RAD52 as a Potential Target for Synthetic Lethality-Based Anticancer Therapies


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
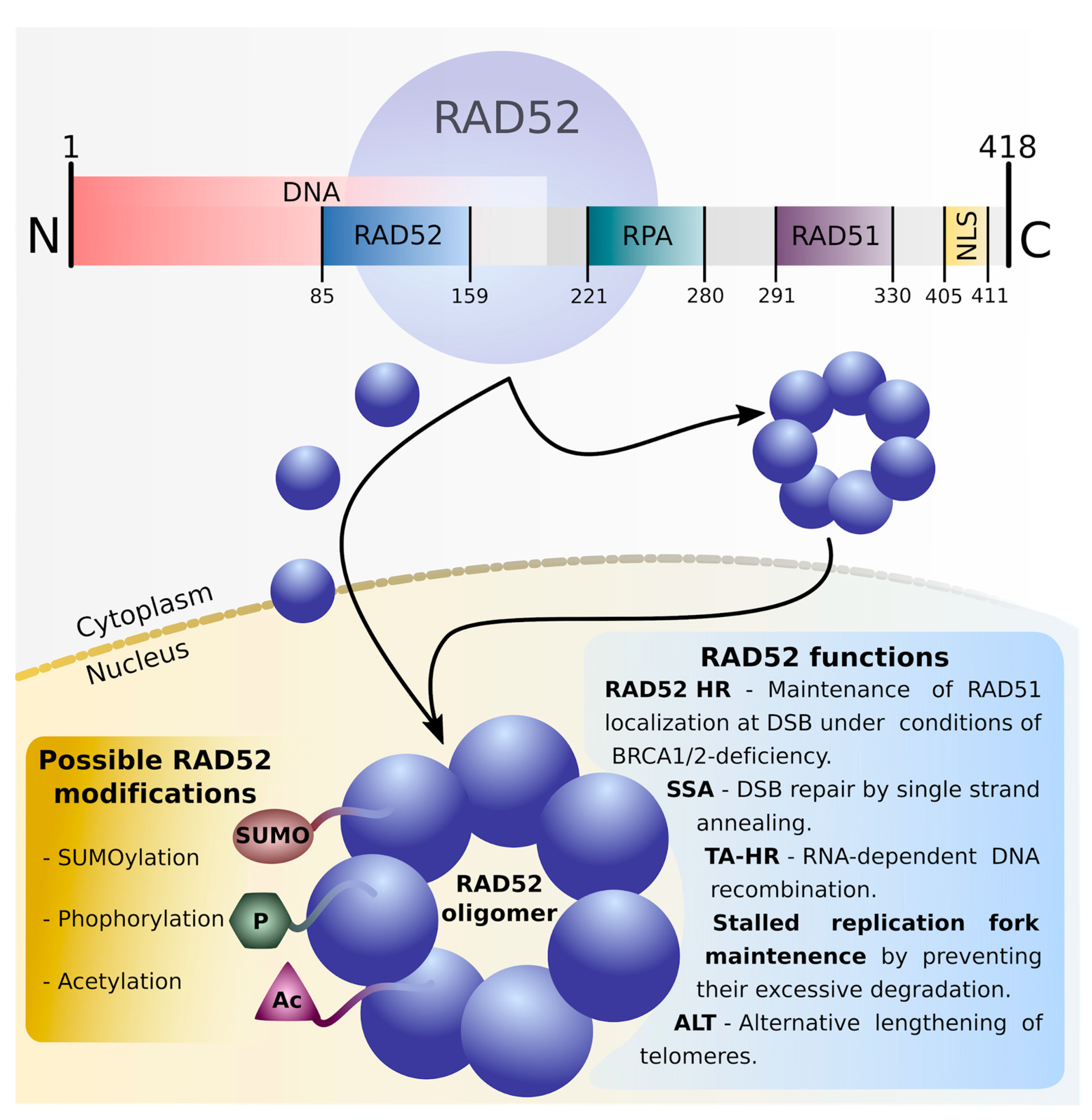
2. RAD52 Protein: Structure and Functional Modifications
3. Role of RAD52 in DNA Repair
3.1. Homologous Recombination (HR)
3.2. Single Strand Annealing (SSA)
3.3. RNA-Dependent DNA Recombination
4. Synthetic Lethality Targeting RAD52
4.1. F79
4.2. 6-OH-dopa
4.3. A5MP and AICAR/ZMP
4.4. D-I03
4.5. ’1’, ‘6’ and NP-004255
4.6. F779-0434
5. Dual Synthetic Lethality as an Aggressive Anticancer Strategy
5.1. Dual Cellular Synthetic Lethality
5.2. Dual Pathway Synthetic Lethality
5.3. Dual Molecular Synthetic Lethality
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALT | alternative lengthening of telomeres |
| APL | acute promyelocytic leukemia |
| B-NHEJ | backup non-homologous end-joining |
| BER | base excision repair |
| BIR | break-induced replication |
| BRCA1/2 HR | BRCA1/2-mediated homologous recombination |
| CML | chronic myeloid leukemia |
| CPC | cancer progenitor cell |
| D-NHEJ | DNA-PK-mediated non-homologous end-joining |
| DDR | DNA damage response |
| D-Loop | displacement loop |
| DSB | DNA double strand break |
| EOC | epithelial ovarian cancer |
| HR | homologous recombination |
| MMEJ | microhomology-mediated end-joining |
| NHEJ | non-homologous end-joining |
| PARP1i | poly(ADP-ribose) polymerase 1 inhibitor |
| RAD52 HR | RAD52-mediated homologous recombination |
| RAD52i | RAD52 inhibitor |
| SSA | single strand annealing |
| SSB | DNA single strand break |
| ssDNA | single stranded DNA |
| TA-HR | transcription-associated homologous recombination |
| TC-HR | transcription-coupled homologous recombination |
| TNBC | triple negative breast cancer |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Taylor, M.R.G.; Boulton, S.J. Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Karanam, K.; Kafri, R.; Loewer, A.; Lahav, G. Quantitative Live Cell Imaging Reveals a Gradual Shift between DNA Repair Mechanisms and a Maximal Use of HR in Mid S Phase. Mol. Cell 2012, 47, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G. The Concept of Synthetic Lethality in the Context of Anticancer Therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Hartwell, L.H. Integrating Genetic Approaches into the Discovery of Anticancer Drugs. Science 1997, 278, 1064–1068. [Google Scholar] [CrossRef]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Toma, M.; Skorski, T.; Sliwinski, T. DNA Double Strand Break Repair—Related Synthetic Lethality. CMC 2019, 26, 1446–1482. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. Mechanisms of Resistance to Therapies Targeting BRCA-Mutant Cancers. Nat. Med. 2013, 19, 1381–1388. [Google Scholar] [CrossRef]
