ErbB3 Phosphorylation as Central Event in Adaptive Resistance to Targeted Therapy in Metastatic Melanoma: Early Detection in CTCs during Therapy and Insights into Regulation by Autocrine Neuregulin

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
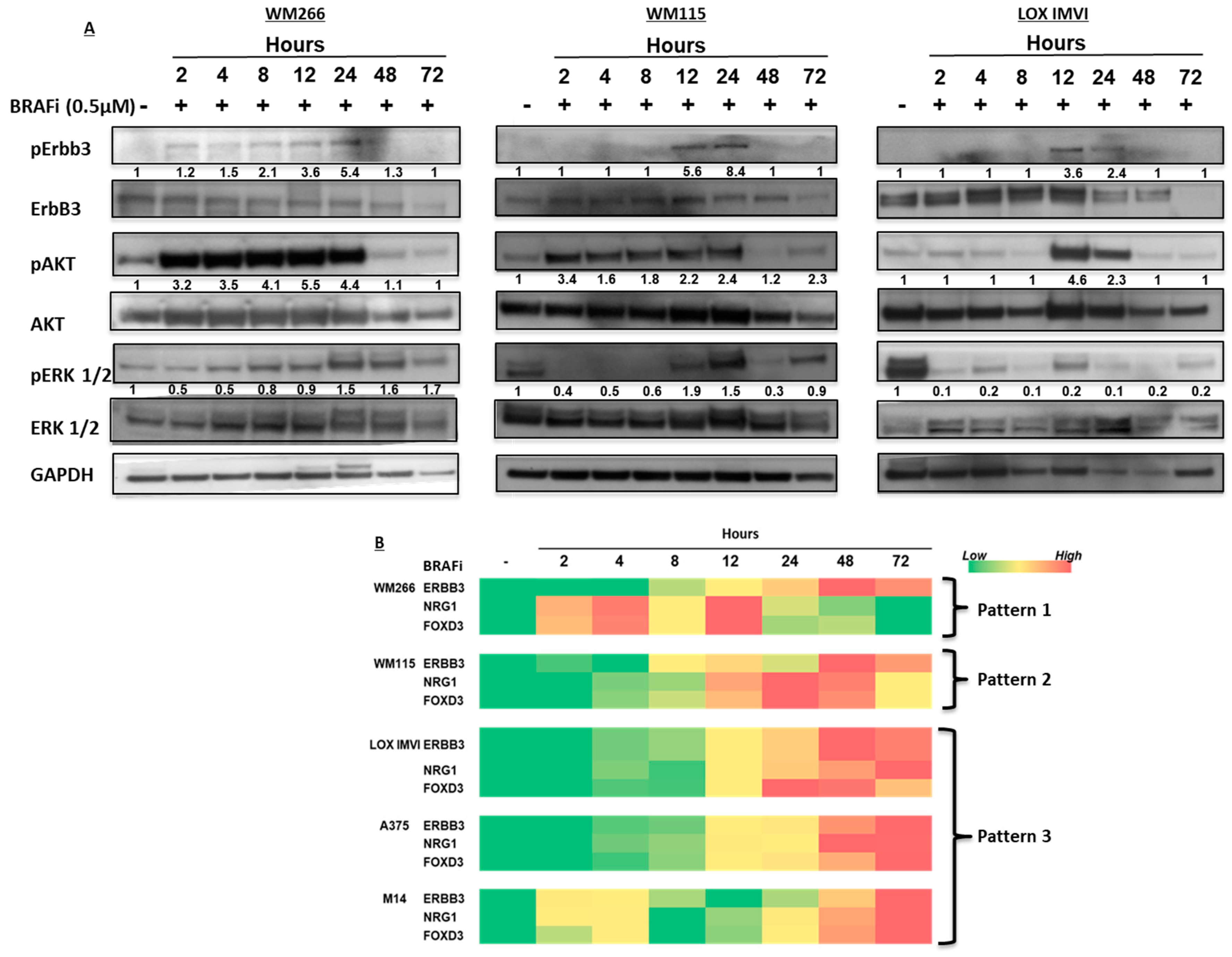
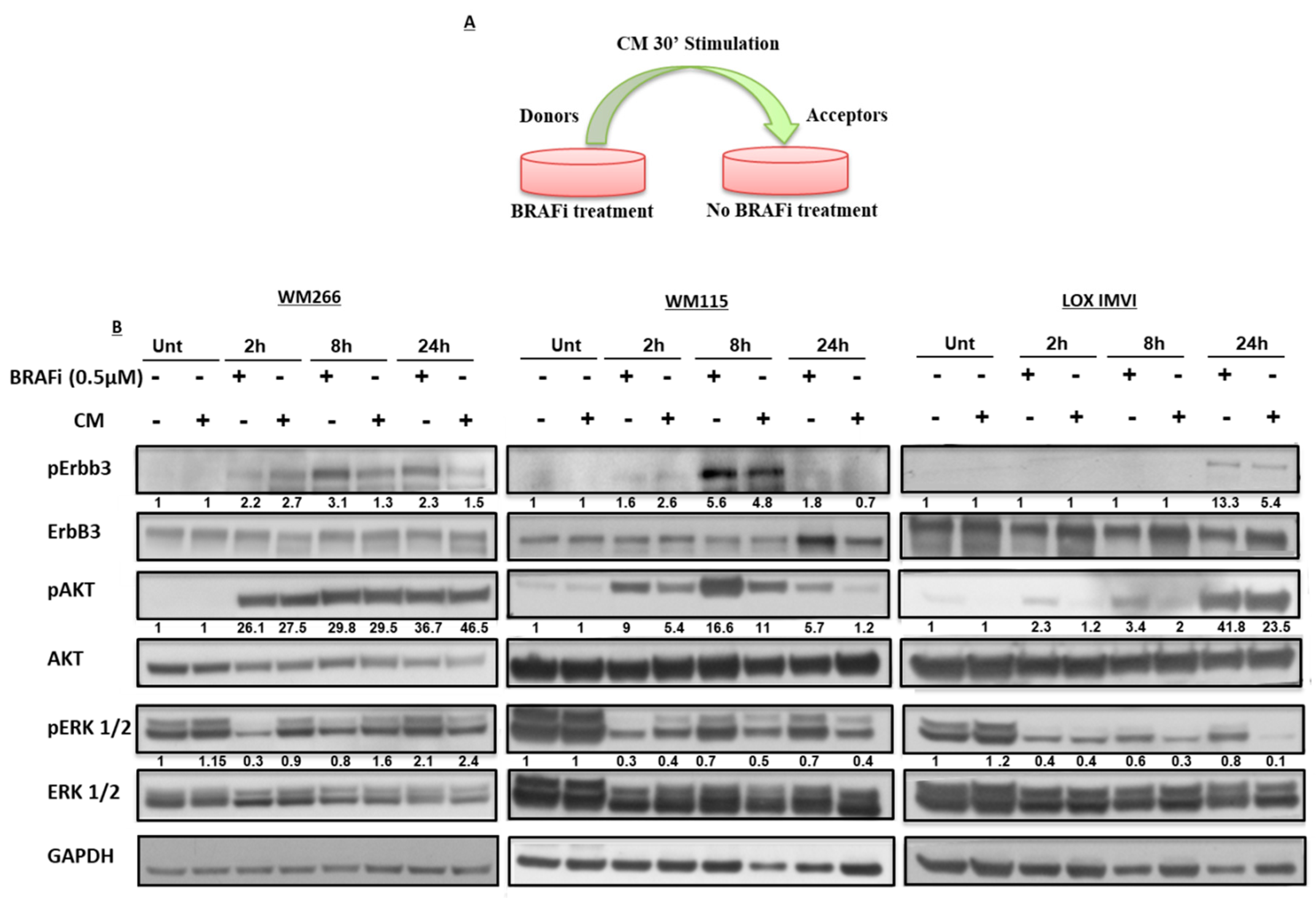
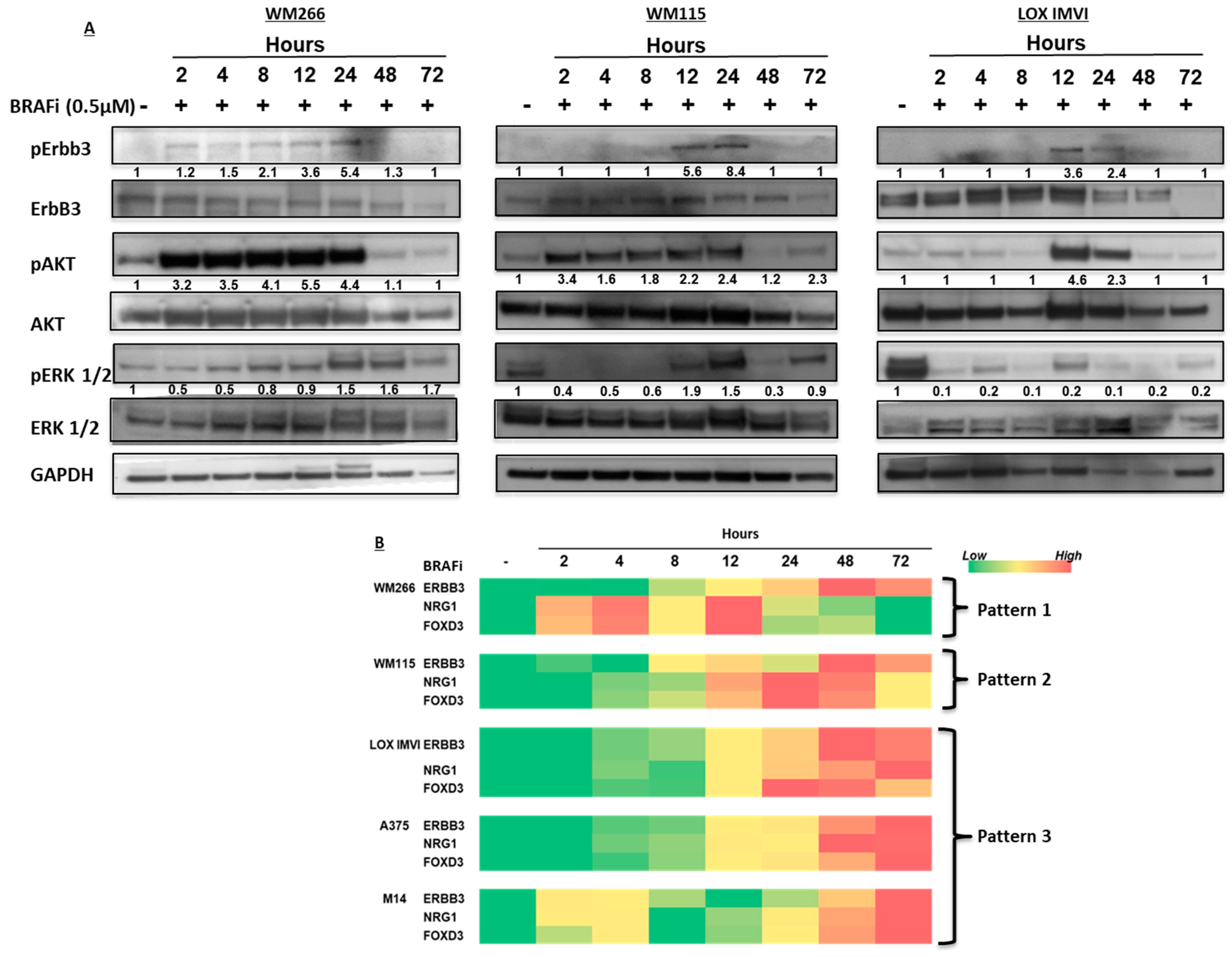
2.1. BRAF-Mutated Melanoma Cell Lines Show Different Activation Kinetics of the pErbB3/pAKT Axis After Exposure to BRAF Inhibitors
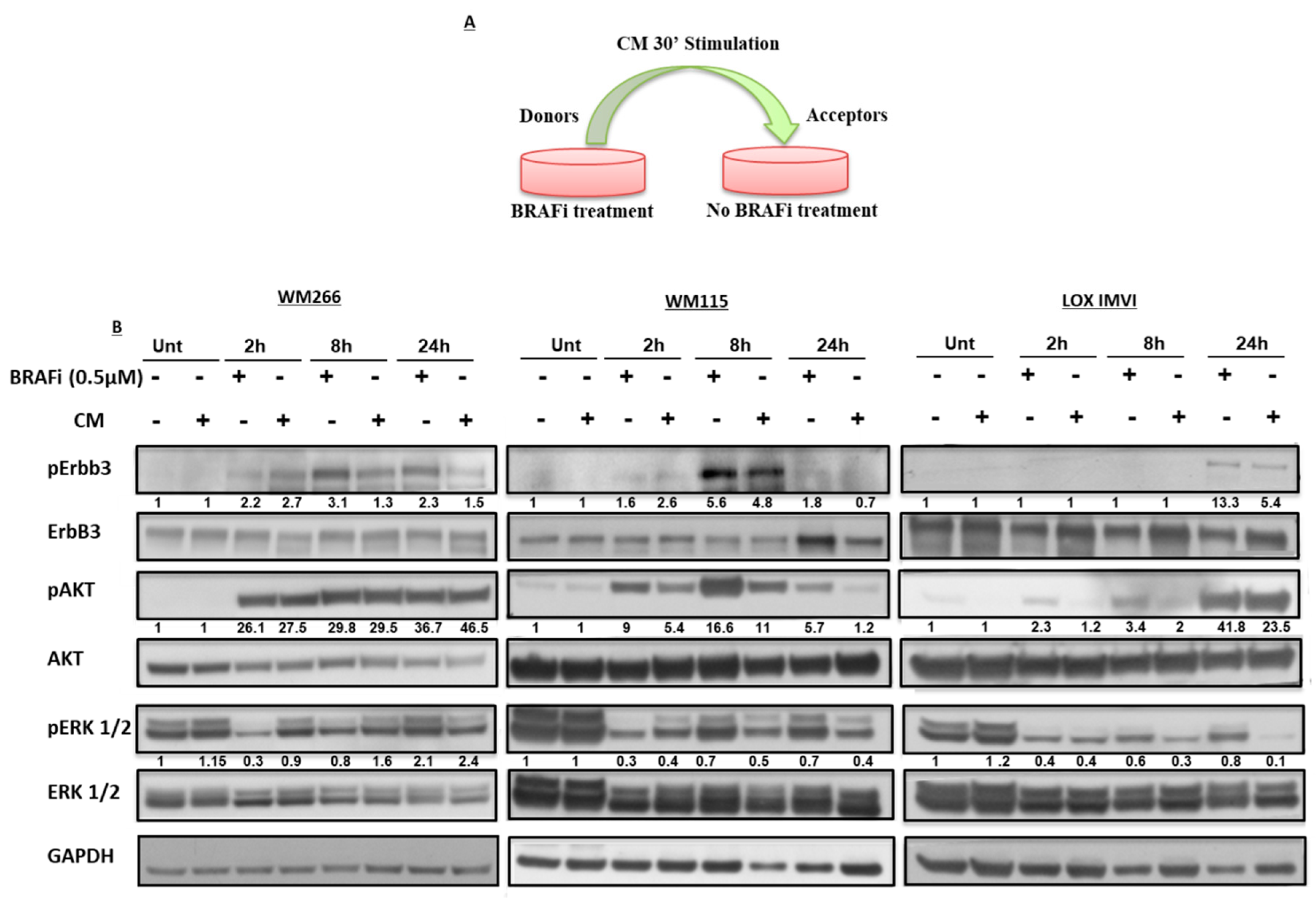
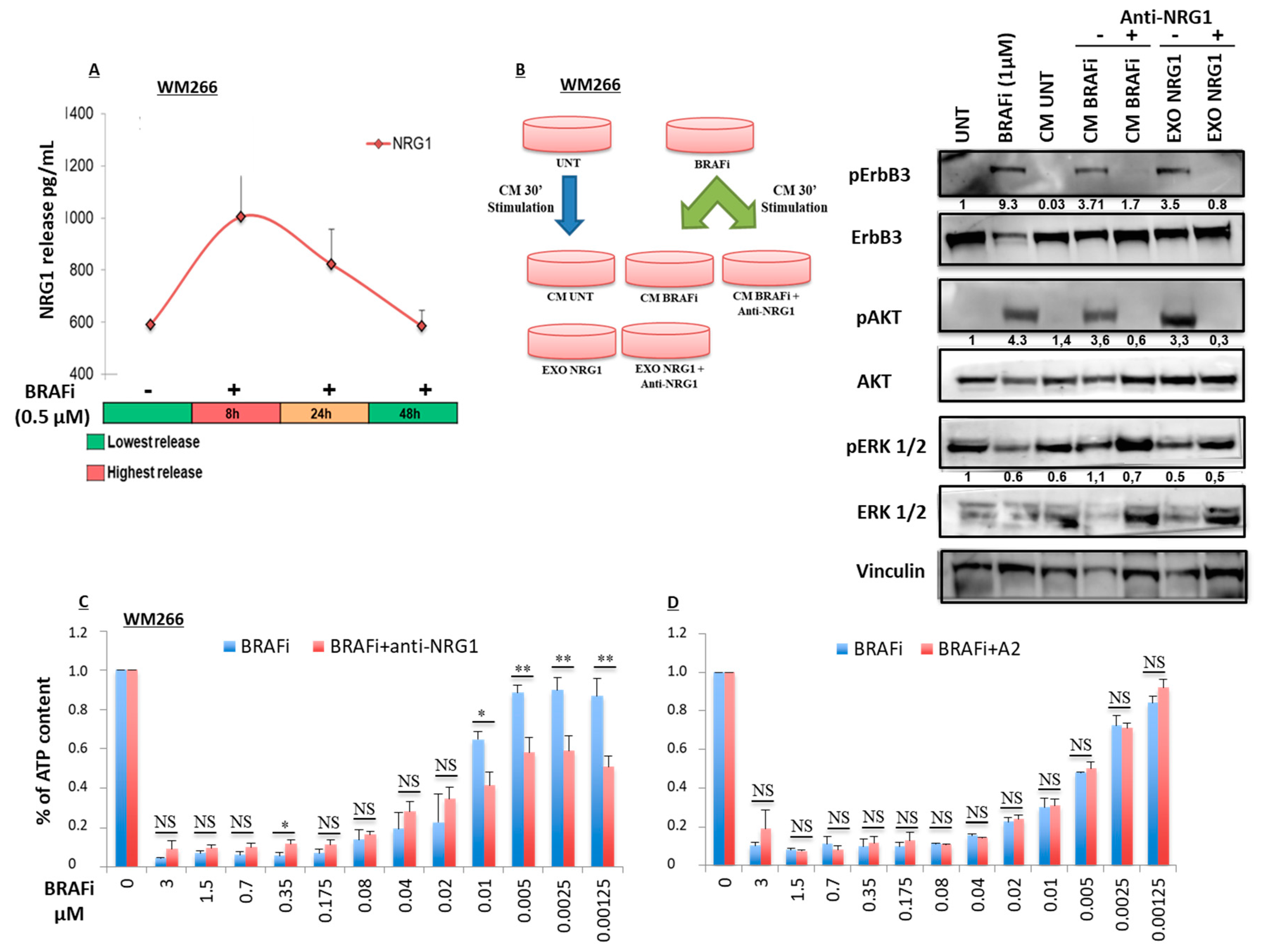
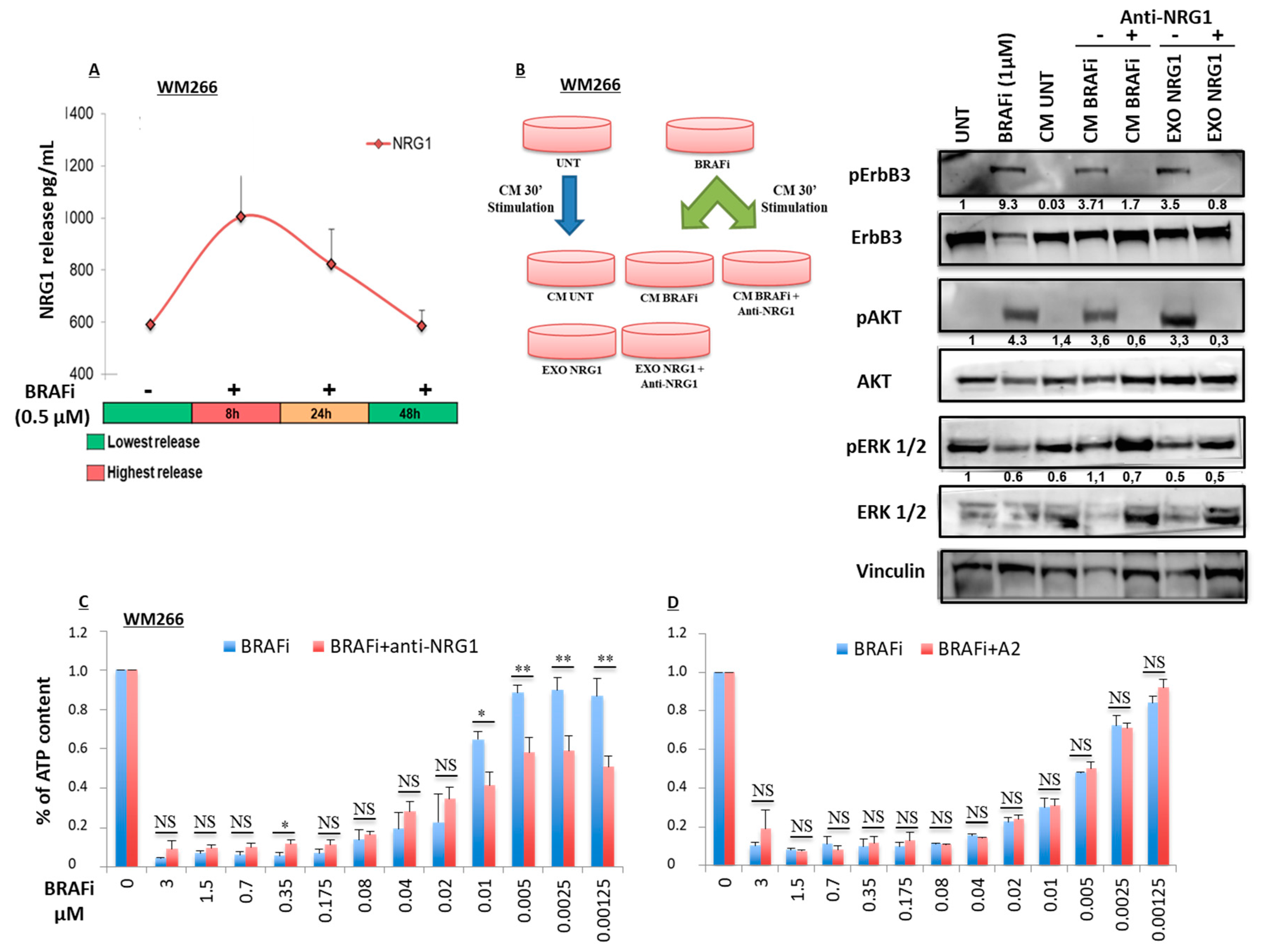
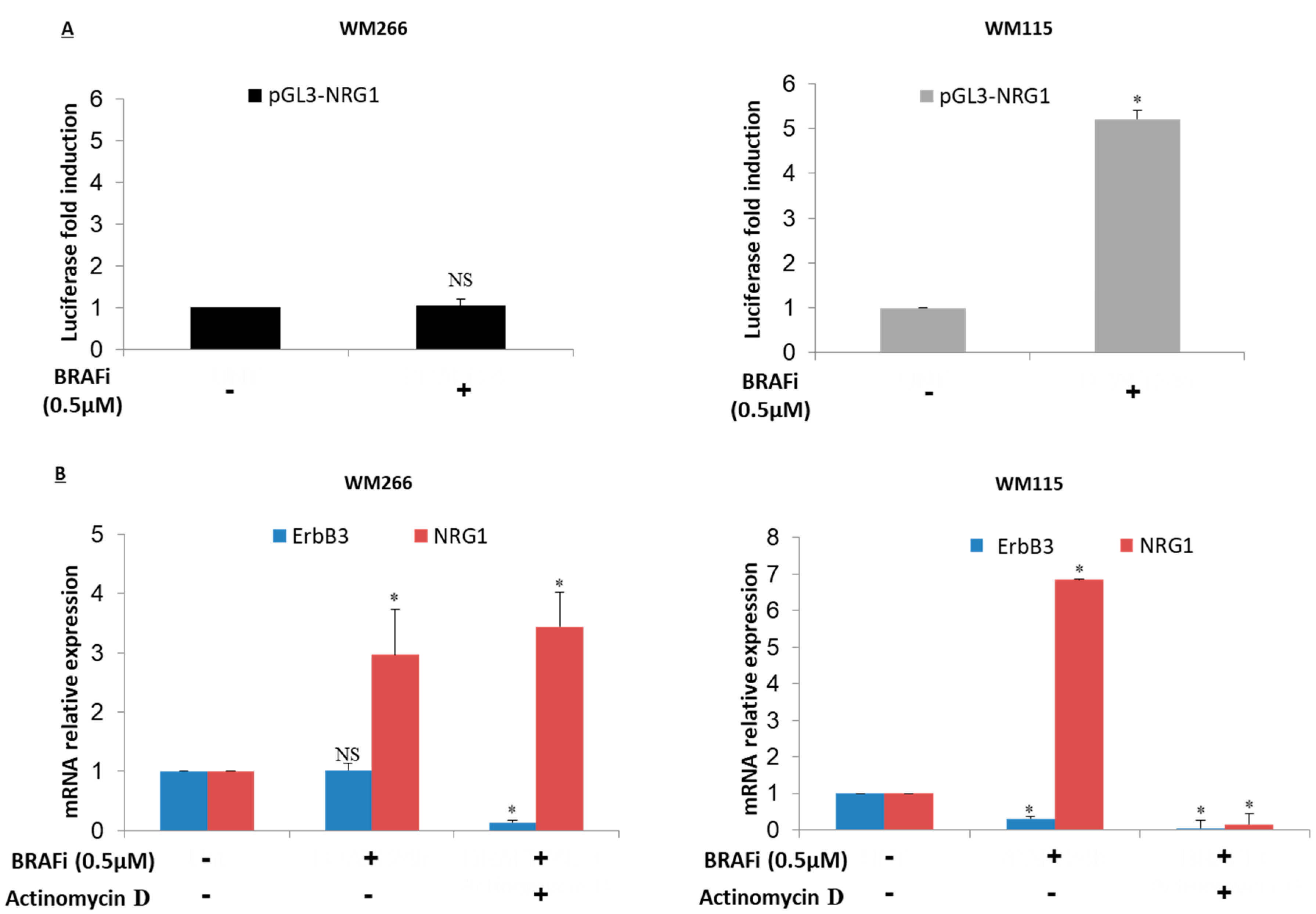
2.2. Early Release of NRG-1 Is Responsible for the Activation of the ErbB3 Receptor and NRG-1 Inhibition Potentiates the Growth Inhibitory Effect of BRAF Inhibition
2.3. BRAF-Mutated Melanoma Cells Show Different Patterns of Changes in ErbB3, NRG-1 and FOXD3 Gene Expression Levels after BRAFi Treatment
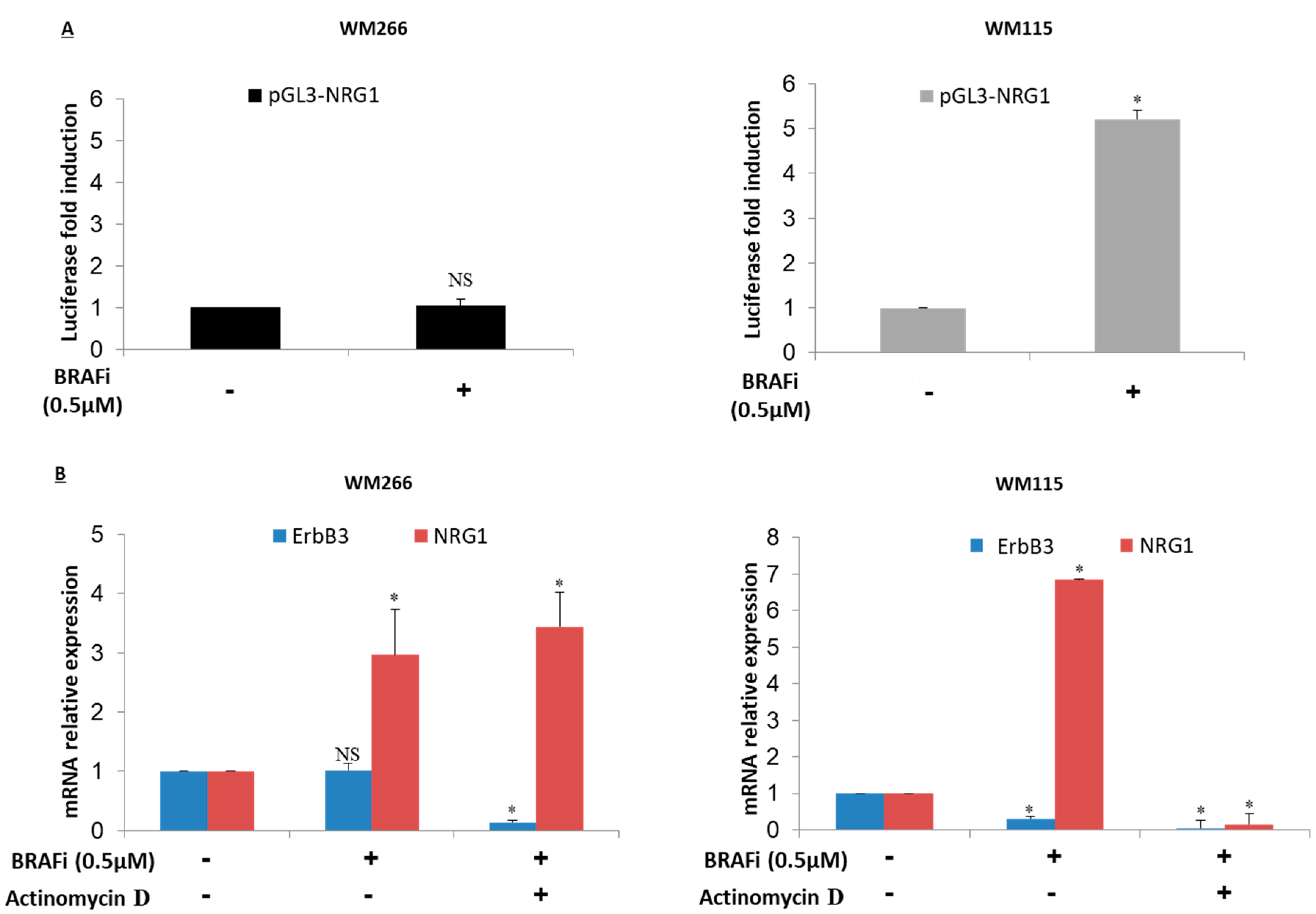
2.4. NRG-1 Activation Occurs via Different Mechanisms
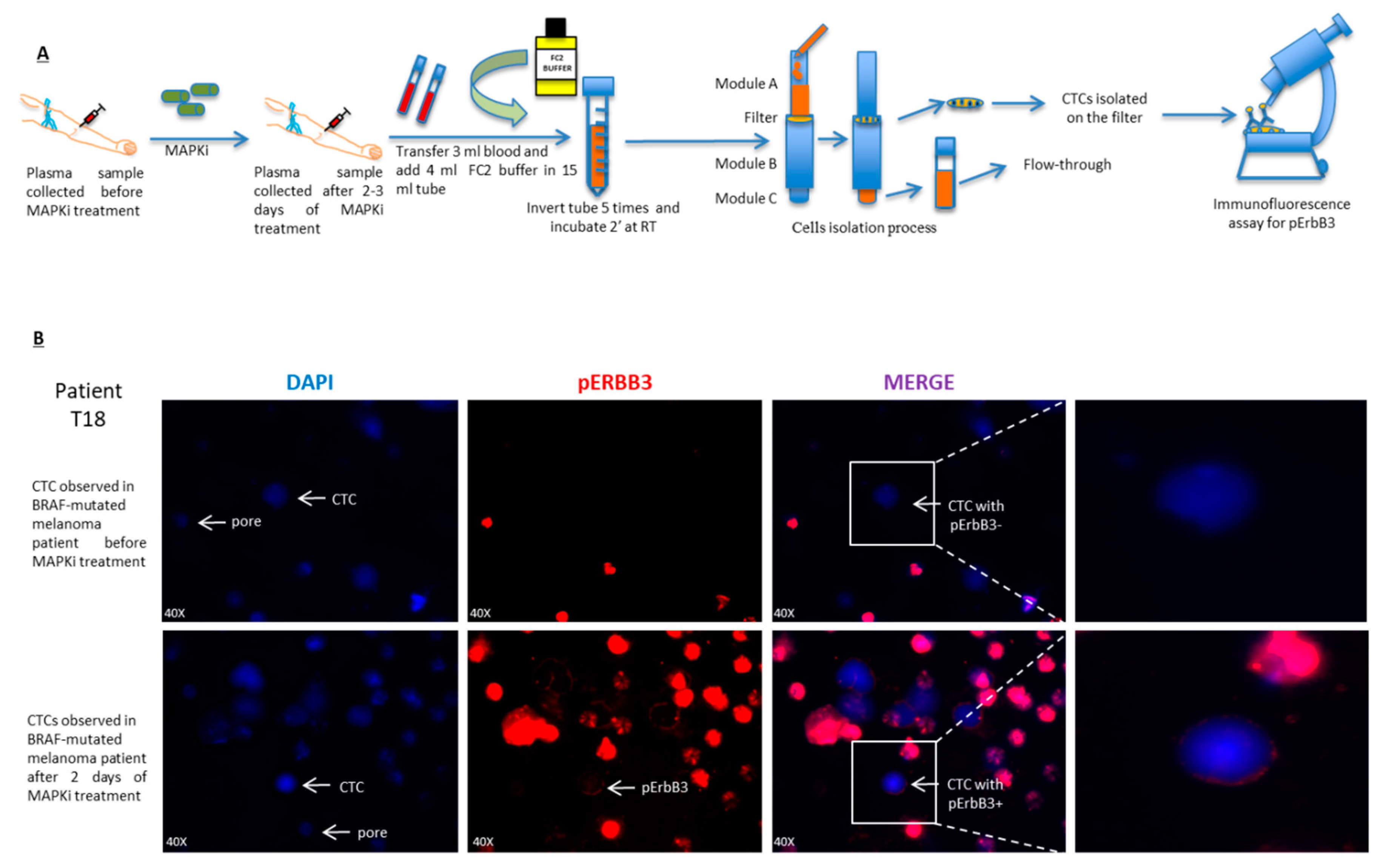
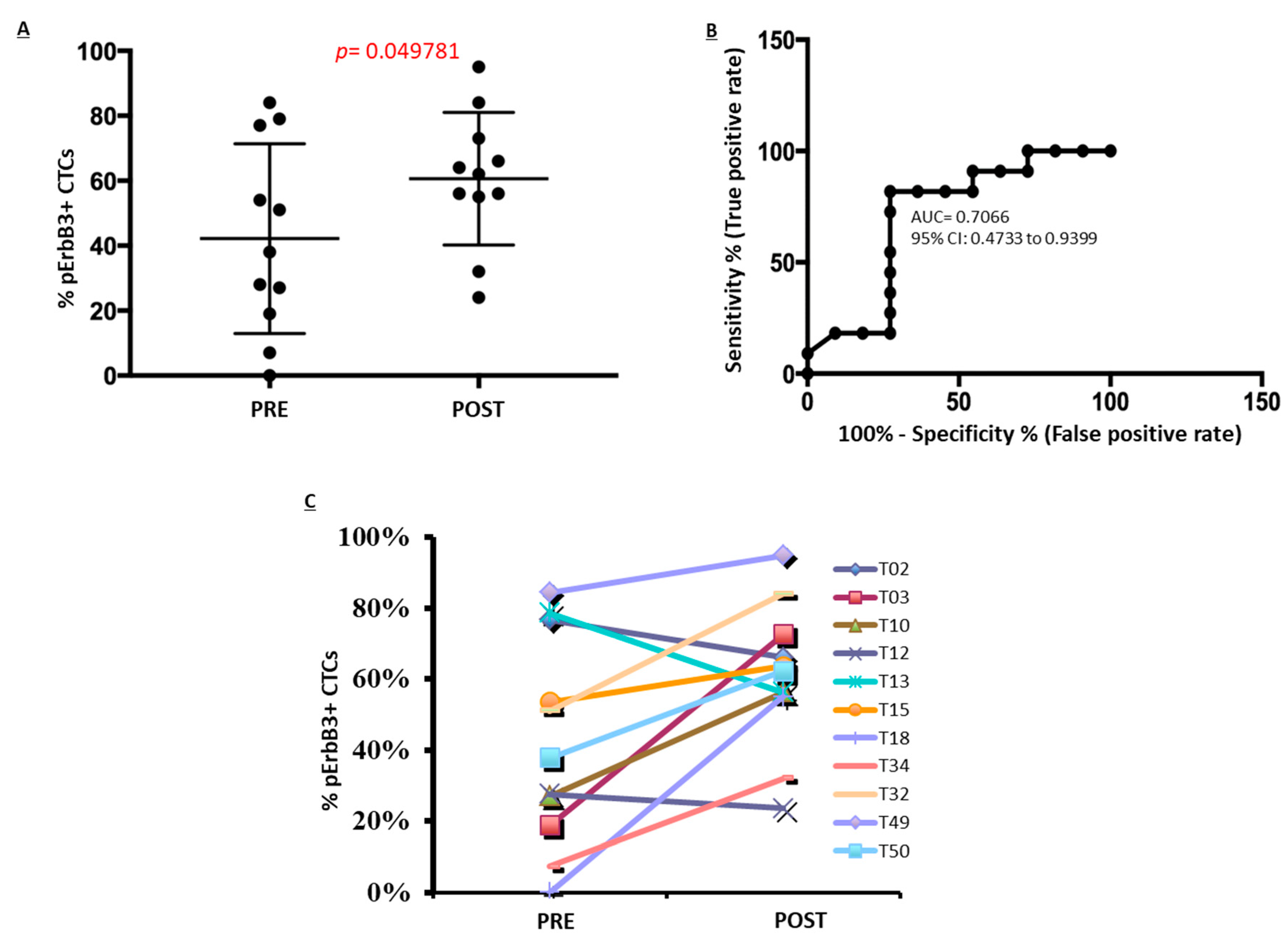
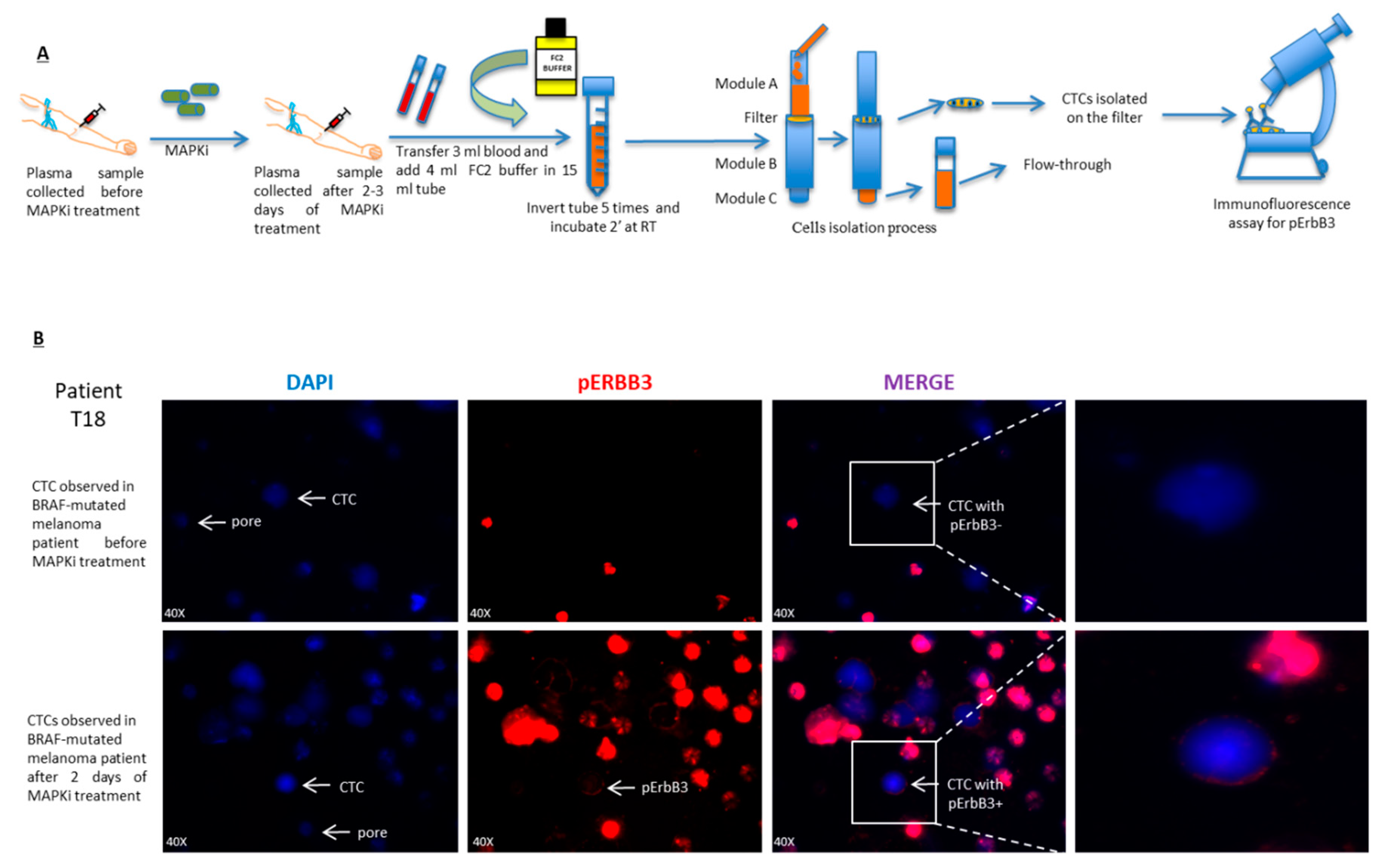
2.5. pErbB3 Upregulation Is Observed in Circulating Melanoma Cells After Treatment with MAPK Inhibitors
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Treatments
4.2. Antibodies and Reagents
4.3. NRG-1 Detection
4.4. Western Blot Analysis
4.5. RNA Extraction and Real-Time PCR Analysis
4.6. Luciferase Assays
4.7. Cell Viability
4.8. Patient Selection
- (1)
- Patients ≥18 years old.
- (2)
- Patients able to understand and willing to sign the informed consent form and able to adhere to the study visit scheme and other protocol requirements. Written informed consent must be obtained before any procedure.
- (3)
- Diagnosis confirmed by locally advanced stage melanoma histology or metastatic (stage IIIB–IV according to the American Joint Committee on Cancer—AJCC).
- (4)
- The patient is a candidate for therapy with BRAF inhibitors and/or MEK inhibitors.
4.9. Circulating Tumor Cells (CTCs) Isolation
4.10. Immunofluorescence
