PKA at a Cross-Road of Signaling Pathways Involved in the Regulation of Glioblastoma Migration and Invasion by the Neuropeptides VIP and PACAP

 and
and 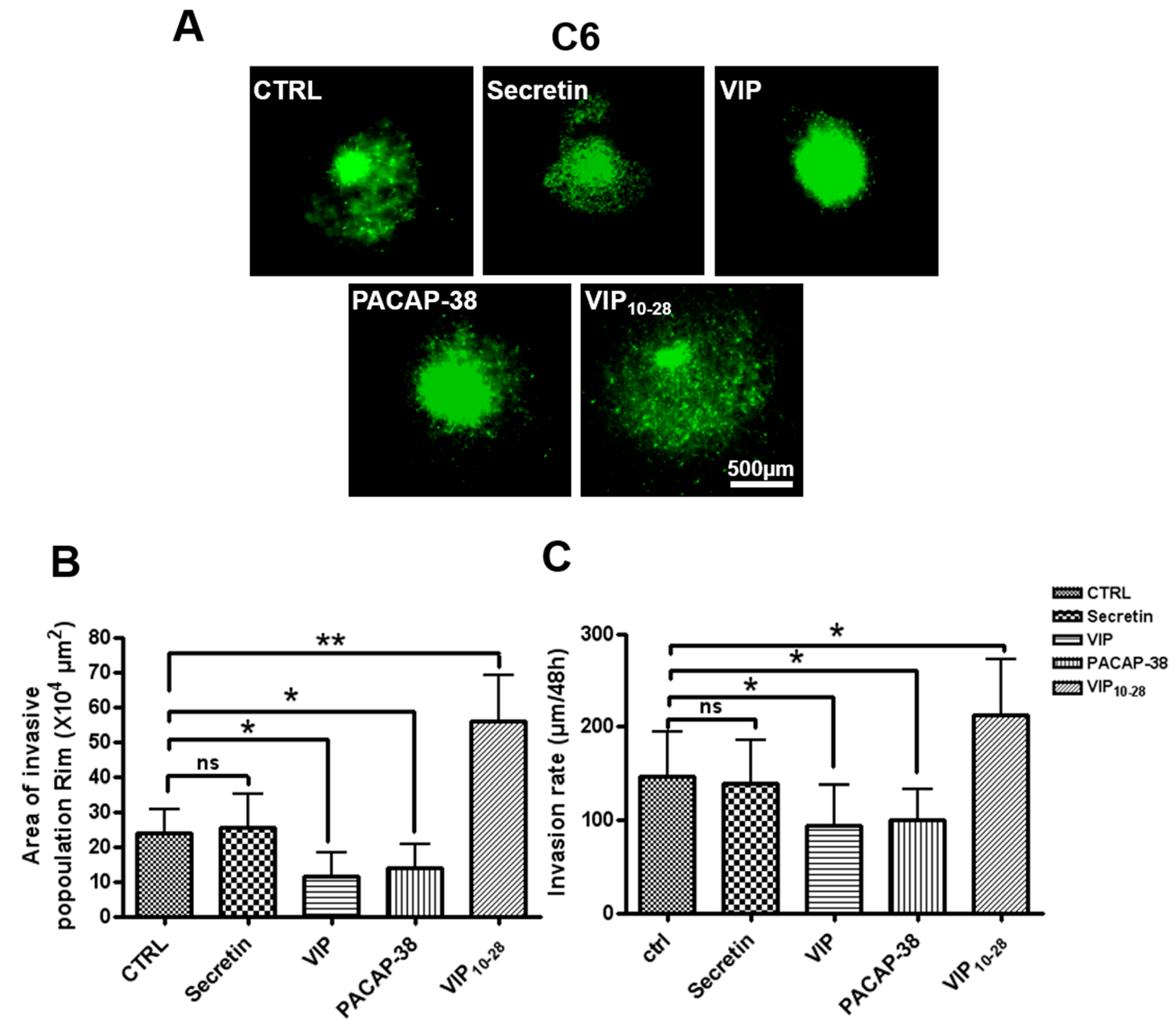{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Components of the VIP-Receptor System Are Expressed in C6 and U87 Cell Lines
2.2. VIP, PACAP-38, and VIP10-28 Effects on Invasion by C6-GFP Cells of the Rat Brain Parenchyma
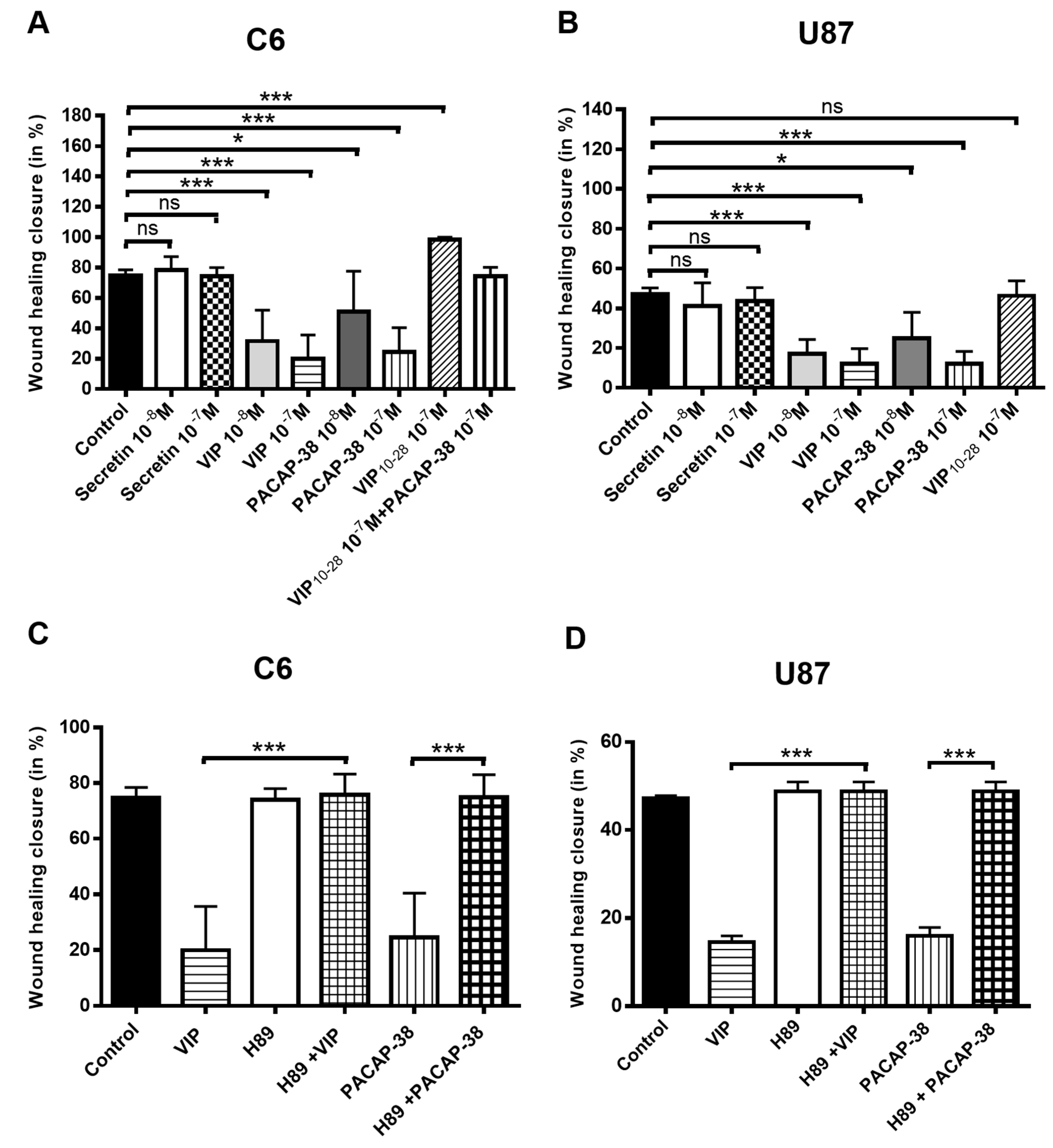
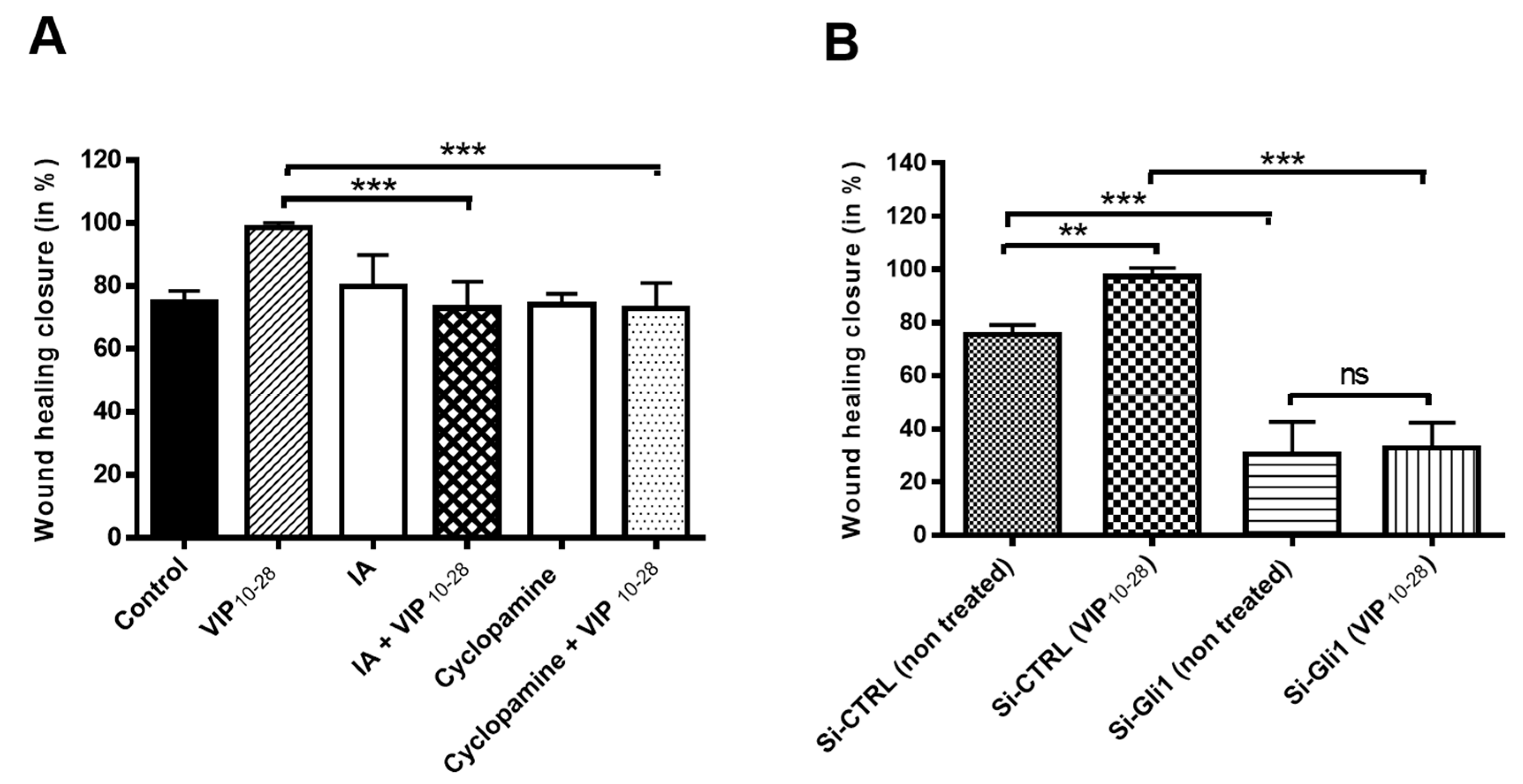
2.3. VIP, PACAP-38, and VIP10-28 Regulation of the Migration Process of C6 and U87 Cells is Affected by Different Signaling Pathways Inhibitors
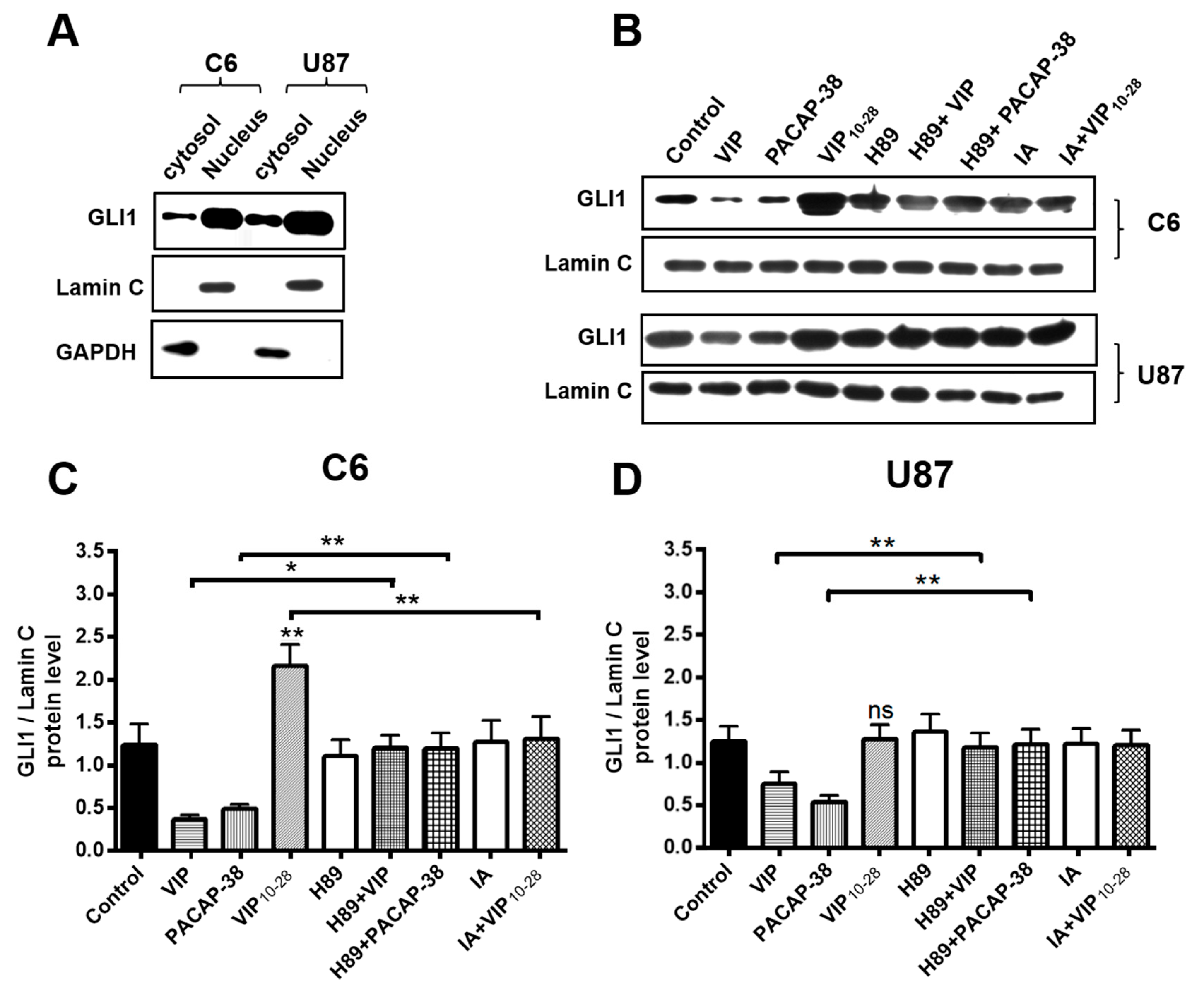
2.4. Inhibitors of PKA (H89) or of Akt (IA) Abolish the Effects of VIP-Related Peptides on the GLI1 Protein Nuclear Expression
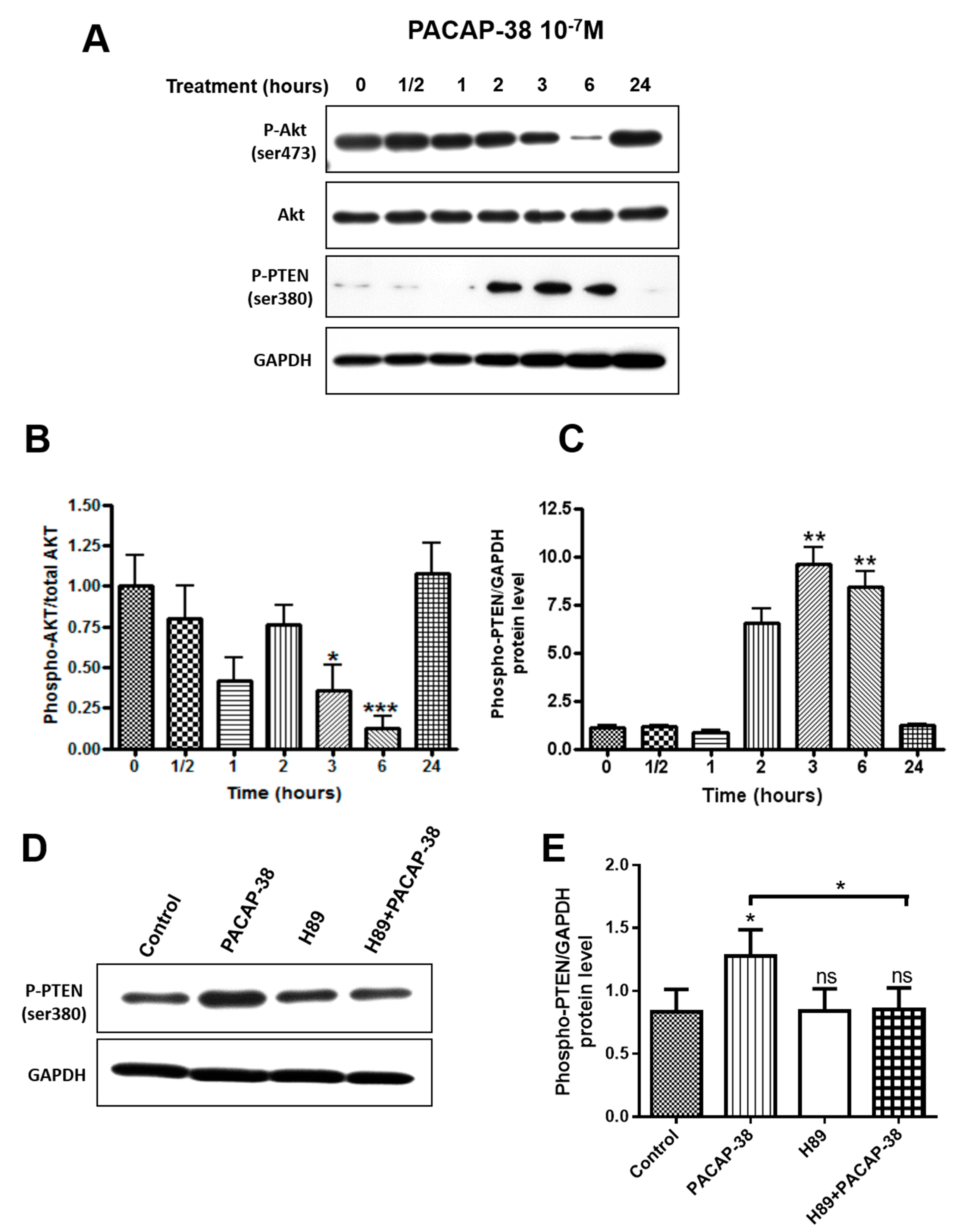
2.5. PACAP-38 Triggers a Time-Dependent Decrease of Phospho-Akt and Elevation of Phospho-PTEN Protein Expression in C6 Cells
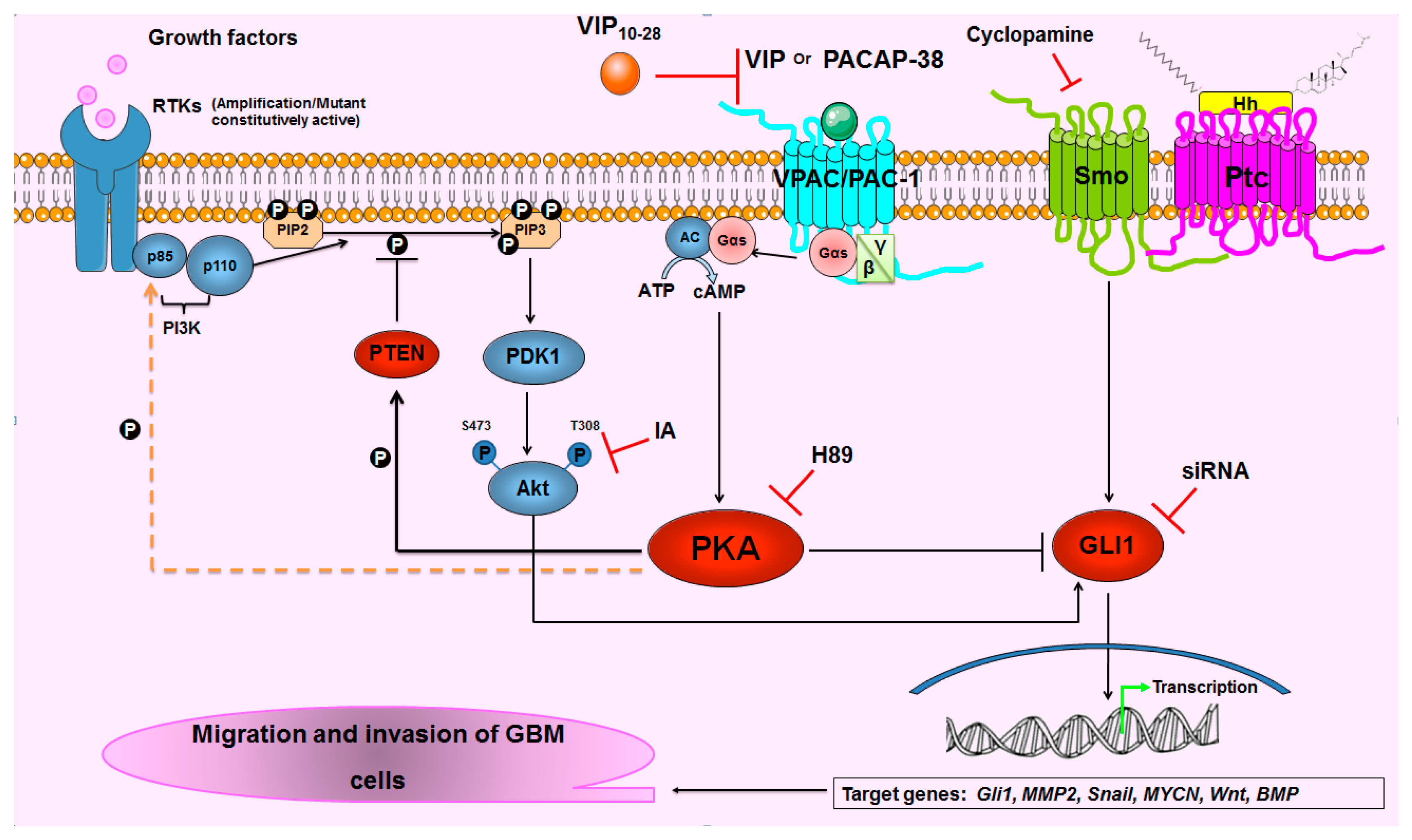
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Transfection with a GFP-Encoding Vector
4.4. siRNA Transfection
4.5. Ex Vivo Invasion Assay on Rat Brain Slices
4.6. Wound Healing
4.7. Western Immunoblotting and Antibodies
4.8. cDNA Synthesis
4.9. Quantitative Polymerase Chain Reaction of Reverse Transcribed mRNA
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zong, H.; Verhaak, R.G.W.; Canoll, P. The cellular origin for malignant glioma and prospects for clinical advancements. Expert Rev. Mol. Diagn. 2012, 12, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, D.M.; Tung, T.; Agnihotri, S.; Singh, S.; Guha, A.; Zadeh, G.; Hawkins, C. Loss of p53 cooperates with K-ras activation to induce glioma formation in a region-independent manner. Glia 2013, 61, 1862–1872. [Google Scholar] [CrossRef]
