Regulators of Oncogenic Mutant TP53 Gain of Function

Abstract
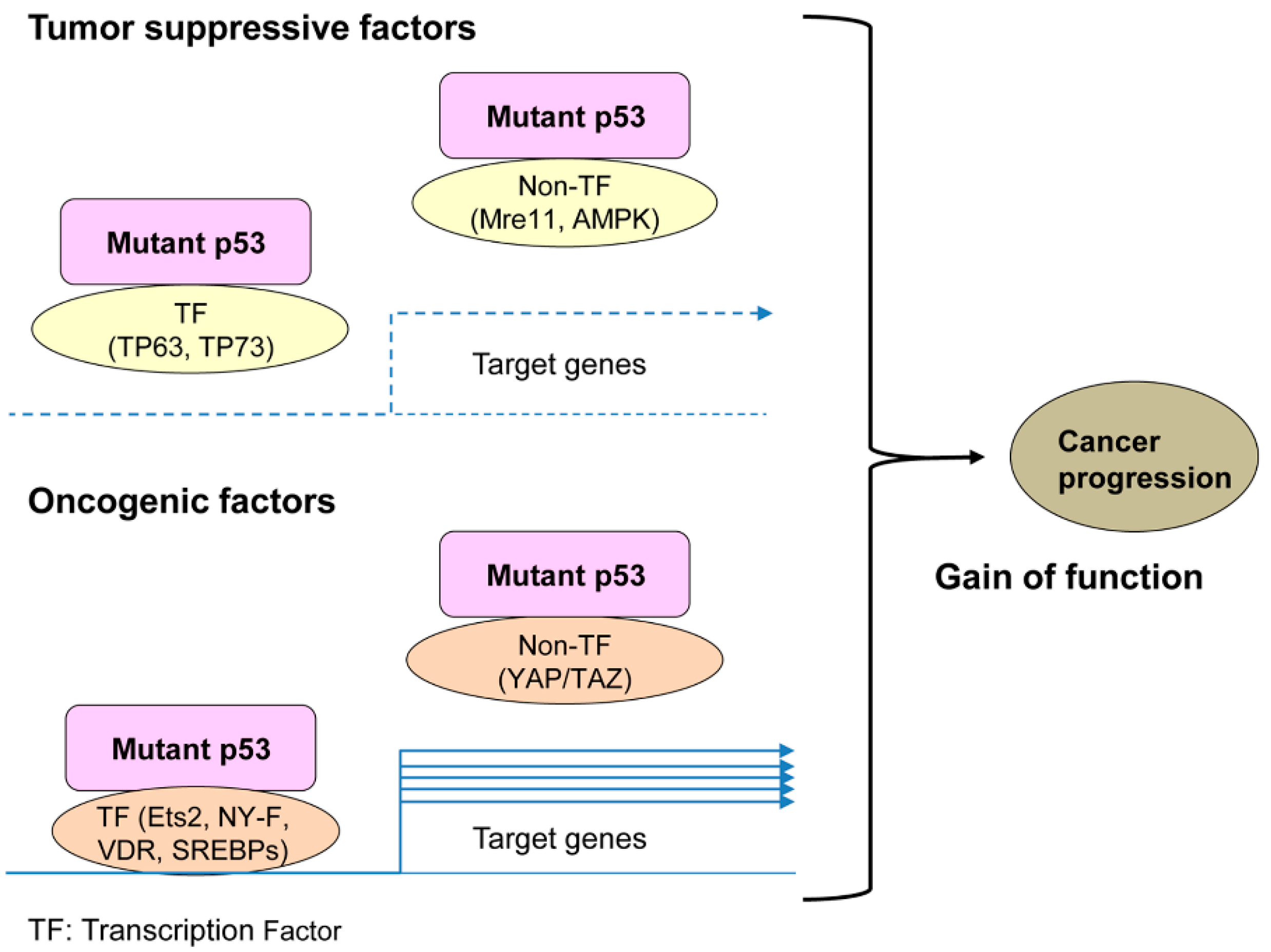
1. Introduction
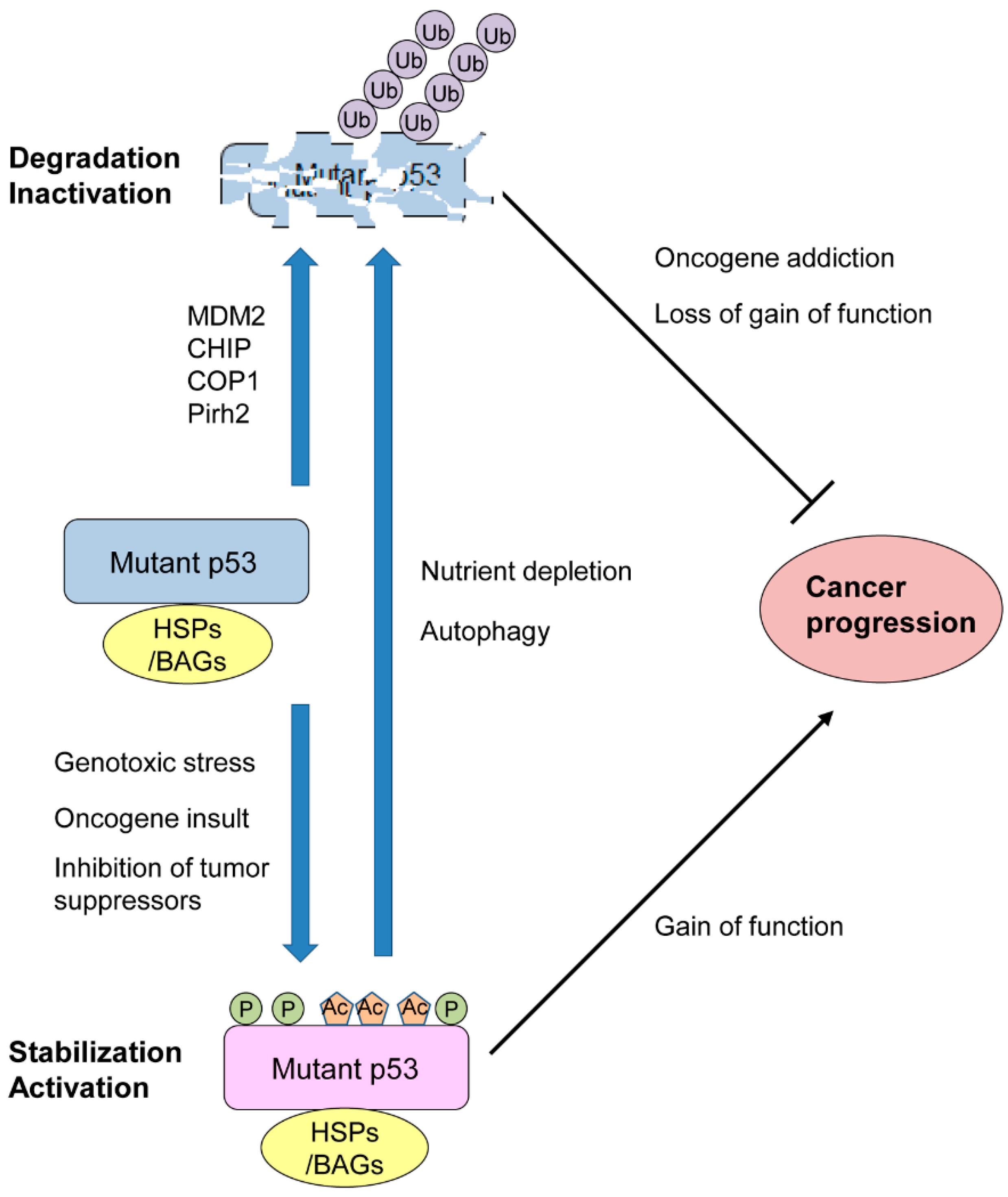
2. Mutp53 Stability and Activity Are Altered by Stress and Chemical Compounds
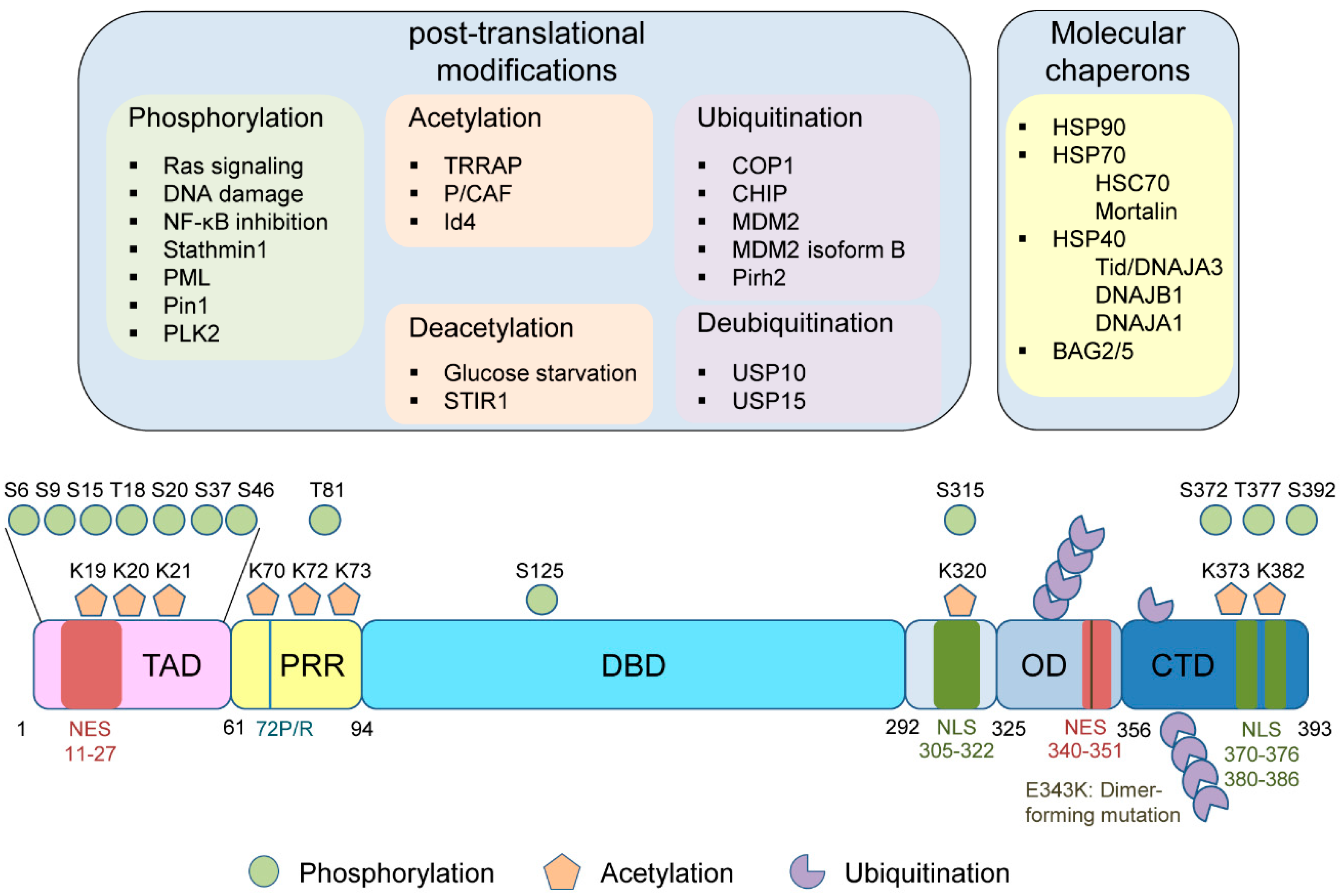
3. PMTs of Mutp53
3.1. Phosphorylation
3.2. Acetylation
3.3. Ubiquitination
4. Molecular Chaperones
5. SNP at Codon 72 in Mutp53
6. Dimer-Forming Mutp53
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T.; Beroud, C. Assessing TP53 status in human tumours to evaluate clinical outcome. Nat. Rev. Cancer 2001, 1, 233–240. [Google Scholar] [CrossRef]
- Powell, B.; Soong, R.; Iacopetta, B.; Seshadri, R.; Smith, D.R. Prognostic significance of mutations to different structural and functional regions of the p53 gene in breast cancer. Clin. Cancer Res. 2000, 6, 443–451. [Google Scholar] [PubMed]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Iwakuma, T. p53 as a Regulator of Lipid Metabolism in Cancer. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef]
- Kern, S.E.; Kinzler, K.W.; Baker, S.J.; Nigro, J.M.; Rotter, V.; Levine, A.J.; Friedman, P.; Prives, C.; Vogelstein, B. Mutant p53 proteins bind DNA abnormally in vitro. Oncogene 1991, 6, 131–136. [Google Scholar]
- Brazdova, M.; Navratilova, L.; Tichy, V.; Nemcova, K.; Lexa, M.; Hrstka, R.; Pecinka, P.; Adamik, M.; Vojtesek, B.; Palecek, E.; et al. Preferential binding of hot spot mutant p53 proteins to supercoiled DNA in vitro and in cells. PLoS ONE 2013, 8, e59567. [Google Scholar] [CrossRef]
- Adhikari, A.S.; Iwakuma, T. Mutant p53 gain of oncogenic function: In vivo evidence, mechanism of action and its clinical implications. Fukuoka Igaku Zasshi 2009, 100, 217–228. [Google Scholar]
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2018, 25, 161–168. [Google Scholar] [CrossRef]
- Sabapathy, K. The Contrived Mutant p53 Oncogene—Beyond Loss of Functions. Front. Oncol. 2015, 5, 276. [Google Scholar] [CrossRef]
- Stambolsky, P.; Tabach, Y.; Fontemaggi, G.; Weisz, L.; Maor-Aloni, R.; Siegfried, Z.; Shiff, I.; Kogan, I.; Shay, M.; Kalo, E.; et al. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell 2010, 17, 273–285. [Google Scholar] [CrossRef]
- Kim, M.P.; Zhang, Y.; Lozano, G. Mutant p53: Multiple Mechanisms Define Biologic Activity in Cancer. Front. Oncol. 2015, 5, 249. [Google Scholar] [CrossRef]
- Goh, A.M.; Coffill, C.R.; Lane, D.P. The role of mutant p53 in human cancer. J. Pathol. 2011, 223, 116–126. [Google Scholar] [CrossRef]
- Gualberto, A.; Aldape, K.; Kozakiewicz, K.; Tlsty, T.D. An oncogenic form of p53 confers a dominant, gain-of-function phenotype that disrupts spindle checkpoint control. Proc. Natl. Acad. Sci. USA 1998, 95, 5166–5171. [Google Scholar] [CrossRef]
- Blandino, G.; Levine, A.J.; Oren, M. Mutant p53 gain of function: Differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene 1999, 18, 477–485. [Google Scholar] [CrossRef]
- Weisz, L.; Oren, M.; Rotter, V. Transcription regulation by mutant p53. Oncogene 2007, 26, 2202–2211. [Google Scholar] [CrossRef]
- Di Agostino, S.; Strano, S.; Emiliozzi, V.; Zerbini, V.; Mottolese, M.; Sacchi, A.; Blandino, G.; Piaggio, G. Gain of function of mutant p53: The mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 2006, 10, 191–202. [Google Scholar] [CrossRef]
- Zalcenstein, A.; Stambolsky, P.; Weisz, L.; Muller, M.; Wallach, D.; Goncharov, T.M.; Krammer, P.H.; Rotter, V.; Oren, M. Mutant p53 gain of function: Repression of CD95(Fas/APO-1) gene expression by tumor-associated p53 mutants. Oncogene 2003, 22, 5667–5676. [Google Scholar] [CrossRef]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef]
- Terzian, T.; Suh, Y.A.; Iwakuma, T.; Post, S.M.; Neumann, M.; Lang, G.A.; Van Pelt, C.S.; Lozano, G. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008, 22, 1337–1344. [Google Scholar] [CrossRef]
- Iwakuma, T.; Lozano, G.; Flores, E.R. Li-Fraumeni syndrome: A p53 family affair. Cell Cycle 2005, 4, 865–867. [Google Scholar] [CrossRef]
- Suh, Y.A.; Post, S.M.; Elizondo-Fraire, A.C.; Maccio, D.R.; Jackson, J.G.; El-Naggar, A.K.; Van Pelt, C.; Terzian, T.; Lozano, G. Multiple stress signals activate mutant p53 in vivo. Cancer Res. 2011, 71, 7168–7175. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. Structural biology of the tumor suppressor p53 and cancer-associated mutants. Adv. Cancer Res. 2007, 97, 1–23. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef]
- Jenkins, J.R.; Rudge, K.; Chumakov, P.; Currie, G.A. The cellular oncogene p53 can be activated by mutagenesis. Nature 1985, 317, 816–818. [Google Scholar] [CrossRef]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef]
- Maan, M.; Pati, U. CHIP promotes autophagy-mediated degradation of aggregating mutant p53 in hypoxic conditions. FEBS J. 2018. [Google Scholar] [CrossRef]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef]
- Dibra, D.; Mitra, A.; Newman, M.; Xia, X.; Cutrera, J.J.; Gagea, M.; Kleinerman, E.S.; Lozano, G.; Li, S. Lack of Immunomodulatory Interleukin-27 Enhances Oncogenic Properties of Mutant p53 In Vivo. Clin. Cancer Res. 2016, 22, 3876–3883. [Google Scholar] [CrossRef]
- Haupt, S.; Mitchell, C.; Corneille, V.; Shortt, J.; Fox, S.; Pandolfi, P.P.; Castillo-Martin, M.; Bonal, D.M.; Cordon-Cardo, C.; Lozano, G.; et al. Loss of PML cooperates with mutant p53 to drive more aggressive cancers in a gender-dependent manner. Cell Cycle 2013, 12, 1722–1731. [Google Scholar] [CrossRef]
- Yallowitz, A.R.; Li, D.; Lobko, A.; Mott, D.; Nemajerova, A.; Marchenko, N. Mutant p53 Amplifies Epidermal Growth Factor Receptor Family Signaling to Promote Mammary Tumorigenesis. Mol. Cancer Res. MCR 2015, 13, 743–754. [Google Scholar] [CrossRef]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.G.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef]