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ryška, A.; Horký, O.; Berkovcová, J.; Tichá, I.; Kalinová, M.; Matějčková, M.; Bóday, A.; Drábek, J.; Martínek, P.; Šimová, J.; et al. Malignant Melanoma—From Classical Histology towards Molecular Genetic Testing. Klin. Onkol. 2017, 30, 182–189. [Google Scholar] [PubMed]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [PubMed]
- Mason, R.; Au, L.; Ingles Garces, A.; Larkin, J. Current and emerging systemic therapies for cutaneous metastatic melanoma. Expert Opin. Pharmacother. 2019, 20, 1135–1152. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; Maclennan, G.T.; Montironi, R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: A move toward precision medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Arozarena, I.; Wellbrock, C. Overcoming resistance to BRAF inhibitors. Ann. Transl. Med. 2017, 5, 387. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Eroglu, Z.; Infante, J.; Patel, S.; Daud, A.; Johnson, D.B.; Gonzalez, R.; Kefford, R.; Hamid, O.; Schuchter, L.; et al. Long-term outcomes in patients with BRAF V600-mutant metastatic melanoma who received dabrafenib combined with trametinib. J. Clin. Oncol. 2018, 36, 667–673. [Google Scholar] [PubMed]
- Lim, S.Y.; Menzies, A.M.; Rizos, H. Mechanisms and strategies to overcome resistance to molecularly targeted therapy for melanoma. Cancer 2017, 123, 2118–2129. [Google Scholar] [CrossRef] [Green Version]
- Belleudi, F.; Marra, E.; Mazzetta, F.; Fattore, L.; Giovagnoli, M.R.; Mancini, R.; Aurisicchio, L.; Torrisi, M.R.; Ciliberto, G. Monoclonal antibody-induced ErbB3 receptor internalization and degradation inhibits growth and migration of human melanoma cells. Cell Cycle 2012, 11, 1455–1467. [Google Scholar] [Green Version]
- Ma, J.; Lyu, H.; Huang, J.; Liu, B. Targeting of erbB3 receptor to overcome resistance in cancer treatment. Mol. Cancer 2014, 13, 108. [Google Scholar] [CrossRef]
- Noto, A.; De Vitis, C.; Roscilli, G.; Fattore, L.; Malpicci, D.; Marra, E.; Luberto, L.; D’Andrilli, A.; Coluccia, P.; Giovagnoli, M.R.; et al. Combination therapy with anti-ErbB3 monoclonal antibodies and EGFR TKIs potently inhibits Non-small Cell Lung Cancer. Oncotarget 2013, 4, 1253–1265. [Google Scholar] [CrossRef]