- Li, Q.-J.; Cai, J.-Q.; Liu, C.-Y. Evolving Molecular Genetics of Glioblastoma. Chin. Med. J. 2016, 129, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.-S.; Qin, X.-L.; Zong, H.-L.; He, X.-G.; Cao, L. Cancer stem cell markers in glioblastoma—An update. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3207–3211. [Google Scholar] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, A.; Symons, M. Signaling determinants of glioma cell invasion. Adv. Exp. Med. Biol. 2013, 986, 121–141. [Google Scholar] [PubMed]
- Vehlow, A.; Cordes, N. Invasion as target for therapy of glioblastoma multiforme. Biochim. Biophys. Acta 2013, 1836, 236–244. [Google Scholar] [CrossRef] [PubMed]
- De Gooijer, M.C.; Guillén Navarro, M.; Bernards, R.; Wurdinger, T.; van Tellingen, O. An Experimenter’s Guide to Glioblastoma Invasion Pathways. Trends Mol. Med. 2018, 24, 763–780. [Google Scholar] [CrossRef]
- Swartz, A.M.; Shen, S.H.; Salgado, M.A.; Congdon, K.L.; Sanchez-Perez, L. Promising vaccines for treating glioblastoma. Expert Opin. Biol. Ther. 2018, 18, 1159–1170. [Google Scholar] [CrossRef]
- Paw, I.; Carpenter, R.C.; Watabe, K.; Debinski, W.; Lo, H.-W. Mechanisms regulating glioma invasion. Cancer Lett. 2015, 362, 1–7. [Google Scholar] [CrossRef]
- Höland, K.; Salm, F.; Arcaro, A. The phosphoinositide 3-kinase signaling pathway as a therapeutic target in grade IV brain tumors. Curr. Cancer Drug Targets 2011, 11, 894–918. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Lee, E.Q.; Reardon, D.A.; Ligon, K.L.; Alfred Yung, W.K. Current clinical development of PI3K pathway inhibitors in glioblastoma. Neuro-Oncology 2012, 14, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.-W.; Weiss, W.A. Targeting the RTK-PI3K-mTOR axis in malignant glioma: Overcoming resistance. Curr. Top. Microbiol. Immunol. 2010, 347, 279–296. [Google Scholar] [PubMed]
