Hepatic Stress Response in HCV Infection Promotes STAT3-Mediated Inhibition of HNF4A-miR-122 Feedback Loop in Liver Fibrosis and Cancer Progression

 ,
,
Abstract
1. Introduction
2. Results
2.1. Persistent HCV Replication Leads to ER Stress and Oxidative Stress
2.2. Oxidative Stress and ER Stress Activate NRF2 Signaling in HCV Infection
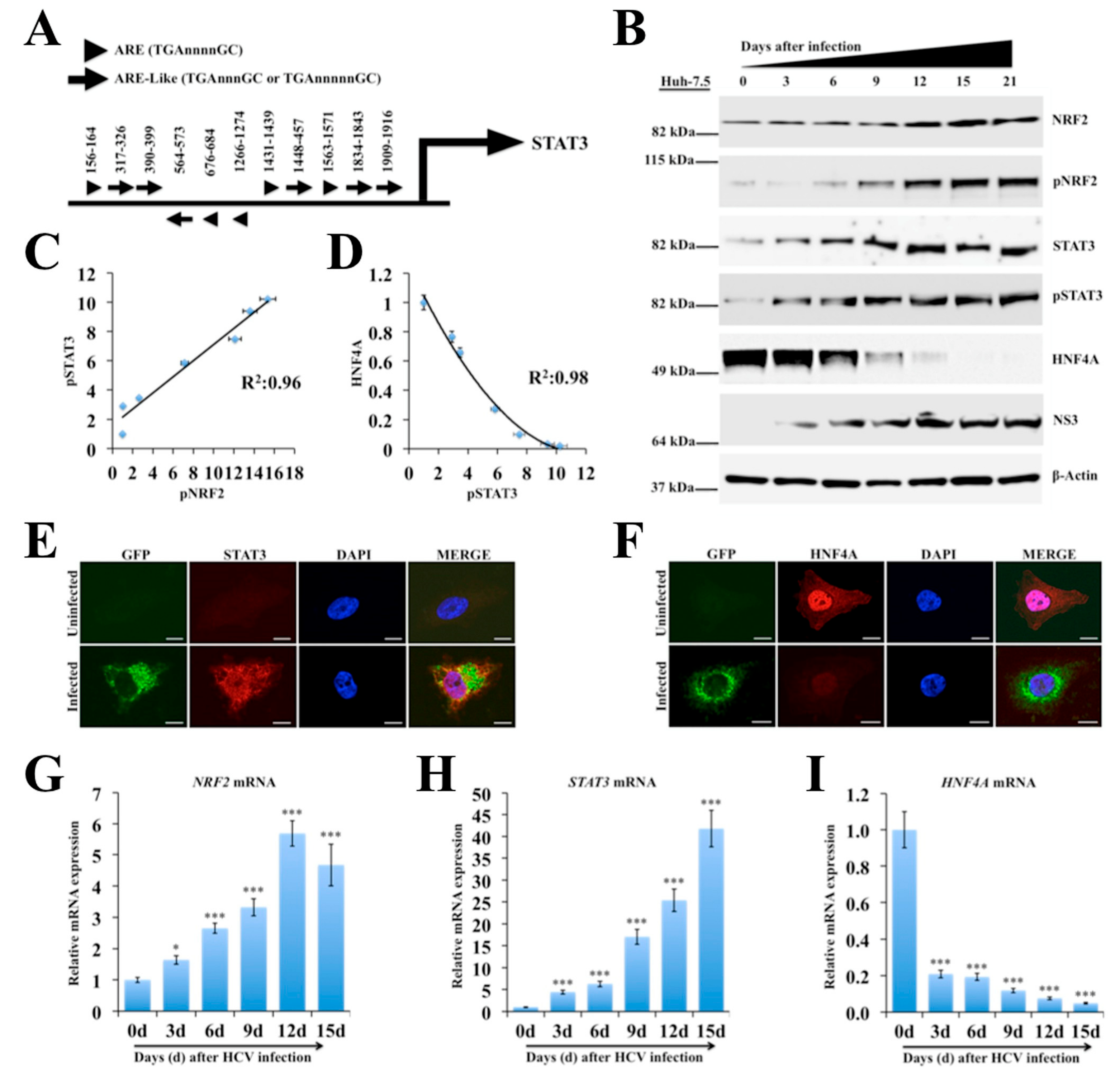
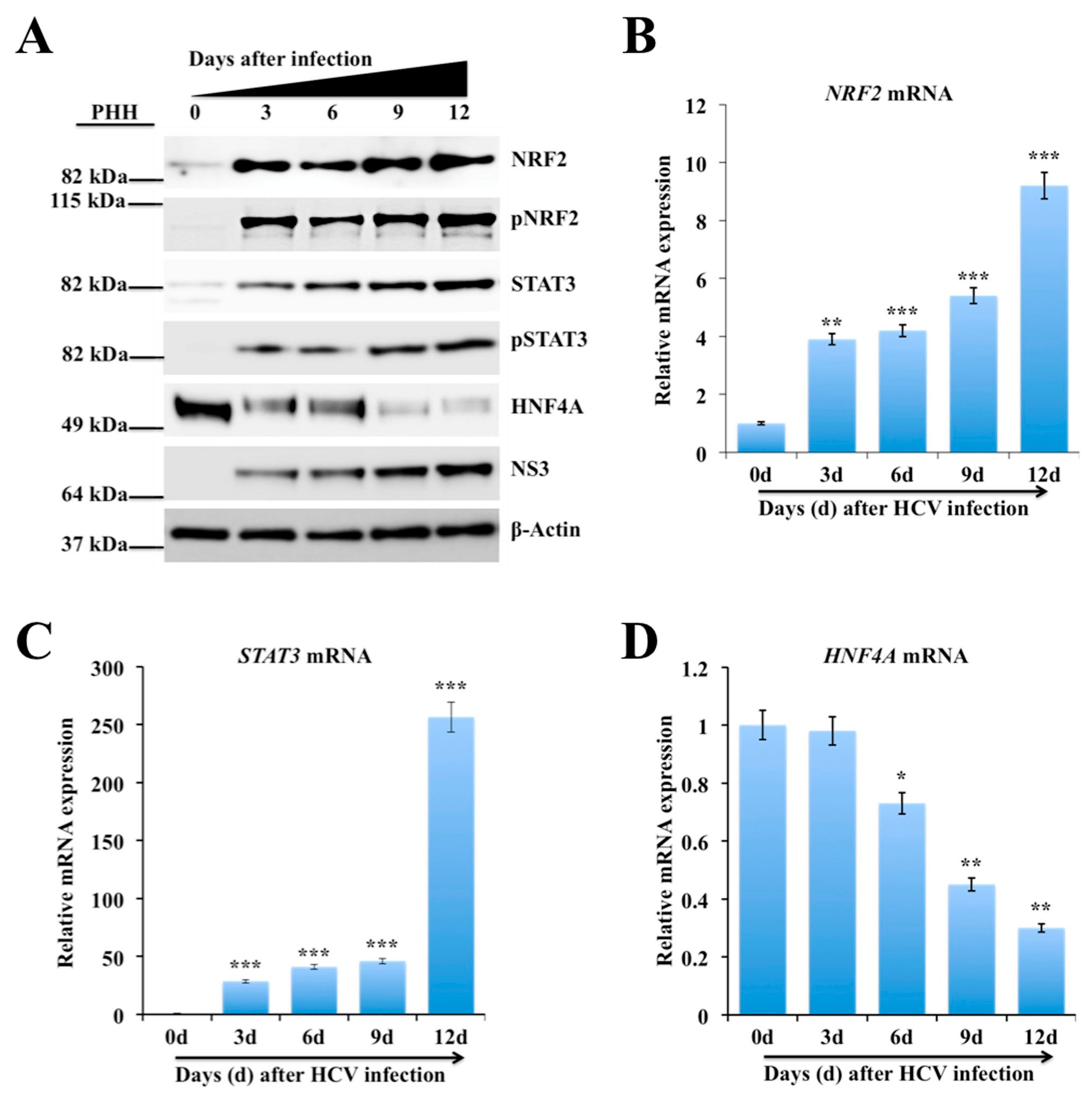
2.3. Persistent HCV Infection Activates the NRF2-STAT3-HNF4A Regulatory Axis as a Cytoprotective Response
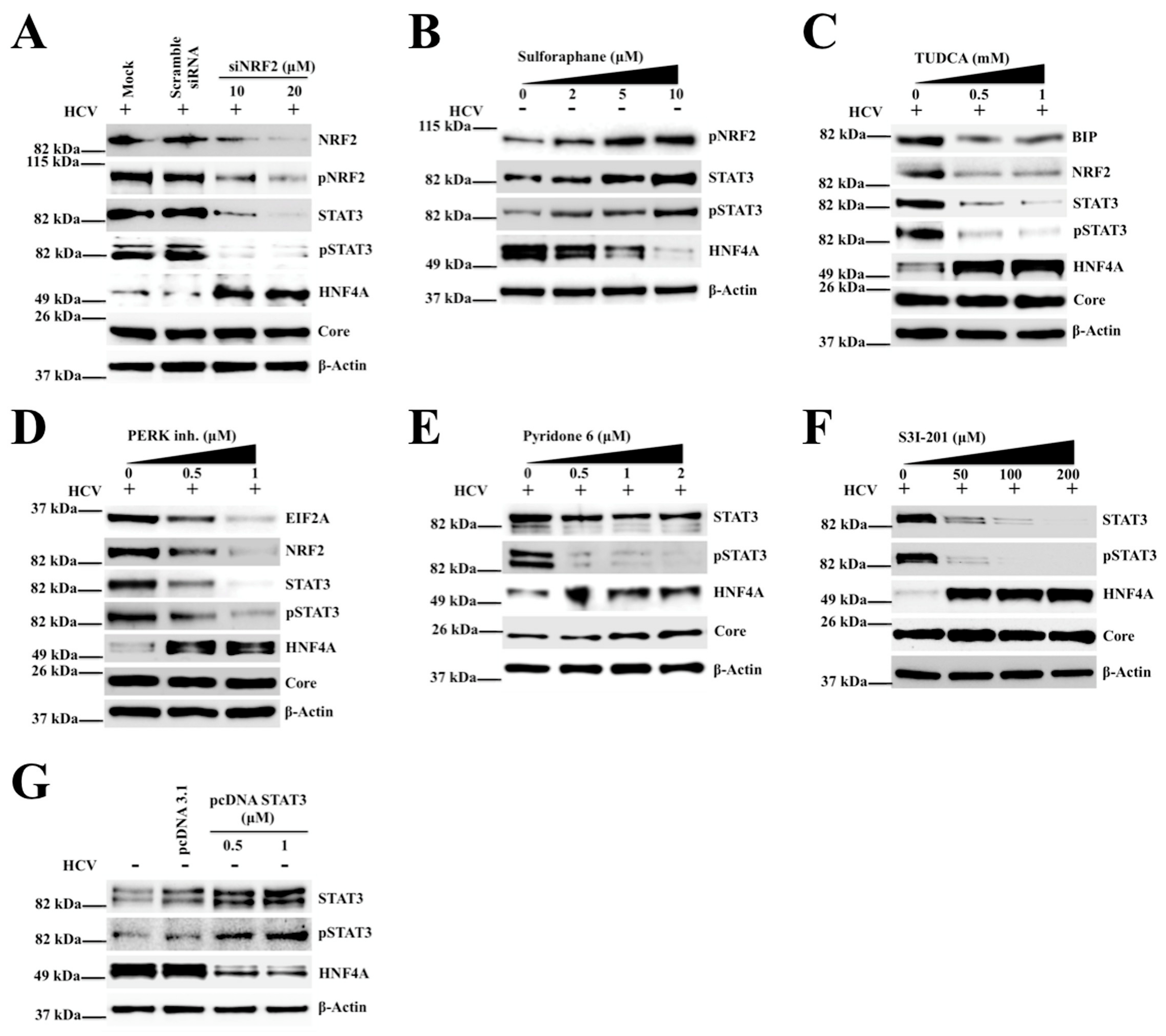
2.4. Silencing PERK and NRF2 Pathway Restore Expression of STAT3-HNF4A Inflammatory Loop
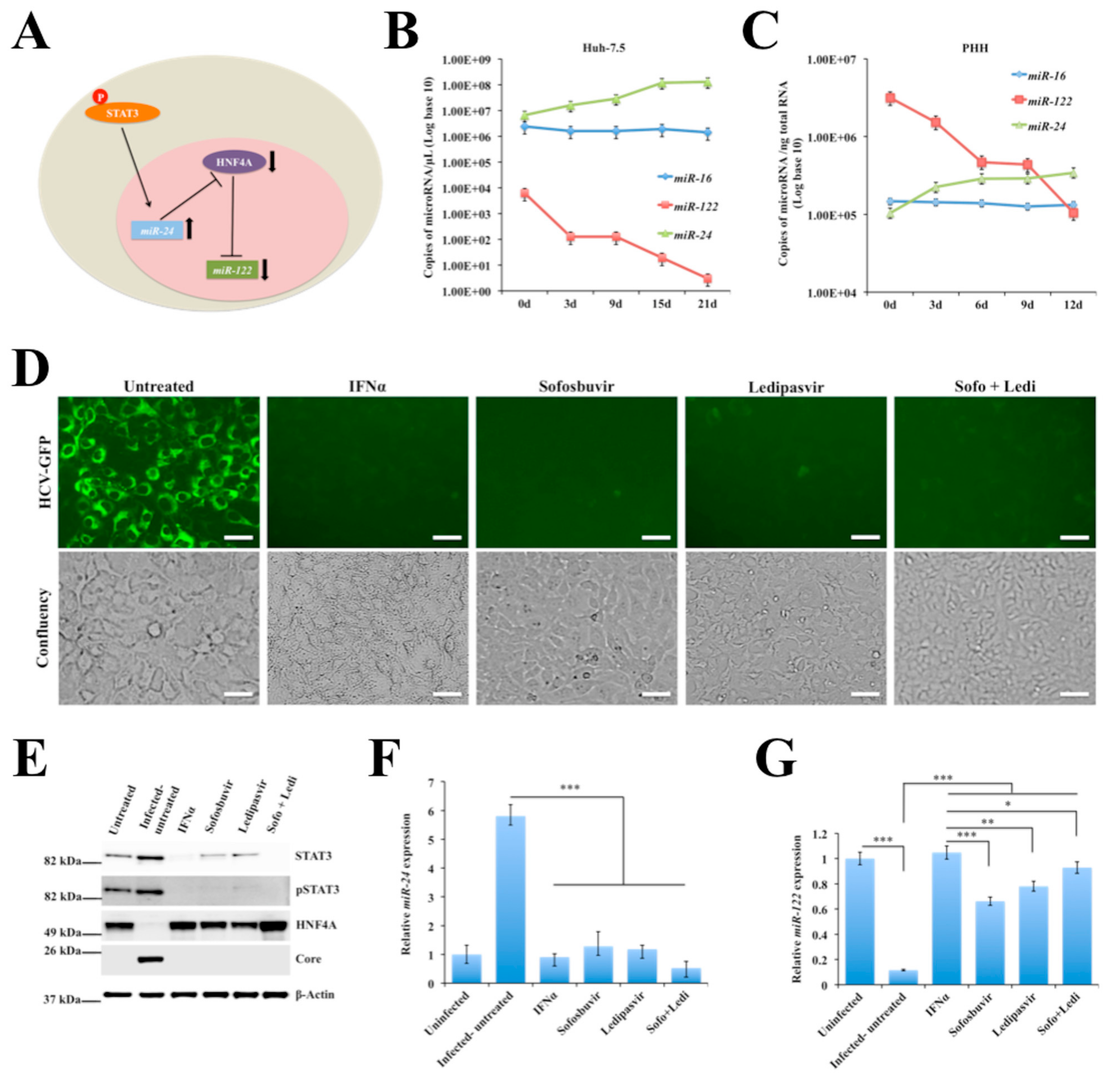
2.5. Persistent HCV Replication in Huh-7.5 Cells Leads to STAT3-HNF4A-Mediated Silencing of miR-122
2.6. Persistent HCV Replication in Huh-7.5 Cells Leads to Decreased miR-122 Promoter Activity
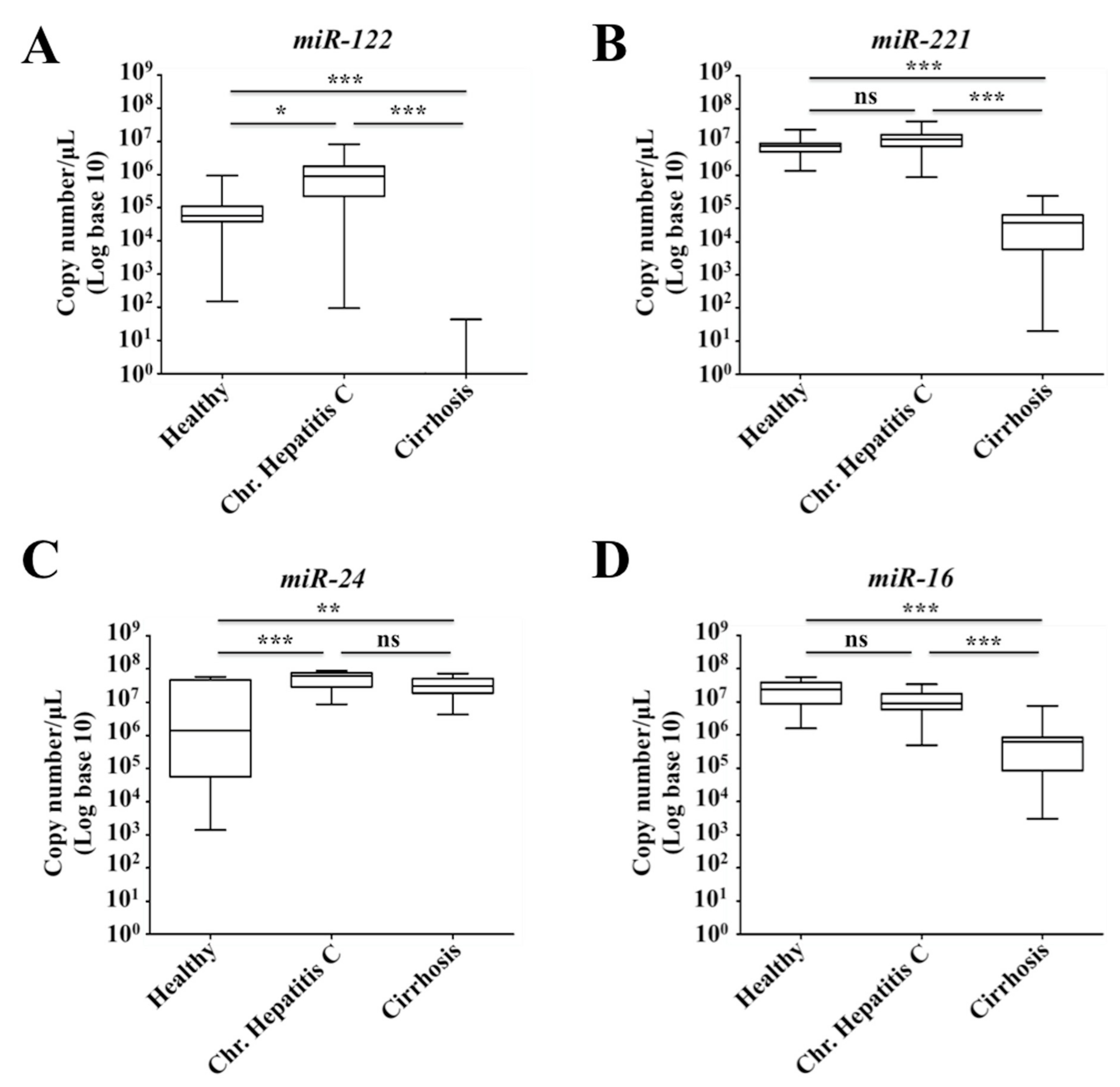
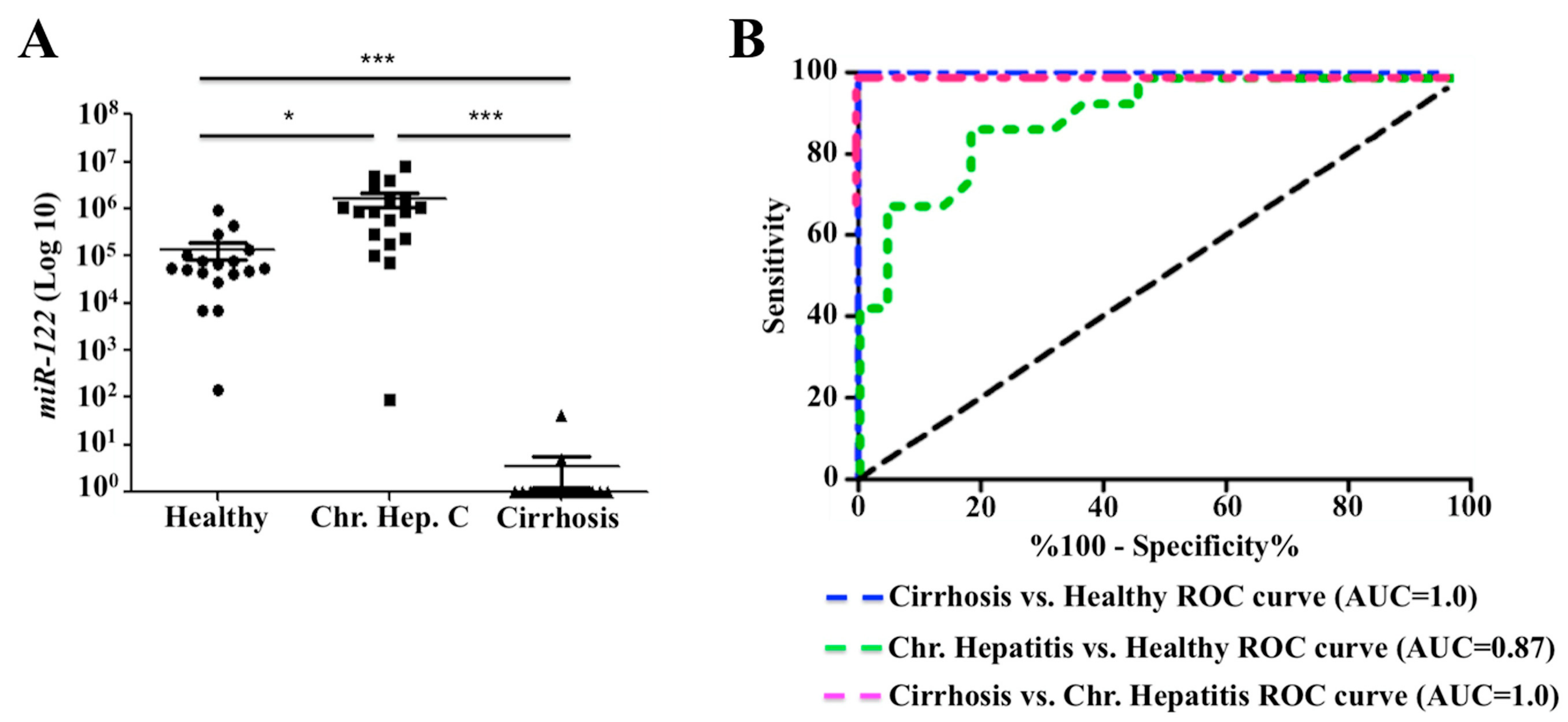
2.7. Decreased Expression of miR-122 Correlates with Patients with Cirrhosis
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Antibodies and Chemicals
4.2. Quantitative Assessment of Misfolded Protein Burden in the ER
4.3. Detection of Reactive Oxygen Species (ROS)
4.4. Immunohistochemical Staining
4.5. SDS-PAGE and Western Blotting
4.6. siRNA Transfection
4.7. Quantification of mRNA Levels by RT-qPCR
4.8. Quantification of Serum microRNA Levels by RT-qPCR
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sayiner, M.; Golabi, P.; Younossi, Z.M. Disease Burden of Hepatocellular Carcinoma: A Global Perspective. Dig. Dis. Sci. 2019, 64, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Allison, R.D.; Tong, X.; Moorman, A.C.; Ly, K.N.; Rupp, L.; Xu, F.; Gordon, S.C.; Holmberg, S.D.; Chronic Hepatitis Cohort Study (CHeCS) Investigators. Increased incidence of cancer and cancer-related mortality among persons with chronic hepatitis C infection, 2006–2010. J. Hepatol. 2015, 63, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Thrift, A.P.; El-Serag, H.B.; Kanwal, F. Global epidemiology and burden of HCV infection and HCV-related disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 14, 122. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.M.; Feld, J.J.; Zeuzem, S.; Hoofnagle, J.H. From non-A, non-B hepatitis to hepatitis C virus cure. J. Hepatol. 2015, 62, S87–S99. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, A.; Mondelli, M.U.; ESCMID Study Group for Viral Hepatitis (ESGVH). Hepatitis C: Is eradication possible? Liver Int. 2019, 39, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.L. Global control of hepatitis C: Where challenge meets opportunity. Nat. Med. 2013, 19, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Thimme, R.; Blum, H.E.; Zoulim, F. Hepatitis C virus-induced hepatocarcinogenesis. J. Hepatol. 2009, 51, 810–820. [Google Scholar] [CrossRef]
- Dash, S.; Aydin, Y.; Wu, T. Integrated stress response in hepatitis C promotes Nrf2-related chaperone-mediated autophagy: A novel mechanism for host-microbe survival and HCC development in liver cirrhosis. Semin. Cell Dev. Biol. 2019. [Google Scholar] [CrossRef]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef]