- Clements, K.E.; Thakar, T.; Nicolae, C.M.; Liang, X.; Wang, H.-G.; Moldovan, G.-L. Loss of E2F7 Confers Resistance to Poly-ADP-Ribose Polymerase(PARP) Inhibitors in BRCA2-Deficient Cells. Nucleic Acids Res. 2018, 46, 8898–8907. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 Loss Rescues BRCA1 Deficiency and Is Associated with Triple-Negative and BRCA-Mutated Breast Cancers. Nat. Struc.t Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Tarsounas, M.; Davies, D.; West, S.C. BRCA2-Dependent and Independent Formation of RAD51 Nuclear Foci. Oncogene 2003, 22, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Cramer-Morales, K.; Nieborowska-Skorska, M.; Scheibner, K.; Padget, M.; Irvine, D.A.; Sliwinski, T.; Haas, K.; Lee, J.; Geng, H.; Roy, D.; et al. Personalized Synthetic Lethality Induced by Targeting RAD52 in Leukemias Identified by Gene Mutation and Expression Profile. Blood 2013, 122, 1293–1304. [Google Scholar] [CrossRef]
- Kumar, A.; Purohit, S.; Sharma, N.K. Aberrant DNA Double-Strand Break Repair Threads in Breast Carcinoma: Orchestrating Genomic Insult Survival. J. Cancer Prev. 2016, 21, 227–234. [Google Scholar] [CrossRef]
- Game, J.C.; Mortimer, R.K. A Genetic Study of X-Ray Sensitive Mutants in Yeast. Mutat. Res. 1974, 24, 281–292. [Google Scholar] [CrossRef]
- Stasiak, A.Z.; Larquet, E.; Stasiak, A.; Müller, S.; Engel, A.; Van Dyck, E.; West, S.C.; Egelman, E.H. The human Rad52 protein exists as a heptameric ring. Curr. Biol. 2000, 10, 337–340. [Google Scholar] [CrossRef]
- Kagawa, W.; Kagawa, A.; Saito, K.; Ikawa, S.; Shibata, T.; Kurumizaka, H.; Yokoyama, S. Identification of a Second DNA Binding Site in the Human Rad52 Protein. J. Biol. Chem. 2008, 283, 24264–24273. [Google Scholar] [CrossRef]
- Hanamshet, K.; Mazina, O.M.; Mazin, A.V. Reappearance from Obscurity: Mammalian Rad52 in Homologous Recombination. Genes 2016, 7, 63. [Google Scholar] [CrossRef]
- Plate, I.; Albertsen, L.; Lisby, M.; Hallwyl, S.C.L.; Feng, Q.; Rothstein, R.; Sung, P.; Mortensen, U.H. RAD52 Multimerization Is Important for Its Nuclear Localization In S. cerevisiae. DNA Repair 2009, 7, 57–66. [Google Scholar] [CrossRef][Green Version]