- Nonagase, Y.; Yonesaka, K.; Kawakami, H.; Watanabe, S.; Haratani, K.; Takahama, T.; Takegawa, N.; Ueda, H.; Tanizaki, J.; Hayashi, H.; et al. Heregulin-expressing HER2-positive breast and gastric cancer exhibited heterogeneous susceptibility to the anti-HER2 agents lapatinib, trastuzumab and T-DM1. Oncotarget 2016, 7, 84860–84871. [Google Scholar] [PubMed] [Green Version]
- Poovassery, J.S.; Kang, J.C.; Kim, D.; Ober, R.J.; Ward, E.S. Antibody targeting of HER2/HER3 signaling overcomes heregulin-induced resistance to PI3K inhibition in prostate cancer. Int. J. Cancer 2015, 137, 267–277. [Google Scholar] [CrossRef]
- Tiwary, S.; Preziosi, M.; Rothberg, P.G.; Zeitouni, N.; Corson, N.; Xu, L. ERBB3 is required for metastasis formation of melanoma cells. Oncogenesis 2014, 3, e110. [Google Scholar] [CrossRef]
- Reschke, M.; Mihic-Probst, D.; Der Van Horst, E.H.; Knyazev, P.; Wild, P.J.; Hutterer, M.; Meyer, S.; Dummer, R.; Moch, H.; Ullrich, A. HER3 Is a determinant for poor prognosis in melanoma. Clin. Cancer Res. 2008, 14, 5188–5197. [Google Scholar]
- Fattore, L.; Malpicci, D.; Marra, E.; Belleudi, F.; Noto, A.; De Vitis, C.; Pisanu, M.E.; Coluccia, P.; Camerlingo, R.; Roscilli, G.; et al. Combination of antibodies directed against different ErbB3 surface epitopes prevents the establishment of resistance to BRAF/MEK inhibitors in melanoma. Oncotarget 2015, 6, 24823–24841. [Google Scholar] [CrossRef] [Green Version]
- Prasetyanti, P.R.; Capone, E.; Barcaroli, D.; D’Agostino, D.; Volpe, S.; Benfante, A.; van Hooff, S.; Iacobelli, V.; Rossi, C.; Iacobelli, S.; et al. ErbB-3 activation by NRG-1β sustains growth and promotes vemurafenib resistance in BRAF-V600E colon cancer stem cells (CSCs). Oncotarget 2015, 6, 16902–16911. [Google Scholar] [PubMed]
- Yang, L.; Li, Y.; Shen, E.; Cao, F.; Li, L.; Li, X.; Wang, X.; Kariminia, S.; Chang, B.; Li, H.; et al. NRG1-dependent activation of HER3 induces primary resistance to trastuzumab in HER2-overexpressing breast cancer cells. Int. J. Oncol. 2017, 51, 1553–1562. [Google Scholar] [PubMed]