- Chava, S.; Lee, C.; Aydin, Y.; Chandra, P.K.; Dash, A.; Chedid, M.; Thung, S.N.; Moroz, K.; Wu, T.; Nayak, N.C.; et al. Chaperone-mediated autophagy compensates for impaired macroautophagy in the cirrhotic liver to promote hepatocellular carcinoma. Oncotarget 2017, 8, 40019–40036. [Google Scholar] [CrossRef]
- Aydin, Y.; Chatterjee, A.; Chandra, P.K.; Chava, S.; Chen, W.; Tandon, A.; Dash, A.; Chedid, M.; Moehlen, M.W.; Regenstein, F.; et al. Interferon-alpha-induced hepatitis C virus clearance restores p53 tumor suppressor more than direct-acting antivirals. Hepatol. Commun. 2017, 1, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Aydin, Y.; Chedid, M.; Chava, S.; Danielle Williams, D.; Liu, S.; Hagedorn, C.H.; Sumitran-Holgersson, S.; Reiss, K.; Moroz, K.; Lu, H.; et al. Activation of PERK-Nrf2 oncogenic signaling promotes Mdm2-mediated Rb degradation in persistently infected HCV culture. Sci. Rep. 2017, 7, 9223. [Google Scholar] [CrossRef] [PubMed]
- Aydin, Y.; Stephens, C.M.; Chava, S.; Heidari, Z.; Panigrahi, R.; Williams, D.D.; Wiltz, K.; Bell, A.; Wilson, W.; Reiss, K.; et al. Chaperone-Mediated Autophagy Promotes Beclin1 Degradation in Persistently Infected Hepatitis C Virus Cell Culture. Am. J. Pathol. 2018, 188, 2339–2355. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, A.; Perra, A.; Cora, D.; Sulas, P.; Menegon, S.; Manca, C.; Migliore, C.; Kowalik, M.A.; Ledda-Columbano, G.M.; Giordano, S.; et al. MicroRNA/gene profiling unveils early molecular changes and nuclear factor erythroid related factor 2 (NRF2) activation in a rat model recapitulating human hepatocellular carcinoma (HCC). Hepatology 2014, 59, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.F.; Wang, S.N.; Wu, C.F.; Yeh, Y.T.; Chai, C.Y.; Chunag, S.C.; Sheen, M.C.; Lee, K.T. Altered p-STAT3 (tyr705) expression is associated with histological grading and intratumour microvessel density in hepatocellular carcinoma. J. Clin. Pathol. 2007, 60, 642–648. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Yu, G.Y.; Temkin, V.; Ogata, H.; Kuntzen, C.; Sakurai, T.; Sieghart, W.; Peck-Radosavljevic, M.; Leffert, H.L.; Karin, M. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 2010, 17, 286–297. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Karin, M. NF-kappaB and STAT3—Key players in liver inflammation and cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef]
- Svinka, J.; Mikulits, W.; Eferl, R. STAT3 in hepatocellular carcinoma: New perspectives. Hepat. Oncol. 2014, 1, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Hatziapostolou, M.; Polytarchou, C.; Aggelidou, E.; Drakaki, A.; Poultsides, G.A.; Jaeger, S.A.; Ogata, H.; Karin, M.; Struhl, K.; Hadzopoulou-Cladaras, M.; et al. An HNF4alpha-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell 2011, 147, 1233–1247. [Google Scholar] [CrossRef]
- Aboulnasr, F.; Hazari, S.; Nayak, S.; Chandra, P.K.; Panigrahi, R.; Ferraris, P.; Chava, S.; Kurt, R.; Song, K.; Dash, A.; et al. IFN-lambda Inhibits MiR-122 Transcription through a Stat3-HNF4alpha Inflammatory Feedback Loop in an IFN-alpha Resistant HCV Cell Culture System. PLoS ONE 2015, 10, e0141655. [Google Scholar] [CrossRef]
