Google Scholar] [CrossRef] [PubMed]
- Kiiski, J.I.; Tervasmaki, A.; Pelttari, L.M.; Khan, S.; Mantere, T.; Pylkas, K.; Mannermaa, A.; Tengstrom, M.; Kvist, A.; Borg, A. FANCM mutation c.5791C>T is a risk factor for triple-negative breast cancer in the Finnish population. Breast Cancer Res. Treat. 2017, 166, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Kiiski, J.I.; Pelttari, L.M.; Khan, S.; Freysteinsdottir, E.S.; Reynisdottir, I.; Hart, S.N.; Shimelis, H.; Vilske, S.; Kallioniemi, A.; Schleutker, J. Exome sequencing identifies FANCM as a susceptibility gene for triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 15172–15177. [Google Scholar] [CrossRef] [PubMed]
- Dicks, E.; Song, H.; Ramus, S.J.; Oudenhove, E.V.; Tyrer, J.P.; Intermaggio, M.P.; Kar, S.; Harrington, P.; Bowtell, D.D.; Group, A.S. Germline whole exome sequencing and large-scale replication identifies FANCM as a likely high grade serous ovarian cancer susceptibility gene. Oncotarget 2017, 8, 50930–50940. [Google Scholar] [CrossRef] [PubMed]
- Wreesmann, V.B.; Estilo, C.; Eisele, D.W.; Singh, B.; Wang, S.J. Downregulation of Fanconi anemia genes in sporadic head and neck squamous cell carcinoma. ORL J. Otorhinolaryngol. Relat. Spec. 2007, 69, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Cramer-Morales, K.; Nieborowska-Skorska, M.; Scheibner, K.; Padget, M.; Irvine, D.A.; Sliwinski, T.; Haas, K.; Lee, J.; Geng, H.; Roy, D.; et al. Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood 2013, 122, 1293–1304. [Google Scholar] [CrossRef]
- Chandramouly, G.; McDevitt, S.; Sullivan, K.; Kent, T.; Luz, A.; Glickman, J.F.; Andrake, M.; Skorski, T.; Pomerantz, R.T. Small-Molecule Disruption of RAD52 Rings as a Mechanism for Precision Medicine in BRCA-Deficient Cancers. Chem Biol. 2015, 22, 1491–1504. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Goyal, N.; Sullivan, K.; Hanamshet, K.; Patel, M.; Mazina, O.M.; Wang, C.X.; An, W.F.; Spoonamore, J.; Metkar, S.; et al. Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. 2016, 44, 4189–4199. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.; Cramer-Morales, K.; McElroy, D.L.; Ostrov, D.A.; Haas, K.; Childers, W.; Hromas, R.; Skorski, T. Identification of a Small Molecule Inhibitor of RAD52 by Structure-Based Selection. PLoS ONE 2016, 11, e0147230. [Google Scholar] [CrossRef] [PubMed]
- Hengel, S.R.; Malacaria, E.; Folly da Silva Constantino, L.; Bain, F.E.; Diaz, A.; Koch, B.G.; Yu, L.; Wu, M.; Pichierri, P.; Spies, M.A.; et al. Small-molecule inhibitors identify the RAD52-ssDNA interaction as critical for recovery from replication stress and for survival of BRCA2 deficient cells. Elife 2016, 5, e14740. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X. Replication Stress Response Links RAD52 to Protecting Common Fragile Sites. Cancers 2019, 11, 1467. https://doi.org/10.3390/cancers11101467
Wu X. Replication Stress Response Links RAD52 to Protecting Common Fragile Sites. Cancers. 2019; 11(10):1467. https://doi.org/10.3390/cancers11101467
Chicago/Turabian StyleWu, Xiaohua. 2019. "Replication Stress Response Links RAD52 to Protecting Common Fragile Sites" Cancers 11, no. 10: 1467. https://doi.org/10.3390/cancers11101467
APA StyleWu, X. (2019). Replication Stress Response Links RAD52 to Protecting Common Fragile Sites. Cancers, 11(10), 1467. https://doi.org/10.3390/cancers11101467