- Van Lengerich, B.; Agnew, C.; Puchner, E.M.; Huang, B.; Jura, N. EGF and NRG induce phosphorylation of HER3/ERBB3 by EGFR using distinct oligomeric mechanisms. Proc. Natl. Acad. Sci. USA 2017, 114, E2836–E2845. [Google Scholar] [PubMed] [Green Version]
- Fattore, L.; Marra, E.; Pisanu, M.E.; Noto, A.; de Vitis, C.; Belleudi, F.; Aurisicchio, L.; Mancini, R.; Torrisi, M.R.; Ascierto, P.A.; et al. Activation of an early feedback survival loop involving phospho-ErbB3 is a general response of melanoma cells to RAF/MEK inhibition and is abrogated by anti-ErbB3 antibodies. J. Transl. Med. 2013, 11, 180. [Google Scholar] [CrossRef]
- Capparelli, C.; Rosenbaum, S.; Berger, A.C.; Aplin, A.E. Fibroblast-derived neuregulin 1 promotes Compensatory ErbB3 receptor signaling in mutant BRAF melanoma. J. Biol. Chem. 2015, 290, 24267–24277. [Google Scholar] [PubMed]
- Capparelli, C.; Rosenbaum, S.; Berman-Booty, L.D.; Salhi, A.; Gaborit, N.; Zhan, T.; Chervoneva, I.; Roszik, J.; Woodman, S.E.; Davies, M.A.; et al. ErbB3-ErbB2 complexes as a therapeutic target in a subset of wild-type BRAF/NRAS cutaneous melanomas. Cancer Res. 2015, 75, 3554–3567. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.V.; Basile, K.J.; Kugel, C.H.; Witkiewicz, A.K.; Le, K.; Amaravadi, R.K.; Karakousis, G.C.; Xu, X.; Xu, W.; Schuchter, L.M.; et al. Melanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3. J. Clin. Investig. 2013, 123, 4609–4618. [Google Scholar]
- Zhang, K.; Wong, P.; Zhang, L.; Jacobs, B.; Borden, E.C.; Aster, J.C.; Bedogni, B. A Notch1neuregulin1 autocrine signaling loop contributes to melanoma growth. Oncogene 2012, 31, 4609–4618. [Google Scholar] [PubMed]
- Geethadevi, A.; Parashar, D.; Bishop, E.; Pradeep, S.; Chaluvally-Raghavan, P. ERBB signaling in CTCs of ovarian cancer and glioblastoma. Genes Cancer 2017, 8, 746–751. [Google Scholar]
- Fabisiewicz, A.; Grzybowska, E. CTC clusters in cancer progression and metastasis. Med. Oncol. 2017, 34, 12. [Google Scholar] [CrossRef] [PubMed]
- Bidard, F.C.; Proudhon, C.; Pierga, J.Y. Circulating tumor cells in breast cancer. Mol. Oncol. 2016, 10, 418–430. [Google Scholar] [Green Version]