- Lau, H.H.; Ng, N.H.J.; Loo, L.S.W.; Jasmen, J.B.; Teo, A.K.K. The molecular functions of hepatocyte nuclear factors—In and beyond the liver. J. Hepatol. 2018, 68, 1033–1048. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.Y.; Yin, C.; Hou, J.L.; Zeng, X.; Chen, Y.X.; Zhong, W.; Hu, P.F.; Deng, X.; Tan, Y.X.; Zhang, J.P.; et al. Hepatocyte nuclear factor 4alpha attenuates hepatic fibrosis in rats. Gut 2010, 59, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Lazarevich, N.L.; Cheremnova, O.A.; Varga, E.V.; Ovchinnikov, D.A.; Kudrjavtseva, E.I.; Morozova, O.V.; Fleishman, D.I.; Engelhardt, N.V.; Duncan, S.A. Progression of HCC in mice is associated with a downregulation in the expression of hepatocyte nuclear factors. Hepatology 2004, 39, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Lazarevich, N.L.; Shavochkina, D.A.; Fleishman, D.I.; Kustova, I.F.; Morozova, O.V.; Chuchuev, E.S.; Patyutko, Y.I. Deregulation of hepatocyte nuclear factor 4 (HNF4) as a marker of epithelial tumors progression. Exp. Oncol. 2010, 32, 167–171. [Google Scholar] [PubMed]
- Ning, B.F.; Ding, J.; Yin, C.; Zhong, W.; Wu, K.; Zeng, X.; Yang, W.; Chen, Y.X.; Zhang, J.P.; Zhang, X.; et al. Hepatocyte nuclear factor 4 alpha suppresses the development of hepatocellular carcinoma. Cancer Res. 2010, 70, 7640–7651. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Jiang, S.; Hotta, H.; Takano, K.; Iwanari, H.; Sumi, K.; Daigo, K.; Ohashi, R.; Sugai, M.; Ikegame, C.; et al. Dysregulated expression of P1 and P2 promoter-driven hepatocyte nuclear factor-4alpha in the pathogenesis of human cancer. J. Pathol. 2006, 208, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Lin, Y.; Zhang, X.; Chen, Y.X.; Zeng, X.; Yue, H.Y.; Hou, J.L.; Deng, X.; Zhang, J.P.; Han, Z.G.; et al. Differentiation therapy of hepatocellular carcinoma in mice with recombinant adenovirus carrying hepatocyte nuclear factor-4alpha gene. Hepatology 2008, 48, 1528–1539. [Google Scholar] [CrossRef]
- Li, C.; Deng, M.; Hu, J.; Li, X.; Chen, L.; Ju, Y.; Hao, J.; Meng, S. Chronic inflammation contributes to the development of hepatocellular carcinoma by decreasing miR-122 levels. Oncotarget 2016, 7, 17021–17034. [Google Scholar] [CrossRef]
- Yang, Y.M.; Lee, C.G.; Koo, J.H.; Kim, T.H.; Lee, J.M.; An, J.; Kim, K.M.; Kim, S.G. Galpha12 overexpressed in hepatocellular carcinoma reduces microRNA-122 expression via HNF4alpha inactivation, which causes c-Met induction. Oncotarget 2015, 6, 19055–19069. [Google Scholar] [CrossRef]
- Li, M.; Tang, Y.; Wu, L.; Mo, F.; Wang, X.; Li, H.; Qi, R.; Zhang, H.; Srivastava, A.; Ling, C. The hepatocyte-specific HNF4alpha/miR-122 pathway contributes to iron overload-mediated hepatic inflammation. Blood 2017, 130, 1041–1051. [Google Scholar] [CrossRef]
- Hsu, S.H.; Wang, B.; Kota, J.; Yu, J.; Costinean, S.; Kutay, H.; Yu, L.; Bai, S.; La Perle, K.; Chivukula, R.R.; et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J. Clin. Investig. 2012, 122, 2871–2883. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Hsu, S.D.; Hsu, C.S.; Lai, T.C.; Chen, S.J.; Shen, R.; Huang, Y.; Chen, H.C.; Lee, C.H.; Tsai, T.F.; et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J. Clin. Investig. 2012, 122, 2884–2897. [Google Scholar] [CrossRef] [PubMed]