- Rodriguez, O.C.; Choudhury, S.; Kolukula, V.; Vietsch, E.E.; Catania, J.; Preet, A.; Reynoso, K.; Bargonetti, J.; Wellstein, A.; Albanese, C.; et al. Dietary downregulation of mutant p53 levels via glucose restriction: Mechanisms and implications for tumor therapy. Cell Cycle 2012, 11, 4436–4446. [Google Scholar] [CrossRef]
- Choudhury, S.; Kolukula, V.K.; Preet, A.; Albanese, C.; Avantaggiati, M.L. Dissecting the pathways that destabilize mutant p53: The proteasome or autophagy? Cell Cycle 2013, 12, 1022–1029. [Google Scholar] [CrossRef]
- Halasi, M.; Pandit, B.; Gartel, A.L. Proteasome inhibitors suppress the protein expression of mutant p53. Cell Cycle 2014, 13, 3202–3206. [Google Scholar] [CrossRef]
- Parrales, A.; Iwakuma, T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front. Oncol. 2015, 5, 288. [Google Scholar] [CrossRef]
- Jung, C.L.; Mun, H.; Jo, S.Y.; Oh, J.H.; Lee, C.; Choi, E.K.; Jang, S.J.; Suh, Y.A. Suppression of gain-of-function mutant p53 with metabolic inhibitors reduces tumor growth in vivo. Oncotarget 2016, 7, 77664–77682. [Google Scholar] [CrossRef]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef] [PubMed]
- Boeckler, F.M.; Joerger, A.C.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.B.; Fersht, A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Tsai, K.Y.; Crowley, D.; Sengupta, S.; Yang, A.; McKeon, F.; Jacks, T. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 2002, 416, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Borresen-Dale, A.L.; Johnsen, H.; Aas, T.; Geisler, J.; Akslen, L.A.; Anker, G.; Lonning, P.E. TP53 gene mutations predict the response to neoadjuvant treatment with 5-fluorouracil and mitomycin in locally advanced breast cancer. Clin. Cancer Res. 2003, 9, 5582–5588. [Google Scholar] [PubMed]
- Geisler, S.; Lonning, P.E.; Aas, T.; Johnsen, H.; Fluge, O.; Haugen, D.F.; Lillehaug, J.R.; Akslen, L.A.; Borresen-Dale, A.L. Influence of TP53 gene alterations and c-erbB-2 expression on the response to treatment with doxorubicin in locally advanced breast cancer. Cancer Res. 2001, 61, 2505–2512. [Google Scholar] [PubMed]
- Do, P.M.; Varanasi, L.; Fan, S.; Li, C.; Kubacka, I.; Newman, V.; Chauhan, K.; Daniels, S.R.; Boccetta, M.; Garrett, M.R.; et al. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev. 2012, 26, 830–845. [Google Scholar] [CrossRef]
- Fiorini, C.; Cordani, M.; Padroni, C.; Blandino, G.; Di Agostino, S.; Donadelli, M. Mutant p53 stimulates chemoresistance of pancreatic adenocarcinoma cells to gemcitabine. Biochim. Biophys. Acta 2015, 1853, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.V.; Parrales, A.; Begani, P.; Narkar, A.; Adhikari, A.S.; Martinez, L.A.; Iwakuma, T. Allele-specific silencing of mutant p53 attenuates dominant-negative and gain-of-function activities. Oncotarget 2016, 7, 5401–5415. [Google Scholar] [CrossRef]
- Schulz-Heddergott, R.; Stark, N.; Edmunds, S.J.; Li, J.; Conradi, L.C.; Bohnenberger, H.; Ceteci, F.; Greten, F.R.; Dobbelstein, M.; Moll, U.M. Therapeutic Ablation of Gain-of-Function Mutant p53 in Colorectal Cancer Inhibits Stat3-Mediated Tumor Growth and Invasion. Cancer Cell 2018, 34, 298–314.e7. [Google Scholar] [CrossRef]
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D.A.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 2015, 523, 352–356. [Google Scholar] [CrossRef]
- Ingallina, E.; Sorrentino, G.; Bertolio, R.; Lisek, K.; Zannini, A.; Azzolin, L.; Severino, L.U.; Scaini, D.; Mano, M.; Mantovani, F.; et al. Mechanical cues control mutant p53 stability through a mevalonate-RhoA axis. Nat. Cell Biol. 2018, 20, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Wiman, K.G. Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett. 2014, 588, 2622–2627. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Blanden, A.R.; Yu, X.; Loh, S.N.; Levine, A.J.; Carpizo, D.R. Reactivating mutant p53 using small molecules as zinc metallochaperones: Awakening a sleeping giant in cancer. Drug Discov. Today 2015, 20, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Cheok, C.F.; Verma, C.S.; Lane, D.P. Reactivation of p53: From peptides to small molecules. Trends Pharmacol. Sci. 2011, 32, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Weinmann, L.; Wischhusen, J.; Demma, M.J.; Naumann, U.; Roth, P.; Dasmahapatra, B.; Weller, M. A novel p53 rescue compound induces p53-dependent growth arrest and sensitises glioma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 2008, 15, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Hiraki, M.; Hwang, S.Y.; Cao, S.; Ramadhar, T.R.; Byun, S.; Yoon, K.W.; Lee, J.H.; Chu, K.; Gurkar, A.U.; Kolev, V.; et al. Small-Molecule Reactivation of Mutant p53 to Wild-Type-like p53 through the p53-Hsp40 Regulatory Axis. Chem. Biol. 2015, 22, 1206–1216. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Lane, D.; Levine, A. p53 Research: The past thirty years and the next thirty years. Cold Spring Harb. Perspect. Biol. 2010, 2, a000893. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef]
- Dai, C.; Gu, W. p53 post-translational modification: Deregulated in tumorigenesis. Trends Mol. Med. 2010, 16, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.A.; Menendez, D.; Resnick, M.A.; Anderson, C.W. Mutant TP53 posttranslational modifications: Challenges and opportunities. Hum. Mutat. 2014, 35, 738–755. [Google Scholar] [CrossRef] [PubMed]
- Wesierska-Gadek, J.; Bugajska-Schretter, A.; Cerni, C. ADP-ribosylation of p53 tumor suppressor protein: Mutant but not wild-type p53 is modified. J. Cell. Biochem. 1996, 62, 90–101. [Google Scholar] [CrossRef]