- Lee, M.; Lee, C.-H.; Demin, A.A.; Munashingha, P.R.; Amangyeld, T.; Kwon, B.; Formosa, T.; Seo, Y.-S. Rad52/Rad59-Dependent Recombination as a Means to Rectify Faulty Okazaki Fragment Processing. J. Biol. Chem. 2014, 289, 15064–15079. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Kagawa, W.; Ogi, T.; Kato, T.A.; Suzuki, T.; Dohmae, N.; Takizawa, K.; Nakazawa, Y.; Genet, M.D.; Saotome, M.; et al. Novel Function of HATs and HDACs in Homologous Recombination through Acetylation of Human RAD52 at Double-Strand Break Sites. Plos Genet 2018, 14, e1007277. [Google Scholar] [CrossRef] [PubMed]
- Altmannova, V.; Eckert-Boulet, N.; Arneric, M.; Kolesar, P.; Chaloupkova, R.; Damborsky, J.; Sung, P.; Zhao, X.; Lisby, M.; Krejci, L. Rad52 SUMOylation Affects the Efficiency of the DNA Repair. Nucleic Acids Res. 2010, 38, 4708–4721. [Google Scholar] [CrossRef] [PubMed]
- Sacher, M.; Pfander, B.; Hoege, C.; Jentsch, S. Control of Rad52 Recombination Activity by Double-Strand Break-Induced SUMO Modification. Nat. Cell Biol. 2006, 8, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Honda, M.; Okuno, Y.; Yoo, J.; Ha, T.; Spies, M. Tyrosine Phosphorylation Enhances RAD52-Mediated Annealing by Modulating Its DNA Binding: RAD52 Phosphorylation Upregulates SsDNA Annealing. Embo. J. 2011, 30, 3368–3382. [Google Scholar] [CrossRef]
- Fernandes, M.S.; Reddy, M.M.; Gonneville, J.R.; DeRoo, S.C.; Podar, K.; Griffin, J.D.; Weinstock, D.M.; Sattler, M. BCR-ABL Promotes the Frequency of Mutagenic Single-Strand Annealing DNA Repair. Blood 2009, 114, 1813–1819. [Google Scholar] [CrossRef]
- Cramer, K.; Nieborowska-Skorska, M.; Koptyra, M.; Slupianek, A.; Penserga, E.T.P.; Eaves, C.J.; Aulitzky, W.; Skorski, T. BCR/ABL and Other Kinases from Chronic Myeloproliferative Disorders Stimulate Single-Strand Annealing, an Unfaithful DNA Double-Strand Break Repair. Cancer Res. 2008, 68, 6884–6888. [Google Scholar] [CrossRef]
- Malacaria, E.; Pugliese, G.M.; Honda, M.; Marabitti, V.; Aiello, F.A.; Spies, M.; Franchitto, A.; Pichierri, P. Rad52 Prevents Excessive Replication Fork Reversal and Protects from Nascent Strand Degradation. Nat. Commun. 2019, 10, 1412. [Google Scholar] [CrossRef]
- Murfuni, I.; Basile, G.; Subramanyam, S.; Malacaria, E.; Bignami, M.; Spies, M.; Franchitto, A.; Pichierri, P. Survival of the Replication Checkpoint Deficient Cells Requires MUS81-RAD52 Function. Plos Genet 2013, 9, e1003910. [Google Scholar] [CrossRef]
- Liao, H.; Ji, F.; Helleday, T.; Ying, S. Mechanisms for Stalled Replication Fork Stabilization: New Targets for Synthetic Lethality Strategies in Cancer Treatments. Embo. Rep. 2018, 19. [Google Scholar] [CrossRef]