- Takaki, Y.; Saito, Y.; Takasugi, A.; Toshimitsu, K.; Yamada, S.; Muramatsu, T.; Kimura, M.; Sugiyama, K.; Suzuki, H.; Arai, E.; et al. Silencing of microRNA-122 is an early event during hepatocarcinogenesis from non-alcoholic steatohepatitis. Cancer Sci. 2014, 105, 1254–1260. [Google Scholar] [CrossRef] [PubMed]
- Shim, S.H.; Xia, C.; Zhong, G.; Babcock, H.P.; Vaughan, J.C.; Huang, B.; Wang, X.; Xu, C.; Bi, G.Q.; Zhuang, X. Super-resolution fluorescence imaging of organelles in live cells with photoswitchable membrane probes. Proc. Natl. Acad. Sci. USA 2012, 109, 13978–13983. [Google Scholar] [CrossRef] [PubMed]
- Burban, A.; Sharanek, A.; Guguen-Guillouzo, C.; Guillouzo, A. Endoplasmic reticulum stress precedes oxidative stress in antibiotic-induced cholestasis and cytotoxicity in human hepatocytes. Free Radic. Biol. Med. 2018, 115, 166–178. [Google Scholar] [CrossRef]
- Beriault, D.R.; Werstuck, G.H. Detection and quantification of endoplasmic reticulum stress in living cells using the fluorescent compound, Thioflavin T. Biochim. Biophys. Acta 2013, 1833, 2293–2301. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Higuchi, K. Amyloid fibril proteins. Mech. Ageing Dev. 2002, 123, 1625–1636. [Google Scholar] [CrossRef]
- Tardif, K.D.; Waris, G.; Siddiqui, A. Hepatitis C virus, ER stress, and oxidative stress. Trends Microbiol. 2005, 13, 159–163. [Google Scholar] [CrossRef]
- Waris, G.; Turkson, J.; Hassanein, T.; Siddiqui, A. Hepatitis C virus (HCV) constitutively activates STAT-3 via oxidative stress: Role of STAT-3 in HCV replication. J. Virol. 2005, 79, 1569–1580. [Google Scholar] [CrossRef]
- Gong, G.; Waris, G.; Tanveer, R.; Siddiqui, A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc. Natl. Acad. Sci. USA 2001, 98, 9599–9604. [Google Scholar] [CrossRef]
- Joyce, M.A.; Walters, K.A.; Lamb, S.E.; Yeh, M.M.; Zhu, L.F.; Kneteman, N.; Doyle, J.S.; Katze, M.G.; Tyrrell, D.L. HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb-uPA mice. PLoS Pathog. 2009, 5, e1000291. [Google Scholar] [CrossRef] [PubMed]
- Burdette, D.; Olivarez, M.; Waris, G. Activation of transcription factor Nrf2 by hepatitis C virus induces the cell-survival pathway. J. Gen. Virol. 2010, 91, 681–690. [Google Scholar] [CrossRef]
- Xu, D.; Xu, M.; Jeong, S.; Qian, Y.; Wu, H.; Xia, Q.; Kong, X. The Role of Nrf2 in Liver Disease: Novel Molecular Mechanisms and Therapeutic Approaches. Front. Pharmacol. 2018, 9, 1428. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Motohashi, H.; Yamamoto, M. The Keap1-Nrf2 system in cancers: Stress response and anabolic metabolism. Front. Oncol. 2012, 2, 200. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef]
- Vallianou, I.; Dafou, D.; Vassilaki, N.; Mavromara, P.; Hadzopoulou-Cladaras, M. Hepatitis C virus suppresses Hepatocyte Nuclear Factor 4 alpha, a key regulator of hepatocellular carcinoma. Int. J. Biochem. Cell Biol. 2016, 78, 315–326. [Google Scholar] [CrossRef]