- Bykov, V.J.; Lambert, J.M.; Hainaut, P.; Wiman, K.G. Mutant p53 rescue and modulation of p53 redox state. Cell Cycle 2009, 8, 2509–2517. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Kamada, R.; Ito, I.; Chuman, Y.; Shimohigashi, Y.; Sakaguchi, K. Oxidation of methionine residue at hydrophobic core destabilizes p53 tetrameric structure. Biopolymers 2009, 91, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Kaar, J.L.; Basse, N.; Joerger, A.C.; Stephens, E.; Rutherford, T.J.; Fersht, A.R. Stabilization of mutant p53 via alkylation of cysteines and effects on DNA binding. Protein Sci. 2010, 19, 2267–2278. [Google Scholar] [CrossRef]
- Yakovlev, V.A.; Bayden, A.S.; Graves, P.R.; Kellogg, G.E.; Mikkelsen, R.B. Nitration of the tumor suppressor protein p53 at tyrosine 327 promotes p53 oligomerization and activation. Biochemistry 2010, 49, 5331–5339. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A. Phosphatases reverse p53-mediated cell cycle checkpoints. Proc. Natl. Acad. Sci. USA 2014, 111, 7172–7173. [Google Scholar] [CrossRef]
- Liu, C.; Zhu, Y.; Lou, W.; Nadiminty, N.; Chen, X.; Zhou, Q.; Shi, X.B.; deVere White, R.W.; Gao, A.C. Functional p53 determines docetaxel sensitivity in prostate cancer cells. Prostate 2013, 73, 418–427. [Google Scholar] [CrossRef]
- Matsumoto, M.; Furihata, M.; Ohtsuki, Y. Posttranslational phosphorylation of mutant p53 protein in tumor development. Med. Mol. Morphol. 2006, 39, 79–87. [Google Scholar] [CrossRef]
- Ullrich, S.J.; Sakaguchi, K.; Lees-Miller, S.P.; Fiscella, M.; Mercer, W.E.; Anderson, C.W.; Appella, E. Phosphorylation at Ser-15 and Ser-392 in mutant p53 molecules from human tumors is altered compared to wild-type p53. Proc. Natl. Acad. Sci. USA 1993, 90, 5954–5958. [Google Scholar] [CrossRef] [PubMed]
- Minamoto, T.; Buschmann, T.; Habelhah, H.; Matusevich, E.; Tahara, H.; Boerresen-Dale, A.L.; Harris, C.; Sidransky, D.; Ronai, Z. Distinct pattern of p53 phosphorylation in human tumors. Oncogene 2001, 20, 3341–3347. [Google Scholar] [CrossRef] [PubMed]
- Adorno, M.; Cordenonsi, M.; Montagner, M.; Dupont, S.; Wong, C.; Hann, B.; Solari, A.; Bobisse, S.; Rondina, M.B.; Guzzardo, V.; et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 2009, 137, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Hollstein, M.; Xu, Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat. Cell Biol. 2007, 9, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Zerbini, L.F.; Wang, Y.; Correa, R.G.; Cho, J.Y.; Libermann, T.A. Blockage of NF-kappaB induces serine 15 phosphorylation of mutant p53 by JNK kinase in prostate cancer cells. Cell Cycle 2005, 4, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Sonego, M.; Schiappacassi, M.; Lovisa, S.; Dall’Acqua, A.; Bagnoli, M.; Lovat, F.; Libra, M.; D’Andrea, S.; Canzonieri, V.; Militello, L.; et al. Stathmin regulates mutant p53 stability and transcriptional activity in ovarian cancer. EMBO Mol. Med. 2013, 5, 707–722. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; di Agostino, S.; Mizrahi, I.; Alsheich-Bartok, O.; Voorhoeve, M.; Damalas, A.; Blandino, G.; Haupt, Y. Promyelocytic leukemia protein is required for gain of function by mutant p53. Cancer Res. 2009, 69, 4818–4826. [Google Scholar] [CrossRef] [PubMed]
- Girardini, J.E.; Napoli, M.; Piazza, S.; Rustighi, A.; Marotta, C.; Radaelli, E.; Capaci, V.; Jordan, L.; Quinlan, P.; Thompson, A.; et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011, 20, 79–91. [Google Scholar] [CrossRef]
- Valenti, F.; Fausti, F.; Biagioni, F.; Shay, T.; Fontemaggi, G.; Domany, E.; Yaffe, M.B.; Strano, S.; Blandino, G.; Di Agostino, S. Mutant p53 oncogenic functions are sustained by Plk2 kinase through an autoregulatory feedback loop. Cell Cycle 2011, 10, 4330–4340. [Google Scholar] [CrossRef]
- Matsumoto, M.; Furihata, M.; Kurabayashi, A.; Sasaguri, S.; Araki, K.; Hayashi, H.; Ohtsuki, Y. Prognostic significance of serine 392 phosphorylation in overexpressed p53 protein in human esophageal squamous cell carcinoma. Oncology 2004, 67, 143–150. [Google Scholar] [CrossRef]
- Furihata, M.; Takeuchi, T.; Matsumoto, M.; Kurabayashi, A.; Ohtsuki, Y.; Terao, N.; Kuwahara, M.; Shuin, T. p53 mutation arising in Arg72 allele in the tumorigenesis and development of carcinoma of the urinary tract. Clin. Cancer Res. 2002, 8, 1192–1195. [Google Scholar] [PubMed]
- Jethwa, A.; Slabicki, M.; Hullein, J.; Jentzsch, M.; Dalal, V.; Rabe, S.; Wagner, L.; Walther, T.; Klapper, W.; Project, M.N.; et al. TRRAP is essential for regulating the accumulation of mutant and wild-type p53 in lymphoma. Blood 2018, 131, 2789–2802. [Google Scholar] [CrossRef]
- Perez, R.E.; Knights, C.D.; Sahu, G.; Catania, J.; Kolukula, V.K.; Stoler, D.; Graessmann, A.; Ogryzko, V.; Pishvaian, M.; Albanese, C.; et al. Restoration of DNA-binding and growth-suppressive activity of mutant forms of p53 via a PCAF-mediated acetylation pathway. J. Cell. Physiol. 2010, 225, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Knowell, A.E.; Patel, D.; Morton, D.J.; Sharma, P.; Glymph, S.; Chaudhary, J. Id4 dependent acetylation restores mutant-p53 transcriptional activity. Mol. Cancer 2013, 12, 161. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.W.; Kang, H.J.; Kim, H.J.; Kong, Y.; Brown, M.L.; Bae, I. Targeting mutant p53 by a SIRT1 activator YK-3-237 inhibits the proliferation of triple-negative breast cancer cells. Oncotarget 2013, 4, 984–994. [Google Scholar] [CrossRef] [PubMed]
- Lukashchuk, N.; Vousden, K.H. Ubiquitination and degradation of mutant p53. Mol. Cell. Biol. 2007, 27, 8284–8295. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Sasaki, M.; Maki, C.G. Regulation of p53 nuclear export through sequential changes in conformation and ubiquitination. J. Biol. Chem. 2007, 282, 14616–14625. [Google Scholar] [CrossRef]
- Frum, R.A.; Love, I.M.; Damle, P.K.; Mukhopadhyay, N.D.; Palit Deb, S.; Deb, S.; Grossman, S.R. Constitutive Activation of DNA Damage Checkpoint Signaling Contributes to Mutant p53 Accumulation via Modulation of p53 Ubiquitination. Mol. Cancer Res. MCR 2016, 14, 423–436. [Google Scholar] [CrossRef]