- McDowell, K.A.; Riggins, G.J.; Gallia, G.L. Targeting the AKT pathway in glioblastoma. Curr. Pharm. Des. 2011, 17, 2411–2420. [Google Scholar] [CrossRef] [PubMed]
- Narayan, R.S.; Fedrigo, C.A.; Stalpers, L.J.A.; Baumert, B.G.; Sminia, P. Targeting the Akt-pathway to improve radiosensitivity in glioblastoma. Curr. Pharm. Des. 2013, 19, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Sami, A.; Karsy, M. Targeting the PI3K/AKT/mTOR signaling pathway in glioblastoma: Novel therapeutic agents and advances in understanding. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2013, 34, 1991–2002. [Google Scholar] [CrossRef]
- Majewska, E.; Szeliga, M. AKT/GSK3β Signaling in Glioblastoma. Neurochem. Res. 2017, 42, 918–924. [Google Scholar] [CrossRef]
- Katoh, Y.; Katoh, M. Hedgehog target genes: Mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef]
- Santoni, M.; Burattini, L.; Nabissi, M.; Morelli, M.B.; Berardi, R.; Santoni, G.; Cascinu, S. Essential role of Gli proteins in glioblastoma multiforme. Curr. Protein Pept. Sci. 2013, 14, 133–140. [Google Scholar] [CrossRef]
- Ng, J.M.Y.; Curran, T. The Hedgehog’s tale: Developing strategies for targeting cancer. Nat. Rev. Cancer 2011, 11, 493–501. [Google Scholar] [CrossRef]
- Sahebjam, S.; Siu, L.L.; Razak, A.A. The utility of hedgehog signaling pathway inhibition for cancer. Oncologist 2012, 17, 1090–1099. [Google Scholar] [CrossRef] [PubMed]
- Sandhiya, S.; Melvin, G.; Kumar, S.S.; Dkhar, S.A. The dawn of hedgehog inhibitors: Vismodegib. J. Pharmacol. Pharmacother. 2013, 4, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Louis, D.N.; Curry, W.T.; Batchelor, T.T.; Dietrich, J. Diagnostic and therapeutic avenues for glioblastoma: No longer a dead end? Nat. Rev. Clin. Oncol. 2013, 10, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.L.; Rodriguez-Cruz, V.; Walker, N.D.; Greco, S.J.; Rameshwar, P. Temozolomide resistance and tumor recurrence: Halting the Hedgehog. Cancer Cell Microenviron. 