- Li, Z.Y.; Xi, Y.; Zhu, W.N.; Zeng, C.; Zhang, Z.Q.; Guo, Z.C.; Hao, D.L.; Liu, G.; Feng, L.; Chen, H.Z.; et al. Positive regulation of hepatic miR-122 expression by HNF4alpha. J. Hepatol. 2011, 55, 602–611. [Google Scholar] [CrossRef]
- Zeng, C.; Wang, R.; Li, D.; Lin, X.J.; Wei, Q.K.; Yuan, Y.; Wang, Q.; Chen, W.; Zhuang, S.M. A novel GSK-3 beta-C/EBP alpha-miR-122-insulin-like growth factor 1 receptor regulatory circuitry in human hepatocellular carcinoma. Hepatology 2010, 52, 1702–1712. [Google Scholar] [CrossRef]
- Kanda, T.; Goto, T.; Hirotsu, Y.; Moriyama, M.; Omata, M. Molecular Mechanisms Driving Progression of Liver Cirrhosis towards Hepatocellular Carcinoma in Chronic Hepatitis B and C Infections: A Review. Int. J. Mol. Sci. 2019, 20, 1358. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Sandhu, S.; Lai, J.P.; Sandhu, D.S. Hepatocellular carcinoma in non-cirrhotic liver: A comprehensive review. World J. Hepatol. 2019, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Stine, J.G.; Wentworth, B.J.; Zimmet, A.; Rinella, M.E.; Loomba, R.; Caldwell, S.H.; Argo, C.K. Systematic review with meta-analysis: Risk of hepatocellular carcinoma in non-alcoholic steatohepatitis without cirrhosis compared to other liver diseases. Aliment. Pharmacol. Ther. 2018, 48, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Chandra, P.K.; Gunduz, F.; Hazari, S.; Kurt, R.; Panigrahi, R.; Poat, B.; Bruce, D.; Cohen, A.J.; Bohorquez, H.E.; Carmody, I.; et al. Impaired expression of type I and type II interferon receptors in HCV-associated chronic liver disease and liver cirrhosis. PLoS ONE 2014, 9, e108616. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Conner, E.A.; Lee, J.S.; Factor, V.M.; Thorgeirsson, S.S. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 2006, 130, 1117–1128. [Google Scholar] [CrossRef]
- Shen, S.; Niso-Santano, M.; Adjemian, S.; Takehara, T.; Malik, S.A.; Minoux, H.; Souquere, S.; Marino, G.; Lachkar, S.; Senovilla, L.; et al. Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol. Cell 2012, 48, 667–680. [Google Scholar] [CrossRef]
- Tacke, R.S.; Tosello-Trampont, A.; Nguyen, V.; Mullins, D.W.; Hahn, Y.S. Extracellular hepatitis C virus core protein activates STAT3 in human monocytes/macrophages/dendritic cells via an IL-6 autocrine pathway. J. Biol. Chem. 2011, 286, 10847–10855. [Google Scholar] [CrossRef]
- Kyrmizi, I.; Hatzis, P.; Katrakili, N.; Tronche, F.; Gonzalez, F.J.; Talianidis, I. Plasticity and expanding complexity of the hepatic transcription factor network during liver development. Genes Dev. 2006, 20, 2293–2305. [Google Scholar] [CrossRef]
- Odom, D.T.; Zizlsperger, N.; Gordon, D.B.; Bell, G.W.; Rinaldi, N.J.; Murray, H.L.; Volkert, T.L.; Schreiber, J.; Rolfe, P.A.; Gifford, D.K.; et al. Control of pancreas and liver gene expression by HNF transcription factors. Science 2004, 303, 1378–1381. [Google Scholar] [CrossRef]