- Verma, P.; Dilley, R.L.; Zhang, T.; Gyparaki, M.T.; Li, Y.; Greenberg, R.A. RAD52 and SLX4 Act Nonepistatically to Ensure Telomere Stability during Alternative Telomere Lengthening. Genes Dev. 2019, 33, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Rijkers, T.; Van Den Ouweland, J.; Morolli, B.; Rolink, A.G.; Baarends, W.M.; Van Sloun, P.P.H.; Lohman, P.H.M.; Pastink, A. Targeted Inactivation of Mouse RAD52 Reduces Homologous Recombination but Not Resistance to Ionizing Radiation. Mol. Cell Biol. 1998, 18, 6423–6429. [Google Scholar] [CrossRef] [PubMed]
- Park, M.S. Expression of Human RAD52 Confers Resistance to Ionizing Radiation in Mammalian Cells. J. Biol. Chem. 1995, 270, 15467–15470. [Google Scholar] [CrossRef]
- Lok, B.H.; Carley, A.C.; Tchang, B.; Powell, S.N. RAD52 Inactivation Is Synthetically Lethal with Deficiencies in BRCA1 and PALB2 in Addition to BRCA2 through RAD51-Mediated Homologous Recombination. Oncogene 2013, 32, 3552–3558. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 Inactivation Is Synthetically Lethal with BRCA2 Deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef]
- Chun, J.; Buechelmaier, E.S.; Powell, S.N. Rad51 Paralog Complexes BCDX2 and CX3 Act at Different Stages in the BRCA1-BRCA2-Dependent Homologous Recombination Pathway. Mol. Cell Biol. 2013, 33, 387–395. [Google Scholar] [CrossRef]
- Ma, C.J.; Kwon, Y.; Sung, P.; Greene, E.C. Human RAD52 Interactions with Replication Protein A and the RAD51 Presynaptic Complex. J. Biol. Chem. 2017, 292, 11702–11713. [Google Scholar] [CrossRef]
- Gibb, B.; Ye, L.F.; Kwon, Y.; Niu, H.; Sung, P.; Greene, E.C. Protein Dynamics during Presynaptic-Complex Assembly on Individual Single-Stranded DNA Molecules. Nat. Struct. Mol. Biol. 2014, 21, 893–900. [Google Scholar] [CrossRef]
- Ivanov, E.L.; Sugawara, N.; Fishman-Lobell’, J.; Haber, J.E. Genetic Requirements for the Single-Strand Annealing Pathway of Double-Strand Break Repair in Saccharomyces Cerevisiae. Genetics 1996, 142, 693–704. [Google Scholar]
- Stark, J.M.; Pierce, A.J.; Oh, J.; Pastink, A.; Jasin, M. Genetic Steps of Mammalian Homologous Repair with Distinct Mutagenic Consequences. Mol. Cell Biol. 2004, 24, 9305–9316. [Google Scholar] [CrossRef]
- Ochs, F.; Somyajit, K.; Altmeyer, M.; Rask, M.-B.; Lukas, J.; Lukas, C. 53BP1 Fosters Fidelity of Homology-Directed DNA Repair. Nat. Struct. Mol. Biol. 2016, 23, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Díaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.F.; Tkáč, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Puget, N.; Shim, I.; Odate, S.; Jarzyna, I.; Bassing, C.H.; Alt, F.W.; Scully, R. Control of Sister Chromatid Recombination by Histone H2AX. Mol. Cell 2004, 16, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.C.; Laulier, C.; Gunn, A.; Cheng, A.; Robbiani, D.F.; Nussenzweig, A.; Stark, J.M. Ring Finger Nuclear Factor RNF168 Is Important for Defects in Homologous Recombination Caused by Loss of the Breast Cancer Susceptibility Factor BRCA1. J. Biol. Chem. 2012, 287, 40618–40628. [Google Scholar] [CrossRef]