- Zheng, T.; Wang, J.; Zhao, Y.; Zhang, C.; Lin, M.; Wang, X.; Yu, H.; Liu, L.; Feng, Z.; Hu, W. Spliced MDM2 isoforms promote mutant p53 accumulation and gain-of-function in tumorigenesis. Nat. Commun. 2013, 4, 2996. [Google Scholar] [CrossRef]
- Yan, W.; Jung, Y.S.; Zhang, Y.; Chen, X. Arsenic trioxide reactivates proteasome-dependent degradation of mutant p53 protein in cancer cells in part via enhanced expression of Pirh2 E3 ligase. PLoS ONE 2014, 9, e103497. [Google Scholar] [CrossRef]
- Yuan, J.; Luo, K.; Zhang, L.; Cheville, J.C.; Lou, Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell 2010, 140, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, A.; Candelaria, N.; Wong, K.K.; Nikolai, B.C.; Lonard, D.M.; O’Malley, B.W.; Richards, J.S. USP15-dependent lysosomal pathway controls p53-R175H turnover in ovarian cancer cells. Nat. Commun. 2018, 9, 1270. [Google Scholar] [CrossRef]
- Peng, Y.; Chen, L.; Li, C.; Lu, W.; Chen, J. Inhibition of MDM2 by hsp90 contributes to mutant p53 stabilization. J. Biol. Chem. 2001, 276, 40583–40590. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. MCR 2011, 9, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.; Hrstka, R.; Coomber, D.; Lane, D.P.; Vojtesek, B. Chaperone-dependent stabilization and degradation of p53 mutants. Oncogene 2008, 27, 3371–3383. [Google Scholar] [CrossRef] [PubMed]
- Wiech, M.; Olszewski, M.B.; Tracz-Gaszewska, Z.; Wawrzynow, B.; Zylicz, M.; Zylicz, A. Molecular mechanism of mutant p53 stabilization: The role of HSP70 and MDM2. PLoS ONE 2012, 7, e51426. [Google Scholar] [CrossRef]
- Lu, W.J.; Lee, N.P.; Kaul, S.C.; Lan, F.; Poon, R.T.; Wadhwa, R.; Luk, J.M. Mortalin-p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy. Cell Death Differ. 2011, 18, 1046–1056. [Google Scholar] [CrossRef]
- Ahn, B.Y.; Trinh, D.L.; Zajchowski, L.D.; Lee, B.; Elwi, A.N.; Kim, S.W. Tid1 is a new regulator of p53 mitochondrial translocation and apoptosis in cancer. Oncogene 2010, 29, 1155–1166. [Google Scholar] [CrossRef]
- Tracz-Gaszewska, Z.; Klimczak, M.; Biecek, P.; Herok, M.; Kosinski, M.; Olszewski, M.B.; Czerwinska, P.; Wiech, M.; Wiznerowicz, M.; Zylicz, A.; et al. Molecular chaperones in the acquisition of cancer cell chemoresistance with mutated TP53 and MDM2 up-regulation. Oncotarget 2017, 8, 82123–82143. [Google Scholar] [CrossRef]
- Yue, X.; Zhao, Y.; Liu, J.; Zhang, C.; Yu, H.; Wang, J.; Zheng, T.; Liu, L.; Li, J.; Feng, Z.; et al. BAG2 promotes tumorigenesis through enhancing mutant p53 protein levels and function. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Zhao, Y.; Huang, G.; Li, J.; Zhu, J.; Feng, Z.; Hu, W. A novel mutant p53 binding partner BAG5 stabilizes mutant p53 and promotes mutant p53 GOFs in tumorigenesis. Cell Discov. 2016, 2, 16039. [Google Scholar] [CrossRef] [PubMed]
- Marin, M.C.; Jost, C.A.; Brooks, L.A.; Irwin, M.S.; O’Nions, J.; Tidy, J.A.; James, N.; McGregor, J.M.; Harwood, C.A.; Yulug, I.G.; et al. A common polymorphism acts as an intragenic modifier of mutant p53 behaviour. Nat. Genet. 2000, 25, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, D.; Gasco, M.; Hiller, L.; Sullivan, A.; Syed, N.; Trigiante, G.; Yulug, I.; Merlano, M.; Numico, G.; Comino, A.; et al. p53 polymorphism influences response in cancer chemotherapy via modulation of p73-dependent apoptosis. Cancer Cell 2003, 3, 387–402. [Google Scholar] [CrossRef]
- Vikhanskaya, F.; Siddique, M.M.; Kei Lee, M.; Broggini, M.; Sabapathy, K. Evaluation of the combined effect of p53 codon 72 polymorphism and hotspot mutations in response to anticancer drugs. Clin. Cancer Res. 2005, 11, 4348–4356. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Gnanapradeepan, K.; Barnoud, T.; Kung, C.P.; Tavecchio, M.; Scott, J.; Watters, A.; Chen, Q.; Kossenkov, A.V.; Murphy, M.E. Mutant p53 controls tumor metabolism and metastasis by regulating PGC-1alpha. Genes Dev. 2018, 32, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Katz, C.; Low-Calle, A.M.; Choe, J.H.; Laptenko, O.; Tong, D.; Joseph-Chowdhury, J.N.; Garofalo, F.; Zhu, Y.; Friedler, A.; Prives, C. Wild-type and cancer-related p53 proteins are preferentially degraded by MDM2 as dimers rather than tetramers. Genes Dev. 2018, 32, 430–447. [Google Scholar] [CrossRef]
- Jenkins, L.M.; Durell, S.R.; Mazur, S.J.; Appella, E. p53 N-terminal phosphorylation: A defining layer of complex regulation. Carcinogenesis 2012, 33, 1441–1449. [Google Scholar] [CrossRef]
- Melnikova, V.O.; Santamaria, A.B.; Bolshakov, S.V.; Ananthaswamy, H.N. Mutant p53 is constitutively phosphorylated at Serine 15 in UV-induced mouse skin tumors: Involvement of ERK1/2 MAP kinase. Oncogene 2003, 22, 5958–5966. [Google Scholar] [CrossRef]
- Ray, D.; Murphy, K.R.; Gal, S. The DNA binding and accumulation of p53 from breast cancer cell lines and the link with serine 15 phosphorylation. Cancer Biol. Ther. 2012, 13, 848–857. [Google Scholar] [CrossRef]
- Sugikawa, E.; Yazaki, N.; Tsunoda, S.; Nakanishi, N.; Ohashi, M. Inhibition of mutant p53 phosphorylation at serine 15 or serine 315 partially restores the function of wild-type p53. Biochem. Biophys. Res. Commun. 1999, 261, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Ferbeyre, G.; de Stanchina, E.; Querido, E.; Baptiste, N.; Prives, C.; Lowe, S.W. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 2000, 14, 2015–2027. [Google Scholar]
- Alsheich-Bartok, O.; Haupt, S.; Alkalay-Snir, I.; Saito, S.; Appella, E.; Haupt, Y. PML enhances the regulation of p53 by CK1 in response to DNA damage. Oncogene 2008, 27, 3653–3661. [Google Scholar] [CrossRef]