- Huang, S.K.; Hoon, D.S.B. Liquid biopsy utility for the surveillance of cutaneous malignant melanoma patients. Mol. Oncol. 2016, 10, 450–463. [Google Scholar]
- Scaini, M.C.; Pigozzo, J.; Pizzi, M.; Manicone, M.; Chiarion-Sileni, V.; Zambenedetti, P.; Rugge, M.; Zanovello, P.; Rossi, E.; Zamarchi, R.; et al. Clonal heterogeneity of melanoma in a paradigmatic case study. Melanoma Res. 2019, 29, 189–194. [Google Scholar]
- Nicolazzo, C.; Colangelo, L.; Corsi, A.; Carpino, G.; Gradilone, A.; Sonato, C.; Raimondi, C.; Gaudio, E.; Gazzaniga, P.; Gianni, W. Liquid biopsy in rare cancers: Lessons from hemangiopericytoma. Anal. Cell. Pathol. 2018, 2018, 9718585. [Google Scholar] [CrossRef]
- Kuvendjiska, J.; Bronsert, P.; Martini, V.; Lang, S.; Pitman, M.B.; Hoeppner, J.; Kulemann, B. Non-metastatic esophageal adenocarcinoma: Circulating tumor cells in the course of multimodal tumor treatment. Cancers 2019, 11, E397. [Google Scholar] [CrossRef]
- Curley, M.D.; Sabnis, G.J.; Wille, L.; Adiwijaya, B.S.; Garcia, G.; Moyo, V.; Kazi, A.A.; Brodie, A.; MacBeath, G. Seribantumab, an Anti-ERBB3 Antibody, Delays the Onset of Resistance and Restores Sensitivity to Letrozole in an Estrogen Receptor-Positive Breast Cancer Model. Mol. Cancer Ther. 2015, 14, 2642–2652. [Google Scholar] [PubMed]
- Yonesaka, K.; Hirotani, K.; Kawakami, H.; Takeda, M.; Kaneda, H.; Sakai, K.; Okamoto, I.; Nishio, K.; Jänne, P.A.; Nakagawa, K. Anti-HER3 monoclonal antibody patritumab sensitizes refractory non-small cell lung cancer to the epidermal growth factor receptor inhibitor erlotinib. Oncogene 2016, 35, 878–886. [Google Scholar] [PubMed]
- Kawakami, H.; Okamoto, I.; Yonesaka, K.; Okamoto, K.; Shibata, K.; Shinkai, Y.; Sakamoto, H.; Kitano, M.; Tamura, T.; Nishio, K.; et al. The anti-HER3 antibody patritumab abrogates cetuximab resistance mediated by heregulin in colorectal cancer cells. Oncotarget 2014, 5, 11847–11856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Yang, J.; Li, S.; Jiang, M.; Cao, G.; Yang, L.; Zhang, X.; Zhou, Y.; Li, K.; Tang, S.T. Effects of RET, NRG1 and NRG3 Polymorphisms in a Chinese Population with Hirschsprung Disease. Sci. Rep. 2017, 7, 43222. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Endo, H.; Inoue, T.; Nishino, K.; Uchida, J.; Kumagai, T.; Kukita, Y.; Kato, K.; Imamura, F.; Inoue, M. Analysis of ERBB ligand-induced resistance mechanism to crizotinib by primary culture of lung adenocarcinoma with EML4-ALK fusion gene. J. Thorac. Oncol. 2015, 10, 527–530. [Google Scholar] [PubMed]
- Mota, J.M.; Collier, K.A.; Costa, R.L.B.; Taxter, T.; Kalyan, A.; Leite, C.A.; Chae, Y.K.; Giles, F.J.; Carneiro, B.A. A comprehensive review of heregulins, HER3, and HER4 as potential therapeutic targets in cancer. Oncotarget 2017, 8, 89284–89306. [Google Scholar] [PubMed] [Green Version]
- Fedorenko, I.V.; Smalley, K.S. The complexity of microenvironment-mediated drug resistance. Genes Cancer 2015, 6, 367–368. [Google Scholar] [PubMed]
- Montero-Conde, C.; Ruiz-Llorente, S.; Dominguez, J.M.; Knauf, J.A.; Viale, A.; Sherman, E.J.; Ryder, M.; Ghossein, R.A.; Rosen, N.; Fagin, J.A. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov. 2013, 3, 520–533. [Google Scholar] [PubMed]
- Han, S.; Ren, Y.; He, W.; Liu, H.; Zhi, Z.; Zhu, X.; Yang, T.; Rong, Y.; Ma, B.; Purwin, T.J.; et al. ERK-mediated phosphorylation regulates SOX10 sumoylation and targets expression in mutant BRAF melanoma. Nat. Commun. 2018, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Ebbing, E.A.; Medema, J.P.; Damhofer, H.; Meijer, S.L.; Krishnadath, K.K.; van Berge Henegouwen, M.I.; Bijlsma, M.F.; van Laarhoven, H.W.M. ADAM10-mediated release of heregulin confers resistance to trastuzumab by activating HER3. Oncotarget 2016, 7, 10243–10254. [Google Scholar] [PubMed]
- Qiu, H.; Tang, X.; Ma, J.; Shaverdashvili, K.; Zhang, K.; Bedogni, B. Notch1 Autoactivation via Transcriptional Regulation of Furin, Which Sustains Notch1 Signaling by Processing Notch1-Activating Proteases ADAM10 and Membrane Type 1 Matrix Metalloproteinase. Mol. Cell. Biol. 2015, 35, 3622–3632. [Google Scholar] [PubMed] [Green Version]