- Rothenberg, E.; Grimme, J.M.; Spies, M.; Ha, T. Human Rad52-Mediated Homology Search and Annealing Occurs by Continuous Interactions between Overlapping Nucleoprotein Complexes. Proc. Natl. Acad. Sci. USA 2008, 105, 20274–20279. [Google Scholar] [CrossRef]
- Sallmyr, A.; Tomkinson, A.E. Repair of DNA Double-Strand Breaks by Mammalian Alternative End-Joining Pathways. J. Biol. Chem. 2018, 293, 10536–10546. [Google Scholar] [CrossRef]
- Wei, L.; Levine, A.S.; Lan, L. Transcription-Coupled Homologous Recombination after Oxidative Damage. DNA Repair 2016, 44, 76–80. [Google Scholar] [CrossRef]
- Vaughn, J.P.; Cirisano, F.D.; Huper, G.; Berchuck, A.; Futreal, P.A.; Marks, J.R.; Igleha, J.D. Cell Cycle Control of BRCA2. Cancer Res. 1996, 56, 4590–4594. [Google Scholar]
- Misra, S.; Sharma, S.; Agarwal, A.; Khedkar, S.V.; Tripathi, M.K.; Mittal, M.K.; Chaudhuri, G. Cell cycle-dependent regulation of the bi-directional overlapping promoter of human BRCA2/ZAR2 genes in breast cancer cells. Mol. Cancer 2010, 9, 50. [Google Scholar] [CrossRef]
- Welty, S.; Teng, Y.; Liang, Z.; Zhao, W.; Sanders, L.H.; Greenamyre, J.T.; Rubio, M.E.; Thathiah, A.; Kodali, R.; Wetzel, R.; et al. RAD52 Is Required for RNA-Templated Recombination Repair in Post-Mitotic Neurons. J. Biol. Chem. 2018, 293, 1353–1362. [Google Scholar] [CrossRef]
- Wei, L.; Nakajima, S.; Böhm, S.; Bernstein, K.A.; Shen, Z.; Tsang, M.; Levine, A.S.; Lan, L. DNA Damage during the G0/G1 Phase Triggers RNA-Templated, Cockayne Syndrome B-Dependent Homologous Recombination. Proc. Natl. Acad. Sci. USA 2015, 112, E3495–E3504. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, T.; Kato, R.; Hagiwara, Y.; Shiotani, B.; Yamauchi, M.; Nakada, S.; Shibata, A.; Miyagawa, K. Human Rad52 Promotes XPG-Mediated R-Loop Processing to Initiate Transcription-Associated Homologous Recombination Repair. Cell 2018, 175, 558–570.e11. [Google Scholar] [CrossRef] [PubMed]
- McDevitt, S.; Rusanov, T.; Kent, T.; Chandramouly, G.; Pomerantz, R.T. How RNA Transcripts Coordinate DNA Recombination and Repair. Nat. Commun. 2018, 9, 1091. [Google Scholar] [CrossRef] [PubMed]
- Keskin, H.; Shen, Y.; Huang, F.; Patel, M.; Yang, T.; Ashley, K.; Mazin, A.V.; Storici, F. Transcript-RNA-Templated DNA Recombination and Repair. Nature 2014, 515, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Stoppa-Lyonnet, D. The Biological Effects and Clinical Implications of BRCA Mutations: Where Do We Go from Here? Eur. J. Hum. Genet. 2016, 24, S3–S9. [Google Scholar] [CrossRef]