- Moller, A.; Sirma, H.; Hofmann, T.G.; Rueffer, S.; Klimczak, E.; Droge, W.; Will, H.; Schmitz, M.L. PML is required for homeodomain-interacting protein kinase 2 (HIPK2)-mediated p53 phosphorylation and cell cycle arrest but is dispensable for the formation of HIPK domains. Cancer Res. 2003, 63, 4310–4314. [Google Scholar]
- Smeenk, L.; van Heeringen, S.J.; Koeppel, M.; Gilbert, B.; Janssen-Megens, E.; Stunnenberg, H.G.; Lohrum, M. Role of p53 serine 46 in p53 target gene regulation. PLoS ONE 2011, 6, e17574. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga-Takenaka, R.; Fukunaga, K.; Tatemichi, M.; Ohshima, H. Nitric oxide prevents UV-induced phosphorylation of the p53 tumor-suppressor protein at serine 46: A possible role in inhibition of apoptosis. Biochem. Biophys. Res. Commun. 2003, 308, 966–974. [Google Scholar] [CrossRef]
- Mayo, L.D.; Seo, Y.R.; Jackson, M.W.; Smith, M.L.; Rivera Guzman, J.; Korgaonkar, C.K.; Donner, D.B. Phosphorylation of human p53 at serine 46 determines promoter selection and whether apoptosis is attenuated or amplified. J. Biol. Chem. 2005, 280, 25953–25959. [Google Scholar] [CrossRef]
- Mantovani, F.; Tocco, F.; Girardini, J.; Smith, P.; Gasco, M.; Lu, X.; Crook, T.; Del Sal, G. The prolyl isomerase Pin1 orchestrates p53 acetylation and dissociation from the apoptosis inhibitor iASPP. Nat. Struct. Mol. Biol. 2007, 14, 912–920. [Google Scholar] [CrossRef]
- Gillotin, S.; Yap, D.; Lu, X. Mutation at Ser392 specifically sensitizes mutant p53H175 to mdm2-mediated degradation. Cell Cycle 2010, 9, 1390–1398. [Google Scholar] [CrossRef]
- Yap, D.B.; Hsieh, J.K.; Zhong, S.; Heath, V.; Gusterson, B.; Crook, T.; Lu, X. Ser392 phosphorylation regulates the oncogenic function of mutant p53. Cancer Res. 2004, 64, 4749–4754. [Google Scholar] [CrossRef]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Su, F.; Chen, D.; Shiloh, A.; Gu, W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 2000, 408, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef]
- Reed, S.M.; Quelle, D.E. p53 Acetylation: Regulation and Consequences. Cancers 2014, 7, 30–69. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2011, 2, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Warnock, L.J.; Raines, S.A.; Milner, J. Aurora A mediates cross-talk between N- and C-terminal post-translational modifications of p53. Cancer Biol. Ther. 2011, 12, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Murr, R.; Vaissiere, T.; Sawan, C.; Shukla, V.; Herceg, Z. Orchestration of chromatin-based processes: Mind the TRRAP. Oncogene 2007, 26, 5358–5372. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structure-function-rescue: The diverse nature of common p53 cancer mutants. Oncogene 2007, 26, 2226–2242. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Hong, D.; Nam, S.H.; Kim, J.M.; Paik, Y.H.; Joh, J.W.; Kwon, C.H.; Park, J.B.; Choi, G.S.; Jang, K.Y.; et al. SIRT1 regulates oncogenesis via a mutant p53-dependent pathway in hepatocellular carcinoma. J. Hepatol. 2015, 62, 121–130. [Google Scholar] [CrossRef]
- Li, M.; Brooks, C.L.; Wu-Baer, F.; Chen, D.; Baer, R.; Gu, W. Mono- versus polyubiquitination: Differential control of p53 fate by Mdm2. Science 2003, 302, 1972–1975. [Google Scholar] [CrossRef]
- Carter, S.; Bischof, O.; Dejean, A.; Vousden, K.H. C-terminal modifications regulate MDM2 dissociation and nuclear export of p53. Nat. Cell Biol. 2007, 9, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Brooks, C.; Kon, N.; Li, M.; Gu, W. Dissecting roles of ubiquitination in the p53 pathway. In Ernst Schering Foundation Symposium Proceedings; Springer: Berlin/Heidelberg, Germany, 2008; pp. 127–136. [Google Scholar]
- Brooks, C.L.; Gu, W. p53 regulation by ubiquitin. FEBS Lett. 2011, 585, 2803–2809. [Google Scholar] [CrossRef] [PubMed]
- Bartel, F.; Taubert, H.; Harris, L.C. Alternative and aberrant splicing of MDM2 mRNA in human cancer. Cancer Cell 2002, 2, 9–15. [Google Scholar] [CrossRef]
- Schuster, K.; Fan, L.; Harris, L.C. MDM2 splice variants predominantly localize to the nucleoplasm mediated by a COOH-terminal nuclear localization signal. Mol. Cancer Res. MCR 2007, 5, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.C.; Viswanathan, M.; Grier, J.D.; Narayana, M.; El-Naggar, A.K.; Lozano, G. An alternatively spliced HDM2 product increases p53 activity by inhibiting HDM2. Oncogene 2001, 20, 4041–4049. [Google Scholar] [CrossRef] [PubMed]
- Chandler, D.S.; Singh, R.K.; Caldwell, L.C.; Bitler, J.L.; Lozano, G. Genotoxic stress induces coordinately regulated alternative splicing of the p53 modulators MDM2 and MDM4. Cancer Res. 2006, 66, 9502–9508. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Ballinger, C.A.; Wu, Y.; Dai, Q.; Cyr, D.M.; Hohfeld, J.; Patterson, C. CHIP is a U-box-dependent E3 ubiquitin ligase: Identification of Hsc70 as a target for ubiquitylation. J. Biol. Chem. 2001, 276, 42938–42944. [Google Scholar] [CrossRef]
- Esser, C.; Scheffner, M.; Hohfeld, J. The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. J. Biol. Chem. 2005, 280, 27443–27448. [Google Scholar] [CrossRef]
- Zou, Q.; Jin, J.; Hu, H.; Li, H.S.; Romano, S.; Xiao, Y.; Nakaya, M.; Zhou, X.; Cheng, X.; Yang, P.; et al. USP15 stabilizes MDM2 to mediate cancer-cell survival and inhibit antitumor T cell responses. Nat. Immunol. 2014, 15, 562–570. [Google Scholar] [CrossRef]
- Jolly, C.; Morimoto, R.I. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J. Natl. Cancer Inst. 2000, 92, 1564–1572. [Google Scholar] [CrossRef]
- Muller, L.; Schaupp, A.; Walerych, D.; Wegele, H.; Buchner, J. Hsp90 regulates the activity of wild type p53 under physiological and elevated temperatures. J. Biol. Chem. 2004, 279, 48846–48854. [Google Scholar] [CrossRef] [PubMed]