- Zhang, K.; Wong, P.; Salvaggio, C.; Salhi, A.; Osman, I.; Bedogni, B. Synchronized Targeting of Notch and ERBB Signaling Suppresses Melanoma Tumor Growth through Inhibition of Notch1 and ERBB3. J. Investig. Dermatol. 2016, 136, 464–472. [Google Scholar] [PubMed]
- Qiu, H.; Zmina, P.M.; Huang, A.Y.; Askew, D.; Bedogni, B. Inhibiting Notch1 enhances immunotherapy efficacy in melanoma by preventing Notch1 dependent immune suppressive properties. Cancer Lett. 2018, 434, 144–151. [Google Scholar] [PubMed]
- Costanzo, P.; Santini, A.; Fattore, L.; Novellino, E.; Ritieni, A. Toxicity of aflatoxin B1 towards the vitamin D receptor (VDR). Food Chem. Toxicol. 2015, 76, 77–79. [Google Scholar] [PubMed] [Green Version]
- Roscilli, G.; Cappelletti, M.; De Vitis, C.; Ciliberto, G.; Di Napoli, A.; Ruco, L.; Mancini, R.; Aurisicchio, L. Circulating MMP11 and specific antibody immune response in breast and prostate cancer patients. J. Transl. Med. 2014, 12, 54. [Google Scholar] [CrossRef]
- Fattore, L.; Ruggiero, C.F.; Pisanu, M.E.; Liguoro, D.; Cerri, A.; Costantini, S.; Capone, F.; Acunzo, M.; Romano, G.; Nigita, G.; et al. Reprogramming miRNAs global expression orchestrates development of drug resistance in BRAF mutated melanoma. Cell Death Differ. 2019, 26, 1267–1282. [Google Scholar]
- Fattore, L.; Mancini, R.; Acunzo, M.; Romano, G.; Laganà, A.; Pisanu, M.E.; Malpicci, D.; Madonna, G.; Mallardo, D.; Capone, M.; et al. miR-579-3p controls melanoma progression and resistance to target therapy. Proc. Natl. Acad. Sci. USA 2016, 113, E5005–E5013. [Google Scholar]
- Pisanu, M.E.; Maugeri-Saccà, M.; Fattore, L.; Bruschini, S.; De Vitis, C.; Tabbì, E.; Bellei, B.; Migliano, E.; Kovacs, D.; Camera, E.; et al. Inhibition of Stearoyl-CoA desaturase 1 reverts BRAF and MEK inhibition-induced selection of cancer stem cells in BRAF-mutated melanoma. J. Exp. Clin. Cancer Res. 2018, 37, 318. [Google Scholar] [CrossRef]
- Fattore, L.; Costantini, S.; Malpicci, D.; Ruggiero, C.F.; Ascierto, P.A.; Croce, C.M.; Mancini, R.; Ciliberto, G. MicroRNAs in melanoma development and resistance to target therapy. Oncotarget 2017, 8, 22262–22278. [Google Scholar] [Green Version]