2015, 2, e747. [Google Scholar] [PubMed]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.M.; Lelievre, V.; Becq-Giraudon, L.; Meunier, A.C. VIP as a cell-growth and differentiation neuromodulator role in neurodevelopment. Mol. Neurobiol. 1995, 10, 115–134. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.-M.; Debaigt, C.; Goursaud, S.; Montoni, A.; Pineau, N.; Meunier, A.-C.; Janet, T. Unconventional binding sites and receptors for VIP and related peptides PACAP and PHI/PHM: An update. Peptides 2007, 28, 1655–1666. [Google Scholar] [CrossRef]
- Gozes, I. VIP, from gene to behavior and back: Summarizing my 25 years of research. J. Mol. Neurosci. 2008, 36, 115–124. [Google Scholar] [CrossRef]
- Moody, T.W.; Chan, D.; Fahrenkrug, J.; Jensen, R.T. Neuropeptides as autocrine growth factors in cancer cells. Curr. Pharm. Des. 2003, 9, 495–509. [Google Scholar] [CrossRef]
- Vaudry, D.; Falluel-Morel, A.; Bourgault, S.; Basille, M.; Burel, D.; Wurtz, O.; Fournier, A.; Chow, B.K.C.; Hashimoto, H.; Galas, L.; et al. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol. Rev. 2009, 61, 283–357. [Google Scholar] [CrossRef]
- Sharma, A.; Walters, J.; Gozes, Y.; Fridkin, M.; Brenneman, D.; Gozes, I.; Moody, T.W. A vasoactive intestinal peptide antagonist inhibits the growth of glioblastoma cells. J. Mol. Neurosci. 2001, 17, 331–339. [Google Scholar] [CrossRef]
- D’Amico, A.G.; Scuderi, S.; Saccone, S.; Castorina, A.; Drago, F.; D’Agata, V. Antiproliferative Effects of PACAP and VIP in Serum-Starved Glioma Cells. J. Mol. Neurosci. 2013, 51, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska, P.; Nowak, J.Z. Cyclic AMP formation in C6 glioma cells: Effect of PACAP and VIP in early and late passages. Ann. N. Y. Acad. Sci. 2006, 1070, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska, P.; Nowak, J.Z. Effects of PACAP and VIP on cAMP-generating system and proliferation of C6 glioma cells. J. Mol. Neurosci. 