- Selkirk, J.K.; Merrick, B.A.; Stackhouse, B.L.; He, C. Multiple p53 protein isoforms and formation of oligomeric complexes with heat shock proteins Hsp70 and Hsp90 in the human mammary tumor, T47D, cell line. Appl. Theor. Electrophor. 1994, 4, 11–18. [Google Scholar] [PubMed]
- Hainaut, P.; Milner, J. Interaction of heat-shock protein 70 with p53 translated in vitro: Evidence for interaction with dimeric p53 and for a role in the regulation of p53 conformation. EMBO J. 1992, 11, 3513–3520. [Google Scholar] [CrossRef] [PubMed]
- Sturzbecher, H.W.; Chumakov, P.; Welch, W.J.; Jenkins, J.R. Mutant p53 proteins bind hsp 72/73 cellular heat shock-related proteins in SV40-transformed monkey cells. Oncogene 1987, 1, 201–211. [Google Scholar] [PubMed]
- Sugito, K.; Yamane, M.; Hattori, H.; Hayashi, Y.; Tohnai, I.; Ueda, M.; Tsuchida, N.; Ohtsuka, K. Interaction between hsp70 and hsp40, eukaryotic homologues of DnaK and DnaJ, in human cells expressing mutant-type p53. FEBS Lett. 1995, 358, 161–164. [Google Scholar] [CrossRef]
- Blagosklonny, M.V.; Toretsky, J.; Bohen, S.; Neckers, L. Mutant conformation of p53 translated in vitro or in vivo requires functional HSP90. Proc. Natl. Acad. Sci. USA 1996, 93, 8379–8383. [Google Scholar] [CrossRef] [PubMed]
- Iwaya, K.; Tsuda, H.; Fujita, S.; Suzuki, M.; Hirohashi, S. Natural state of mutant p53 protein and heat shock protein 70 in breast cancer tissues. Lab. Investig. 1995, 72, 707–714. [Google Scholar] [PubMed]
- Whitesell, L.; Sutphin, P.D.; Pulcini, E.J.; Martinez, J.D.; Cook, P.H. The physical association of multiple molecular chaperone proteins with mutant p53 is altered by geldanamycin, an hsp90-binding agent. Mol. Cell. Biol. 1998, 18, 1517–1524. [Google Scholar] [CrossRef]
- Nagata, Y.; Anan, T.; Yoshida, T.; Mizukami, T.; Taya, Y.; Fujiwara, T.; Kato, H.; Saya, H.; Nakao, M. The stabilization mechanism of mutant-type p53 by impaired ubiquitination: The loss of wild-type p53 function and the hsp90 association. Oncogene 1999, 18, 6037–6049. [Google Scholar] [CrossRef]
- Lin, K.; Rockliffe, N.; Johnson, G.G.; Sherrington, P.D.; Pettitt, A.R. Hsp90 inhibition has opposing effects on wild-type and mutant p53 and induces p21 expression and cytotoxicity irrespective of p53/ATM status in chronic lymphocytic leukaemia cells. Oncogene 2008, 27, 2445–2455. [Google Scholar] [CrossRef]
- Kovacs, J.J.; Murphy, P.J.; Gaillard, S.; Zhao, X.; Wu, J.T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Finlay, C.A.; Hinds, P.W.; Tan, T.H.; Eliyahu, D.; Oren, M.; Levine, A.J. Activating mutations for transformation by p53 produce a gene product that forms an hsc70-p53 complex with an altered half-life. Mol. Cell. Biol. 1988, 8, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Black, J.D.; Rezvani, K. Heat Shock Protein 70s as Potential Molecular Targets for Colon Cancer Therapeutics. Curr. Med. Chem. 2016, 23, 3171–3188. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.; Bottger, S.; Low, B. Mortalin-based cytoplasmic sequestration of p53 in a nonmammalian cancer model. Am. J. Pathol. 2006, 168, 1526–1530. [Google Scholar] [CrossRef]
- King, F.W.; Wawrzynow, A.; Hohfeld, J.; Zylicz, M. Co-chaperones Bag-1, Hop and Hsp40 regulate Hsc70 and Hsp90 interactions with wild-type or mutant p53. EMBO J. 2001, 20, 6297–6305. [Google Scholar] [CrossRef] [PubMed]
- Trinh, D.L.; Elwi, A.N.; Kim, S.W. Direct interaction between p53 and Tid1 proteins affects p53 mitochondrial localization and apoptosis. Oncotarget 2010, 1, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Dumont, P.; Leu, J.I.; Della Pietra, A.C., 3rd; George, D.L.; Murphy, M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat. Genet. 2003, 33, 357–365. [Google Scholar] [CrossRef]
- Gaiddon, C.; Lokshin, M.; Ahn, J.; Zhang, T.; Prives, C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell. Biol. 2001, 21, 1874–1887. [Google Scholar] [CrossRef]
- Di Como, C.J.; Gaiddon, C.; Prives, C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol. Cell. Biol. 1999, 19, 1438–1449. [Google Scholar] [CrossRef]
- Strano, S.; Munarriz, E.; Rossi, M.; Cristofanelli, B.; Shaul, Y.; Castagnoli, L.; Levine, A.J.; Sacchi, A.; Cesareni, G.; Oren, M.; et al. Physical and functional interaction between p53 mutants and different isoforms of p73. J. Biol. Chem. 2000, 275, 29503–29512. [Google Scholar] [CrossRef]
- Strano, S.; Rossi, M.; Fontemaggi, G.; Munarriz, E.; Soddu, S.; Sacchi, A.; Blandino, G. From p63 to p53 across p73. FEBS Lett. 2001, 490, 163–170. [Google Scholar] [CrossRef]
- Luo, J.L.; Yang, Q.; Tong, W.M.; Hergenhahn, M.; Wang, Z.Q.; Hollstein, M. Knock-in mice with a chimeric human/murine p53 gene develop normally and show wild-type p53 responses to DNA damaging agents: A new biomedical research tool. Oncogene 2001, 20, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.I.; Murphy, M.E.; George, D.L. The p53 Codon 72 Polymorphism Modifies the Cellular Response to Inflammatory Challenge in the Liver. J. Liver 2013, 2, 117. [Google Scholar] [PubMed]
- Kamada, R.; Toguchi, Y.; Nomura, T.; Imagawa, T.; Sakaguchi, K. Tetramer formation of tumor suppressor protein p53: Structure, function, and applications. Biopolymers 2016, 106, 598–612. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Upstream Regulators | Mutant TP53 | Modified Amino Acids | Samples/Cell Lines | Outcomes/Effects | References |
|---|---|---|---|---|---|
| Phosphorylation | |||||
| Ras signaling | R280K | S6, S9 | MDA-MB-231 | Mutp53/Smad2/TP63 Complex inhibit TP63’s metastasis suppressor function. | [73] |
| DNA damage | R248W, R273H | S15 | MEFs, PANC1 | Constitutively activated DNA damage could account for mutp53 stabilization or nuclear accumulation via S15 phosphorylation. | [41,47,74] |
| NF-κB inhibition by IκB overexpression | P223L/V274V | S15 | DU145 | NF-κB inhibition in DU145 cells leads to S15 phosphorylation of mutp53 via GADD45α-mediated JNK1 activation and potential restoration of wtp53. | [75] |