- Guzman-Lepe, J.; Cervantes-Alvarez, E.; Collin de l’Hortet, A.; Wang, Y.; Mars, W.M.; Oda, Y.; Bekki, Y.; Shimokawa, M.; Wang, H.; Yoshizumi, T.; et al. Liver-enriched transcription factor expression relates to chronic hepatic failure in humans. Hepatol. Commun. 2018, 2, 582–594. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yannam, G.R.; Nishikawa, T.; Yamamoto, T.; Basma, H.; Ito, R.; Nagaya, M.; Dutta-Moscato, J.; Stolz, D.B.; Duan, F.; et al. The microenvironment in hepatocyte regeneration and function in rats with advanced cirrhosis. Hepatology 2012, 55, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Safdar, H.; Cheung, K.L.; Vos, H.L.; Gonzalez, F.J.; Reitsma, P.H.; Inoue, Y.; van Vlijmen, B.J. Modulation of mouse coagulation gene transcription following acute in vivo delivery of synthetic small interfering RNAs targeting HNF4alpha and C/EBPalpha. PLoS ONE 2012, 7, e38104. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.W.; Kudaravalli, R.; Russell, T.T.; DiMichele, D.M.; Gibb, C.; Russell, J.E.; Margaritis, P.; Pollak, E.S. Mutation in the factor VII hepatocyte nuclear factor 4alpha-binding site contributes to factor VII deficiency. Blood Coagul. Fibrinolysis 2011, 22, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Peters, L.L.; Yim, S.H.; Inoue, J.; Gonzalez, F.J. Role of hepatocyte nuclear factor 4alpha in control of blood coagulation factor gene expression. J. Mol. Med. 2006, 84, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Bandiera, S.; Pfeffer, S.; Baumert, T.F.; Zeisel, M.B. miR-122-a key factor and therapeutic target in liver disease. J. Hepatol. 2015, 62, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Trebicka, J.; Anadol, E.; Elfimova, N.; Strack, I.; Roggendorf, M.; Viazov, S.; Wedemeyer, I.; Drebber, U.; Rockstroh, J.; Sauerbruch, T.; et al. Hepatic and serum levels of miR-122 after chronic HCV-induced fibrosis. J. Hepatol. 2013, 58, 234–239. [Google Scholar] [CrossRef]
- Cermelli, S.; Ruggieri, A.; Marrero, J.A.; Ioannou, G.N.; Beretta, L. Circulating microRNAs in patients with chronic hepatitis C and non-alcoholic fatty liver disease. PLoS ONE 2011, 6, e23937. [Google Scholar] [CrossRef]
- Sarasin-Filipowicz, M.; Krol, J.; Markiewicz, I.; Heim, M.H.; Filipowicz, W. Decreased levels of microRNA miR-122 in individuals with hepatitis C responding poorly to interferon therapy. Nat. Med. 2009, 15, 31–33. [Google Scholar] [CrossRef]
- Hamdane, N.; Juhling, F.; Crouchet, E.; El Saghire, H.; Thumann, C.; Oudot, M.A.; Bandiera, S.; Saviano, A.; Ponsolles, C.; Roca Suarez, A.A.; et al. HCV-Induced Epigenetic Changes Associated with Liver Cancer Risk Persist After Sustained Virologic Response. Gastroenterology 2019, 156, 2313–2329. [Google Scholar] [CrossRef]
- Perez, S.; Kaspi, A.; Domovitz, T.; Davidovich, A.; Lavi-Itzkovitz, A.; Meirson, T.; Alison Holmes, J.; Dai, C.Y.; Huang, C.F.; Chung, R.T.; et al. Hepatitis C virus leaves an epigenetic signature post cure of infection by direct-acting antivirals. PLoS Genet. 2019, 15, e1008181. [Google Scholar] [CrossRef] [PubMed]