2008, 36, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Robberecht, P.; Woussen-Colle, M.C.; Vertongen, P.; De Neef, P.; Hou, X.; Salmon, I.; Brotchi, J. Expression of pituitary adenylate cyclase activating polypeptide (PACAP) receptors in human glial cell tumors. Peptides 1994, 15, 661–665. [Google Scholar] [CrossRef]
- Cochaud, S.; Chevrier, L.; Meunier, A.-C.; Brillet, T.; Chadéneau, C.; Muller, J.-M. The vasoactive intestinal peptide-receptor system is involved in human glioblastoma cell migration. Neuropeptides 2010, 44, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Barbarin, A.; Séité, P.; Godet, J.; Bensalma, S.; Muller, J.-M.; Chadéneau, C. Atypical nuclear localization of VIP receptors in glioma cell lines and patients. Biochem. Biophys. Res. Commun. 2014, 454, 524–530. [Google Scholar] [CrossRef]
- Cochaud, S.; Meunier, A.-C.; Monvoisin, A.; Bensalma, S.; Muller, J.-M.; Chadéneau, C. Neuropeptides of the VIP family inhibit glioblastoma cell invasion. J. Neurooncol. 2015, 122, 63–73. [Google Scholar] [CrossRef]
- Lelievre, V.; Seksenyan, A.; Nobuta, H.; Yong, W.H.; Chhith, S.; Niewiadomski, P.; Cohen, J.R.; Dong, H.; Flores, A.; Liau, L.M.; et al. Disruption of the PACAP gene promotes medulloblastoma in ptc1 mutant mice. Dev. Biol. 2008, 313, 359–370. [Google Scholar] [CrossRef]
- Cohen, J.R.; Resnick, D.Z.; Niewiadomski, P.; Dong, H.; Liau, L.M.; Waschek, J.A. Pituitary adenylyl cyclase activating polypeptide inhibits gli1 gene expression and proliferation in primary medulloblastoma derived tumorsphere cultures. BMC Cancer 2010, 10, 676. [Google Scholar] [CrossRef]