| Stathmin1 | R175H, R273H | S15, S37 | TOV112D, MDAH-2774 | Stathmin1 enhances interaction of mutp53 with DNA-PK, phosphorylation of mutp53 at S15 and S37 by DNA-PK, and mutp53 stabilization, leading to increased viable cell proliferation. | [76] |
| PML | R175H, R273H, R273H/P309S | Not specified (maybe via phosphorylation at T18 and S46 in mutp53 likewise wtp53) | SKBR3, HT29, SW48 | PML interacts with mutp53 and is required for proliferation and colony formation of cancer cells bearing mutp53; however, it is unclear whether PML promotes phosphorylation of mutp53 like wtp53. | [77] |
| Pin1 | Mouse R172H, R280K | Not specified (maybe via isomerization of phosphorylated S46-P47 site in mutp53 likewise wtp53) | MEFs, MDA-MB-231 | Homozygous deletion of Pin1 attenuates tumor progression in TP53R172H/R172H mice, while Pin1 enhances migration and lung colonization of MDA-MB-231 cells in a manner dependent on mutp53 by enhancing inhibitory interaction of mutp53 with TP63; however, it is unclear whether observed phenotypes by Pin1 are mediated through its detection of phosphorylated S46-P47 site in mutp53. | [78] |
| PLK2 | R175H, R273H | T377 | H1299, SKBR3 | PLK2 phosphorylates mutp53 at T377 and enhances binding of mutp53 with p300, acetylation of mutp53, and mutp53 GOF activity, including increased cell proliferation, NF-Y’s transcriptional activity, and Adriamycin resistance. | [79] |
| Not specified/oncogenic signaling | Not Specified | S392 | Cancer tissues (esophageal squamous cell carcinoma, urothelial transitional cell carcinoma) | S392 phosphorylation in mutp53 is correlated with high levels of Ki67 staining, lymphatic invasion, and poor prognosis, as well as enhanced hetero-oligomerization with wtp53 and dominant-negative activities. | [70,80,81] |
| Acetylation | |||||
| TRRAP | R248Q, R248W, R283H/G245SS, R213Q/Y234H, I254D, R273H | Not specified | BL-41, BL-60, CA-46, DG-75, Namalwa, Raji, Ramos, SUDHL-4, Colo320 | TRAPP recruits HATs to chromatin and increases acetylation and accumulation of mutp53 through inhibition of MDM2-mediated degradation. | [82] |
| P/CAF | R175H, G245A, D281G | K320, K373 | H1299 | Treatment of cells with TSA acetylates mutp53 at K320 and K373 by P/CAF to increase apoptosis with enhanced DNA binding of mutp53 to the p21 and PUMA promoters. | [83] |
| Id4 | P223L/V274F | K320, K373 | DU145 | Id4 increases interaction of mutp53 with CBP/p300, acetylation at K320 and K373 on mutp53, and apoptosis with upregulation of p21, BAX, and PUMA. | [84] |
| Deacetylation | |||||
| Glucose restriction | G245A | Not specified (6Q mutations in K319, K320, K321, K370, K372, K373) | H1299 | G245A-K6Q shows resistance to mutp53 degradation and cell death induced by glucose restriction. | [36] |
| SIRT1 | R249S, R273H, V157F, M237I | K382 | BT549, MDA-MB-468, HS578T, SUM149PT | Activation of SIRT1 deacetylase by YK-3-237 decreases mutp53 levels with reduced acetylation at K382, leading to induction of apoptotic cell death and G2/M cell cycle arrest with induction of wtp53 target genes. | [85] |
| Ubiquitination | |||||
| COP1, CHIP (independent of MDM2) | R175H | Not specified | U2OS, H1299, MDM2−/−TP53−/− MEFs | Downregulation of COP1 or CHIP reduces ubiquitination of mutp53 independent of MDM2. | [86] |
| Not specified/MDM2-independent ubiquitination | C135Y, V143A, H179E | Not specified | U2OS, MDM2−/−TP53−/− MEFs | Misfolded mutp53 is more efficiently ubiquitinated and localizes to the cytoplasm to a greater extent than a DNA contact mutp53 (R248W). | [87] |
| MDM2 | R273H | Not specified | H1048, H1299, WI38 | S15 phosphorylation of mutp53 by ATM following DNA damage inhibits MDM2-mediated polyubiquitination and degradation of mutp53 with allowing its monoubiquitination and accumulation. | [88] |
| MDM2 isoform B (MDM2-B) | R175H, R248W, R273H, Y220C S241F | Not specified | H1299, HCT116, T47D, Huh7, DLD-1 | MDM2-B binds to and inhibits full-length MDM2 (MDM2-FL)-mediated mutp53 degradation, leading to enhanced mutp53 GOF activities to promote tumor growth and metastasis. | [89] |
| CHIP | R156P, R175H, Y220C | Not specified | KHOS/NP, SKBR3, CAL33, BxPC3 | Cholesterol-lowering drugs “statins,” knockdown of mevalonate kinase, and DNAJA1 knockdown induce CHIP-mediated nuclear export and degradation of mainly conformational/misfolded mutp53. | [29] |
| CHIP | R110P, R175H | Not specified | HCT116, CAL33 | Aggregating TP53 mutants (R110P, R175H), but not non-aggregating mutants (R248W, R273H), are ubiquitinated and degraded by CHIP via K63-linked polyubiquitination in a manner dependent on autophagy. | [30] |
| Pirh2 | R175H, R248W, H179Y/R282W, R273H | Not specified | SW480, MiaPaCa-2, HaCaT, HCT116 | Arsenic trioxide (ATO) induces Pirh2-mediated degradation of multiple TP53 mutants. | [90] |
| Deubiquitination | |||||
| USP10 | Not specified | Not specified | 786-O | USP10 overexpression inhibits MDM2-mediated mutp53 ubiquitination leading to stabilization of mutp53 as well as increased colony formation and cell proliferation. | [91] |
| USP15 | R175H | Not specified | TYK-Nu, TOV112D, SKOV3 | MCB-613, a stimulator of steroid receptor coactivators (SRCs), enhances nuclear export, ubiquitination, and lysosome-mediated degradation of mutp53 (R175H), but not R273H, through inhibition of USP15, leading to reduced viability of ovarian cancer cells. | [92] |
| Molecular Chaperons | |||||
| HSP90 | L194F, S241F, R273C, R273H | N/A | T47D, DLD1, C33A, MDA-MB-468 | HSP90 forms a complex with MDM2 and mutp53 to block ubiquitination of both MDM2 and mutp53, while HSP90 inhibition by geldanamycin induces MDM2-mediated mutp53 degradation. | [93] |