- El-Araby, R.E.; Khalifa, M.A.; Zoheiry, M.M.; Zahran, M.Y.; Rady, M.I.; Ibrahim, R.A.; El-Talkawy, M.D.; Essawy, F.M. The interaction between microRNA-152 and DNA methyltransferase-1 as an epigenetic prognostic biomarker in HCV-induced liver cirrhosis and HCC patients. Cancer Gene Ther. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chandra, P.K.; Bao, L.; Song, K.; Aboulnasr, F.M.; Baker, D.P.; Shores, N.; Wimley, W.C.; Liu, S.; Hagedorn, C.H.; Fuchs, S.Y.; et al. HCV infection selectively impairs type I but not type III IFN signaling. Am. J. Pathol. 2014, 184, 214–229. [Google Scholar] [CrossRef] [PubMed]
- Florczyk, U.; Czauderna, S.; Stachurska, A.; Tertil, M.; Nowak, W.; Kozakowska, M.; Poellinger, L.; Jozkowicz, A.; Loboda, A.; Dulak, J. Opposite effects of HIF-1alpha and HIF-2alpha on the regulation of IL-8 expression in endothelial cells. Free Radic. Biol. Med. 2011, 51, 1882–1892. [Google Scholar] [CrossRef]
- Son, Y.O.; Pratheeshkumar, P.; Roy, R.V.; Hitron, J.A.; Wang, L.; Zhang, Z.; Shi, X. Nrf2/p62 signaling in apoptosis resistance and its role in cadmium-induced carcinogenesis. J. Biol. Chem. 2014, 289, 28660–28675. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Xu, S.J.; Xie, S.J.; Zhang, Y.; Yang, J.H.; Zhang, W.Q.; Zheng, M.N.; Zhou, H.; Qu, L.H. MicroRNA-122 supports robust innate immunity in hepatocytes by targeting the RTKs/STAT3 signaling pathway. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Van Renne, N.; Suarez, A.A.R.; Duong, F.H.T.; Gondeau, C.; Calabrese, D.; Fontaine, N.; Ababsa, A.; Bandiera, S.; Croonenborghs, T.; Pochet, N.; et al. miR-135a-5p-mediated downregulation of protein tyrosine phosphatase receptor delta is a candidate driver of HCV-associated hepatocarcinogenesis. Gut 2018, 67, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.N.; Shen, A.; Rider, P.J.; Yu, Y.; Wu, K.L.; Mu, Y.X.; Hao, Q.; Liu, Y.L.; Gong, H.; Zhu, Y.; et al. A liver-specific microRNA binds to a highly conserved RNA sequence of hepatitis B virus and negatively regulates viral gene expression and replication. FASEB J. 2011, 25, 4511–4521. [Google Scholar] [CrossRef] [PubMed]
- Luna, J.M.; Scheel, T.K.H.; Danino, T.; Shaw, K.S.; Mele, A.; Fak, J.J.; Nishiuchi, E.; Takacs, C.N.; Catanese, M.T.; de Jong, Y.P.; et al. Hepatitis C Virus RNA Functionally Sequesters miR-122. Cell 2015, 160, 1099–1110. [Google Scholar] [CrossRef]
- Hyrina, A.; Olmstead, A.D.; Steven, P.; Krajden, M.; Tam, E.; Jean, F. Treatment-Induced Viral Cure of Hepatitis C Virus-Infected Patients Involves a Dynamic Interplay among three Important Molecular Players in Lipid Homeostasis: Circulating microRNA (miR)-24, miR-223, and Proprotein Convertase Subtilisin/Kexin Type 9. Ebiomedicine 2017, 23, 68–78. [Google Scholar] [CrossRef]
- Jopling, C.L.; Yi, M.K.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Mylroie, H.; Dumont, O.; Bauer, A.; Thornton, C.C.; Mackey, J.; Calay, D.; Hamdulay, S.S.; Choo, J.R.; Boyle, J.J.; Samarel, A.M.; et al. PKC epsilon-CREB-Nrf2 signalling induces HO-1 in the vascular endothelium and enhances resistance to inflammation and apoptosis. Cardiovasc. Res. 2015, 106, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.W.; Guo, R.F.; Speyer, C.L.; Reuben, J.; Neff, T.A.; Hoesel, L.M.; Riedemann, N.C.; McClintock, S.A.; Sarma, J.V.; Van Rooijen, N.; et al. Stat3 activation in acute lung injury. J. Immunol. 2004, 172, 7703–7712. [Google Scholar] [CrossRef] [PubMed]