- Liu, J.; Doty, T.; Gibson, B.; Heyer, W.-D. Human BRCA2 Protein Promotes RAD51 Filament Formation on RPA-Covered Single-Stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1260–1262. [Google Scholar] [CrossRef]
- Yamaguchi-Iwai, Y.; Sonoda, E.; Buerstedde, J.-M.; Bezzubova, O.; Morrison, C.; Takata, M.; Shinohara, A.; Takeda, S. Homologous Recombination, but Not DNA Repair, Is Reduced in Vertebrate Cells Deficient in RAD52. Mol. Cell Biol. 1998, 18, 6430–6435. [Google Scholar] [CrossRef]
- Hromas, R.; Kim, H.-S.; Sidhu, G.; Williamson, E.; Jaiswal, A.; Totterdale, T.A.; Nole, J.; Lee, S.-H.; Nickoloff, J.A.; Kong, K.Y. The Endonuclease EEPD1 Mediates Synthetic Lethality in RAD52-Depleted BRCA1 Mutant Breast Cancer Cells. Breast Cancer Res. 2017, 19, 122. [Google Scholar] [CrossRef]
- Kim, H.-S.; Nickoloff, J.A.; Wu, Y.; Williamson, E.A.; Sidhu, G.S.; Reinert, B.L.; Jaiswal, A.S.; Srinivasan, G.; Patel, B.; Kong, K.; et al. Endonuclease EEPD1 Is a Gatekeeper for Repair of Stressed Replication Forks. J. Biol. Chem. 2017, 292, 2795–2804. [Google Scholar] [CrossRef]
- Bass, K.L.; Murray, J.M.; O’Connell, M.J. Brc1-Dependent Recovery from Replication Stress. J. Cell Sci. 2012, 125, 2753–2764. [Google Scholar] [CrossRef]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Podszywalow-Bartnicka, P.; Wolczyk, M.; Kusio-Kobialka, M.; Wolanin, K.; Skowronek, K.; Nieborowska-Skorska, M.; Dasgupta, Y.; Skorski, T.; Piwocka, K. Downregulation of BRCA1 Protein in BCR-ABL1 Leukemia Cells Depends on Stress-Triggered TIAR-Mediated Suppression of Translation. Cell Cycle 2014, 13, 3727–3741. [Google Scholar] [CrossRef] [PubMed]
- Chandramouly, G.; McDevitt, S.; Sullivan, K.; Kent, T.; Luz, A.; Glickman, J.F.; Andrake, M.; Skorski, T.; Pomerantz, R.T. Small-Molecule Disruption of RAD52 Rings as a Mechanism for Precision Medicine in BRCA-Deficient Cancers. Chem. Biol. 2015, 22, 1491–1504. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W.; Zorumski, C.F.; Stewart, G.R.; Price, M.T.; Wang, G.; Labruyere, J. Excitotoxicity of L-DOPA and 6-OH-DOPA: Implications for Parkinson’s and Huntington’s Diseases. Exp. Neurol. 1990, 108, 269–272. [Google Scholar] [CrossRef]
- Sullivan, K.; Cramer-Morales, K.; McElroy, D.L.; Ostrov, D.A.; Haas, K.; Childers, W.; Hromas, R.; Skorski, T. Identification of a Small Molecule Inhibitor of RAD52 by Structure-Based Selection. Plos ONE 2016, 11, e0147230. [Google Scholar] [CrossRef]