| HSP90 | R175H, L194F, P223L/V274F, R273H, R273H/P309S, R280K, R280T | N/A | SKBR3, T47D, DU145, MDA-MB-468, MDA-MB-231, SW480, 5637 | HSP90 forms a complex with mutp53 to prevent mutp53’s aggregation by inhibiting MDM2 and CHIP activities. | [94] |
| HSP90 (HDAC6-HSP90-mutp53 complex) | R175H, L194F, P223L/V274F, S241F, R273H/P309S, R280K | N/A | SKRB3, T47D, DU145, ES2, SW480, MDA-MB-231 | Inhibition of HSP90 activity by SAHA releases mutp53 from the HDAC6-HSP90-mutp53 complex, leading to mutp53 degradation by MDM2 and CHIP to enhance cell death by a chemotherapy agent, Camptothesin. | [95] |
| HSP90 | mouse R172H and R248Q | N/A | T-lymphomas | Inhibition of HSP90 by ganetespib results in degradation of mutp53 in lymphomas and prolongs survival of TP53R172H/R172H and TP53R248Q/− mice with minimal effects on TP53-null mice. | [50] |
| HSP90 (HDAC6-HSP90-mutp53 complex) | R175H, L194F, M237I, R249S, R273H | N/A | SKBR3, T47D, SUM149, Mahlavu, BT549, MDA-MB-468 | Inhibition of the mevalonate–RhoA axis by cerivastatin or GGTI-298 reduces HDAC6 activity, leading to HSP90 hyperacetylation and dissociation of HSP90 from mutp53, which induces degradation of mutp53 by MDM2. | [51] |
| HSP70/HSC70 | R175H, R273H | N/A | MDA-MB-468, H1299 | HSP70 selectively recognizes unfolded/misfolded TP53 proteins and promotes its CHIP-dependent ubiquitination and degradation when HSP90 is inhibited and mutp53 folding is blocked. | [96] |
| HSP70 (not HSC70) | V143A, R175H | N/A | MDM2−/−TP53−/− MEFs, H1299, SKBR3 | HSP70 accelerates CHIP-mediated degradation of mutp53, whereas it partially inhibits MDM2-mediated ubiquitination and degradation of mutp53 to enhance nuclear aggregates which can be inhibited by HSC70. | [97] |
| HSC70 | R248Q, S241F, R158Inf, R280L, G266Q, S227K, S227R, E258K, A161T, R273L, R273H, R280L, R175H, R175D, R175C, R248W, R248L, R282W, P151H, P98S, G245C, L194F | N/A | OVCAR-3, ES2, SUM159, MDA-MB-231, MDA-MB-435, HCT116 | Chaperone-mediated autophagy (CMA), which is induced in cells treated with an autophagy inhibitor (spautin-1) under confluency or nutrient deprivation conditions, facilitates nuclear export of mutp53 and promotes interaction of mutp53 with HSC70, leading to mutp53 degradation through the lysosome. | [35] |
| Mortalin (mtHSP70/Grp75) | R249S | N/A | PLC/PRF/5 | Mortalin knockdown induces nuclear translocation and apoptosis in a mutp53-dependent manner. | [98] |
| Tid/DNAJA3 (HSP40) | R175H, L194F, R273H, E285K | N/A | SKBR3, T47D, U373, BT474 | Overexpression of Tid/DNAJA3 restores mitochondrial localization and pro-apoptotic activities of TP53 in cells treated with desferroxamine (DFX). | [99] |
| DNAJB1 (HSP40) | R175H | N/A | H1299, CAL-33, HuCCT1, FAMPAC, KLE, TOV112D | Chetomin (CTM) binds to HSP40 and increases the interaction between HSP40 and R175H mutp53, leading to restoration of wtp53 activity and inhibition of proliferation and tumor growth of multiple cancer cells expressing R175H with upregulation of TP53 target genes. | [57] |
| DNAJB1 (HSP40), HSP70 | V143A, R158L, R175H, Y220C, G245S, D281G, R282W | N/A | H1299 | DNAJB1 and HSP70 facilitate binding of mutp53 to TAp73α. | [100] |
| DNAJA1 (HSP40) | R156P, R175H, Y220C | N/A | KHOS/NP, SKBR3, CAL33, BxPC3 | DNAJA1 binds to and stabilizes mainly conformational or misfolded mutp53 by competitively binding to CHIP. | [29] |
| BAG2 | mouse R172H, R175H, R248W, R273H | N/A | R172H MEFs, H1299, Saos2, HCT116 | BAG2 binds to mutp53 (mouse R172H) and inhibits MDM2’s activity to accumulate mutp53, while BAG2 knockdown increases chemosensitivity and reduces tumor growth and metastasis. | [101] |
| BAG5 | mouse R172H, R175H, R248W, R273H | N/A | R172H MEFs, H1299, Saos2, HCT116 | BAG5 binds to mutp53 (mouse R172H) and inhibits ubiquitination and degradation of mutp53 by MDM2 and CHIP to accumulate mutp53 and enhance mutp53 GOF, including cell proliferation, migration, chemoresistance, and tumor growth. | [102] |
| SNP | |||||
| 72P/R polymorphism | V143A, V173L, R175H | 72P, 72R | Saos2 | 72R-mutp53 binds more efficiently to TP73 to inactivate TP73-induced apoptosis than 72P-mutp53. | [103] |
| 72P/R polymorphism | V173L, R175H, C176Y, H179R, Y220C, C242Y, G245S/D, R249S, R282W, R273C | 72P, 72R | Saos2 | 72R-mutp53 shows higher efficiency on inhibition of apoptosis induced by chemotherapy drugs, as compared to 72P-mutp53, mainly due to mutp53’s inhibitory binding with TP73. | [104] |
| 72P/R polymorphism | R249S | 72P, 72R | H1299 | Polymorphism at codon 72 in several TP53 mutants (R175H, G245S, R248W, R249S, R273H, R282W) do not show any significant differences in resistance to many chemotherapy drugs, whereas only 72R in R249S mutp53 makes cells more resistance to doxorubicin than 72P in R249S. | [105] |
| 72P/R polymorphism | R175H, R273H | 72P, 72R | H1299, PC13, HT29 (72P-R273H), SW620 (72R-R273H) | 72R-mutp53 less efficiently binds to and inhibits activity of PCG-1α, leading to higher mitochondrial function and metastatic potential than 72P-mutp53. | [106] |
| Dimer | |||||
| Mutations in the TP53 oligomerization domain | R175H, R248W | E343K | U2OS, H1299 | E343K mutation in mutp53 enhances degradation of mutp53 by MDM2, leading to inhibited migration. | [107] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamoto, S.; Iwakuma, T. Regulators of Oncogenic Mutant TP53 Gain of Function. Cancers 2019, 11, 4. https://doi.org/10.3390/cancers11010004
Yamamoto S, Iwakuma T. Regulators of Oncogenic Mutant TP53 Gain of Function. Cancers. 2019; 11(1):4. https://doi.org/10.3390/cancers11010004
Chicago/Turabian StyleYamamoto, Satomi, and Tomoo Iwakuma. 2019. "Regulators of Oncogenic Mutant TP53 Gain of Function" Cancers 11, no. 1: 4. https://doi.org/10.3390/cancers11010004
APA StyleYamamoto, S., & Iwakuma, T. (2019). Regulators of Oncogenic Mutant TP53 Gain of Function. Cancers, 11(1), 4. https://doi.org/10.3390/cancers11010004