- Niewiadomski, P.; Zhujiang, A.; Youssef, M.; Waschek, J.A. Interaction of PACAP with Sonic hedgehog reveals complex regulation of the hedgehog pathway by PKA. Cell. Signal. 2013, 25, 2222–2230. [Google Scholar] [CrossRef] [PubMed]
- Sheng, T.; Chi, S.; Zhang, X.; Xie, J. Regulation of Gli1 localization by the cAMP/protein kinase A signaling axis through a site near the nuclear localization signal. J. Biol. Chem. 2006, 281, 9–12. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Lu, Q.R. G-Protein Gαs controls medulloblastoma initiation by suppressing sonic hedgehog signaling. Mol. Cell. Oncol. 2015, 2, e975070. [Google Scholar] [CrossRef] [PubMed]
- Warrington, N.M.; Sun, T.; Rubin, J.B. Targeting brain tumor cAMP: The case for sex-specific therapeutics. Front. Pharmacol. 2015, 6, 153. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Salloum, R.; Xin, M.; Lu, Q.R. The G protein Gαs acts as a tumor suppressor in sonic hedgehog signaling-driven tumorigenesis. Cell Cycle 2016, 15, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Mucignat-Caretta, C.; Denaro, L.; D’Avella, D.; Caretta, A. Protein Kinase A Distribution Differentiates Human Glioblastoma from Brain Tissue. Cancers 2017, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Sapio, L.; Di Maiolo, F.; Illiano, M.; Esposito, A.; Chiosi, E.; Spina, A.; Naviglio, S. Targeting protein kinase A in cancer therapy: An update. EXCLI J. 2014, 13, 843–855. [Google Scholar]
- Sapio, L.; Gallo, M.; Illiano, M.; Chiosi, E.; Naviglio, D.; Spina, A.; Naviglio, S. The Natural cAMP Elevating Compound Forskolin in Cancer Therapy: Is It Time? J. Cell. Physiol. 2017, 232, 922–927. [Google Scholar] [CrossRef]
- Grobben, B.; De Deyn, P.P.; Slegers, H. Rat C6 glioma as experimental model system for the study of glioblastoma growth and invasion. Cell Tissue Res. 2002, 310, 257–270. [Google Scholar] [CrossRef]