- Fattore, L.; Sacconi, A.; Mancini, R.; Ciliberto, G. MicroRNA-driven deregulation of cytokine expression helps development of drug resistance in metastatic melanoma. Cytokine Growth Factor Rev. 2017, 36, 39–48. [Google Scholar] [PubMed]
- Fattore, L.; Mancini, R.; Ascierto, P.A.; Ciliberto, G. The potential of BRAF-associated non-coding RNA as a therapeutic target in melanoma. Expert Opin. Ther. Targets 2019, 23, 53–68. [Google Scholar] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruggiero, C.F.; Malpicci, D.; Fattore, L.; Madonna, G.; Vanella, V.; Mallardo, D.; Liguoro, D.; Salvati, V.; Capone, M.; Bedogni, B.; et al. ErbB3 Phosphorylation as Central Event in Adaptive Resistance to Targeted Therapy in Metastatic Melanoma: Early Detection in CTCs during Therapy and Insights into Regulation by Autocrine Neuregulin. Cancers 2019, 11, 1425. https://doi.org/10.3390/cancers11101425
Ruggiero CF, Malpicci D, Fattore L, Madonna G, Vanella V, Mallardo D, Liguoro D, Salvati V, Capone M, Bedogni B, et al. ErbB3 Phosphorylation as Central Event in Adaptive Resistance to Targeted Therapy in Metastatic Melanoma: Early Detection in CTCs during Therapy and Insights into Regulation by Autocrine Neuregulin. Cancers. 2019; 11(10):1425. https://doi.org/10.3390/cancers11101425
Chicago/Turabian StyleRuggiero, Ciro Francesco, Debora Malpicci, Luigi Fattore, Gabriele Madonna, Vito Vanella, Domenico Mallardo, Domenico Liguoro, Valentina Salvati, Mariaelena Capone, Barbara Bedogni, and et al. 2019. "ErbB3 Phosphorylation as Central Event in Adaptive Resistance to Targeted Therapy in Metastatic Melanoma: Early Detection in CTCs during Therapy and Insights into Regulation by Autocrine Neuregulin" Cancers 11, no. 10: 1425. https://doi.org/10.3390/cancers11101425
APA StyleRuggiero, C. F., Malpicci, D., Fattore, L., Madonna, G., Vanella, V., Mallardo, D., Liguoro, D., Salvati, V., Capone, M., Bedogni, B., Ascierto, P. A., Mancini, R., & Ciliberto, G. (2019). ErbB3 Phosphorylation as Central Event in Adaptive Resistance to Targeted Therapy in Metastatic Melanoma: Early Detection in CTCs during Therapy and Insights into Regulation by Autocrine Neuregulin. Cancers, 11(10), 1425. https://doi.org/10.3390/cancers11101425