- Tawani, A.; Amanullah, A.; Mishra, A.; Kumar, A. Evidences for Piperine inhibiting cancer by targeting human G-quadruplex DNA sequences. Sci. Rep. 2016, 6, 39239. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Number | Age | Sex | HCV RNA IU/mL | HCV Genotype | Metavir Score |
|---|---|---|---|---|---|
| 1 | 48 | F | 2,000,000 | 1a | 3 |
| 2 | 46 | F | 29,000 | 1b | 0 |
| 3 | 27 | F | 1,000,000 | 1a | 1.2 |
| 4 | 59 | F | 553,000 | 1a | 3 |
| 5 | 64 | M | 2,000,000 | 1a | 1.2 |
| 6 | 34 | F | 7,000,000 | 1a | 1,2 |
| 7 | 59 | F | 1,000,000 | 2 | 0 |
| 8 | 44 | M | 170,000 | 1a | 1 |
| 9 | 50 | M | 3,000,000 | 1b | 0 |
| 10 | 33 | F | 3,000,000 | 1b | 1.2 |
| 11 | 53 | M | 904,000 | 1a | 1 |
| 12 | 62 | M | 7062 | 1a | 0 |
| 13 | 69 | F | 10,000,000 | 1b | 2 |
| 14 | 47 | F | 2,000,000 | 2 | 0 |
| 15 | 50 | F | 10,000,000 | 1a | 0 |
| 16 | 48 | M | 2,000,000 | 1b | 0 |
| 17 | 40 | F | 200,000 | 1a | 1 |
| 18 | 53 | M | 815,000 | 1a | 1 |
| Patient Number | Age | Sex | HCV RNA IU/mL | HCV Genotype | Metavir Score |
|---|---|---|---|---|---|
| 1 | 57 | M | + | 1a | 4 |
| 2 | 52 | M | + | 1a | 4 |
| 3 | 60 | F | + | 1a | 4 |
| 4 | 64 | F | + | N/A | 4 |
| 5 | 62 | M | 2,700,000 | 1b | 4 |
| 6 | 66 | M | + | 1a | 4 |
| 7 | 54 | M | + | 1a | 4 |
| 8 | 54 | M | + | 1b | 4 |
| 9 | 62 | F | + | N/A | 4 |
| 10 | 58 | F | + | N/A | 4 |
| 11 | 64 | M | + | 2 | 4 |
| 12 | 66 | F | + | N/A | 4 |
| 13 | 57 | F | + | N/A | 4 |
| 14 | 56 | M | 5,940,000 | N/A | 4 |
| 15 | 60 | M | 7,200,000 | 1a | 4 |
| 16 | 60 | M | 2,860,000 | 1b | 4 |
| 17 | 56 | M | 4,310,000 | 1a | 4 |
| 18 | 60 | M | 6,480,000 | 1a | 4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aydin, Y.; Kurt, R.; Song, K.; Lin, D.; Osman, H.; Youngquist, B.; Scott, J.W.; Shores, N.J.; Thevenot, P.; Cohen, A.; et al. Hepatic Stress Response in HCV Infection Promotes STAT3-Mediated Inhibition of HNF4A-miR-122 Feedback Loop in Liver Fibrosis and Cancer Progression. Cancers 2019, 11, 1407. https://doi.org/10.3390/cancers11101407
Aydin Y, Kurt R, Song K, Lin D, Osman H, Youngquist B, Scott JW, Shores NJ, Thevenot P, Cohen A, et al. Hepatic Stress Response in HCV Infection Promotes STAT3-Mediated Inhibition of HNF4A-miR-122 Feedback Loop in Liver Fibrosis and Cancer Progression. Cancers. 2019; 11(10):1407. https://doi.org/10.3390/cancers11101407
Chicago/Turabian StyleAydin, Yucel, Ramazan Kurt, Kyoungsub Song, Dong Lin, Hanadi Osman, Brady Youngquist, John W. Scott, Nathan J. Shores, Paul Thevenot, Ari Cohen, and et al. 2019. "Hepatic Stress Response in HCV Infection Promotes STAT3-Mediated Inhibition of HNF4A-miR-122 Feedback Loop in Liver Fibrosis and Cancer Progression" Cancers 11, no. 10: 1407. https://doi.org/10.3390/cancers11101407
APA StyleAydin, Y., Kurt, R., Song, K., Lin, D., Osman, H., Youngquist, B., Scott, J. W., Shores, N. J., Thevenot, P., Cohen, A., & Dash, S. (2019). Hepatic Stress Response in HCV Infection Promotes STAT3-Mediated Inhibition of HNF4A-miR-122 Feedback Loop in Liver Fibrosis and Cancer Progression. Cancers, 11(10), 1407. https://doi.org/10.3390/cancers11101407