- Bensalma, S.; Chadeneau, C.; Legigan, T.; Renoux, B.; Gaillard, A.; de Boisvilliers, M.; Pinet-Charvet, C.; Papot, S.; Muller, J.M. Evaluation of cytotoxic properties of a cyclopamine glucuronide prodrug in rat glioblastoma cells and tumors. J. Mol. Neurosci. 2015, 55, 51–61. [Google Scholar] [CrossRef]
- Lamouche, S.; Yamaguchi, N. Role of PAC(1) receptor in adrenal catecholamine secretion induced by PACAP and VIP in vivo. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 280, R510–R518. [Google Scholar] [CrossRef]
- Summers, M.A.; O’Dorisio, M.S.; Cox, M.O.; Lara-Marquez, M.; Goetzl, E.J. A lymphocyte-generated fragment of vasoactive intestinal peptide with VPAC1 agonist activity and VPAC2 antagonist effects. J. Pharmacol. Exp. Ther. 2003, 306, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Lutz, E.M.; Ronaldson, E.; Shaw, P.; Johnson, M.S.; Holland, P.J.; Mitchell, R. Characterization of novel splice variants of the PAC1 receptor in human neuroblastoma cells: Consequences for signaling by VIP and PACAP. Mol. Cell. Neurosci. 2006, 31, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, Q.; Yen, C.-J.; Xia, W.; Izzo, J.G.; Lang, J.-Y.; Li, C.-W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Ruiz i Altaba, A. Hedgehog signaling and the Gli code in stem cells, cancer, and metastases. Sci. Signal. 2011, 4, pt9. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Xu, Z. Cross-signaling among phosphinositide-3 kinase, mitogen-activated protein kinase and sonic hedgehog pathways exists in esophageal cancer. Int. J. Cancer 2011, 129, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz i Altaba, A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Lin, H.; Huang, H.-J.S.; Cavenee, W.K. Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc. Natl. Acad. Sci. USA 1997, 94, 12479–12484. [Google Scholar] [CrossRef]