- Corton, J.M.; Gillespie, J.G.; Hawley, S.A.; Hardie, D.G. 5-Aminoimidazole-4-Carboxamide Ribonucleoside. A Specific Method for Activating AMP-Activated Protein Kinase in Intact Cells? Eur. J. Biochem. 1995, 229, 558–565. [Google Scholar] [CrossRef]
- Huang, F.; Goyal, N.; Sullivan, K.; Hanamshet, K.; Patel, M.; Mazina, O.M.; Wang, C.X.; An, W.F.; Spoonamore, J.; Metkar, S.; et al. Targeting BRCA1- and BRCA2-Deficient Cells with RAD52 Small Molecule Inhibitors. Nucleic. Acids. Res. 2016, 44, 4189–4199. [Google Scholar] [CrossRef]
- Hengel, S.R.; Malacaria, E.; Folly da Silva Constantino, L.; Bain, F.E.; Diaz, A.; Koch, B.G.; Yu, L.; Wu, M.; Pichierri, P.; Spies, M.A.; et al. Small-Molecule Inhibitors Identify the RAD52-SsDNA Interaction as Critical for Recovery from Replication Stress and for Survival of BRCA2 Deficient Cells. eLife 2016, 5, e14740. [Google Scholar] [CrossRef]
- Li, J.; Yang, Q.; Zhang, Y.; Huang, K.; Sun, R.; Zhao, Q. Compound F779-0434 Causes Synthetic Lethality in BRCA2-Deficient Cancer Cells by Disrupting RAD52–SsDNA Association. Rsc Adv. 2018, 8, 18859–18869. [Google Scholar] [CrossRef]
- Nieborowska-Skorska, M.; Sullivan, K.; Dasgupta, Y.; Podszywalow-Bartnicka, P.; Hoser, G.; Maifrede, S.; Martinez, E.; Di Marcantonio, D.; Bolton-Gillespie, E.; Cramer-Morales, K.; et al. Gene Expression and Mutation-Guided Synthetic Lethality Eradicates Proliferating and Quiescent Leukemia Cells. J. Clin. Investig. 2017, 127, 2392–2406. [Google Scholar] [CrossRef]
- Sullivan-Reed, K.; Bolton-Gillespie, E.; Dasgupta, Y.; Langer, S.; Siciliano, M.; Nieborowska-Skorska, M.; Hanamshet, K.; Belyaeva, E.A.; Bernhardy, A.J.; Lee, J.; et al. Simultaneous Targeting of PARP1 and RAD52 Triggers Dual Synthetic Lethality in BRCA-Deficient Tumor Cells. Cell Rep. 2018, 23, 3127–3136. [Google Scholar] [CrossRef] [PubMed]
- Nieborowska-Skorska, M.; Maifrede, S.; Ye, M.; Toma, M.; Hewlett, E.; Gordon, J.; Le, B.V.; Sliwinski, T.; Zhao, H.; Piwocka, K.; et al. Non-NAD-like PARP1 Inhibitor Enhanced Synthetic Lethal Effect of NAD-like PARP Inhibitors against BRCA1-Deficient Leukemia. Leuk. Lymphoma 2019, 60, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Gasch, C.; Ffrench, B.; O’Leary, J.J.; Gallagher, M.F. Catching Moving Targets: Cancer Stem Cell Hierarchies, Therapy-Resistance & Considerations for Clinical Intervention. Mol. Cancer 2017, 16, 43. [Google Scholar] [CrossRef] [PubMed]
- Black, S.J.; Kashkina, E.; Kent, T.; Pomerantz, R.T. DNA Polymerase θ: A Unique Multifunctional End-Joining Machine. Genes 2016, 7, 67. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.R.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-Recombination-Deficient Tumours Are Dependent on Polθ-Mediated Repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef]
- Kawamura, K.; Bahar, R.; Seimiya, M.; Chiyo, M.; Wada, A.; Okada, S.; Hatano, M.; Tokuhisa, T.; Kimura, H.; Watanabe, S.; et al. DNA Polymeraseθ is Preferentially Expressed in Lymphoid Tissues and Upregulated in Human Cancers. Int. J. Cancer 2004, 109, 9–16. [Google Scholar] [CrossRef]
- Higgins, G.S.; Harris, A.L.; Prevo, R.; Helleday, T.; McKenna, W.G.; Buffa, F.M. Overexpression of POLQ Confers a Poor Prognosis in Early Breast Cancer Patients. Oncotarget 2010, 1. [Google Scholar] [CrossRef]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian Polymerase θ Promotes Alternative NHEJ and Suppresses Recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef]
- Murai, J.; Shar-yin, N.H.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Wahlberg, E.; Karlberg, T.; Kouznetsova, E.; Markova, N.; Macchiarulo, A.; Thorsell, A.-G.; Pol, E.; Frostell, Å.; Ekblad, T.; Öncü, D.; et al. Family-Wide Chemical Profiling and Structural Analysis of PARP and Tankyrase Inhibitors. Nat. Biotechnol. 2012, 30, 283–288. [Google Scholar] [CrossRef]
- Thomas, C.; Ji, Y.; Lodhi, N.; Kotova, E.; Pinnola, A.D.; Golovine, K.; Makhov, P.; Pechenkina, K.; Kolenko, V.; Tulin, A.V. Non-NAD-Like Poly(ADP-Ribose) Polymerase-1 Inhibitors Effectively Eliminate Cancer in Vivo. EBioMedicine 2016, 13, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Pinnola, A.; Naumova, N.; Shah, M.; Tulin, A.V. Nucleosomal Core Histones Mediate Dynamic Regulation of Poly(ADP-Ribose) Polymerase 1 Protein Binding to Chromatin and Induction of Its Enzymatic Activity. J. Biol. Chem. 2007, 282, 32511–32519. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toma, M.; Sullivan-Reed, K.; Śliwiński, T.; Skorski, T. RAD52 as a Potential Target for Synthetic Lethality-Based Anticancer Therapies. Cancers 2019, 11, 1561. https://doi.org/10.3390/cancers11101561
Toma M, Sullivan-Reed K, Śliwiński T, Skorski T. RAD52 as a Potential Target for Synthetic Lethality-Based Anticancer Therapies. Cancers. 2019; 11(10):1561. https://doi.org/10.3390/cancers11101561
Chicago/Turabian StyleToma, Monika, Katherine Sullivan-Reed, Tomasz Śliwiński, and Tomasz Skorski. 2019. "RAD52 as a Potential Target for Synthetic Lethality-Based Anticancer Therapies" Cancers 11, no. 10: 1561. https://doi.org/10.3390/cancers11101561
APA StyleToma, M., Sullivan-Reed, K., Śliwiński, T., & Skorski, T. (2019). RAD52 as a Potential Target for Synthetic Lethality-Based Anticancer Therapies. Cancers, 11(10), 1561. https://doi.org/10.3390/cancers11101561