- Cosentino, C.; Di Domenico, M.; Porcellini, A.; Cuozzo, C.; De Gregorio, G.; Santillo, M.R.; Agnese, S.; Di Stasio, R.; Feliciello, A.; Migliaccio, A.; et al. p85 regulatory subunit of PI3K mediates cAMP-PKA and estrogens biological effects on growth and survival. Oncogene 2007, 26, 2095–2103. [Google Scholar] [CrossRef]
- Lochner, A.; Moolman, J.A. The many faces of H89: A review. Cardiovasc. Drug Rev. 2006, 24, 261–274. [Google Scholar] [CrossRef]
- Murray, A.J. Pharmacological PKA Inhibition: All May Not Be What It Seems. Sci. Signal. 2008, 1, re4. [Google Scholar] [CrossRef] [PubMed]
- Bourgault, S.; Vaudry, D.; Botia, B.; Couvineau, A.; Laburthe, M.; Vaudry, H.; Fournier, A. Novel stable PACAP analogs with potent activity towards the PAC1 receptor. Peptides 2008, 29, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Bourgault, S.; Vaudry, D.; Dejda, A.; Doan, N.D.; Vaudry, H.; Fournier, A. Pituitary adenylate cyclase-activating polypeptide: Focus on structure-activity relationships of a neuroprotective Peptide. Curr. Med. Chem. 2009, 16, 4462–4480. [Google Scholar] [CrossRef] [PubMed]
- Doan, N.-D.; Bourgault, S.; Dejda, A.; Létourneau, M.; Detheux, M.; Vaudry, D.; Vaudry, H.; Chatenet, D.; Fournier, A. Design and in vitro characterization of PAC1/VPAC1-selective agonists with potent neuroprotective effects. Biochem. Pharmacol. 2011, 81, 552–561. [Google Scholar] [CrossRef] [PubMed]
- De Boisvilliers, M.; Perrin, F.; Hebache, S.; Balandre, A.-C.; Bensalma, S.; Garnier, A.; Vaudry, D.; Fournier, A.; Festy, F.; Muller, J.-M.; et al. VIP and PACAP analogs regulate therapeutic targets in high-risk neuroblastoma cells. Peptides 2016, 78, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Benda, P.; Lightbody, J.; Sato, G.; Levine, L.; Sweet, W. Differentiated rat glial cell strain in tissue culture. Science 1968, 161, 370–371. [Google Scholar] [CrossRef] [PubMed]
- Nakada, M.; Anderson, E.M.; Demuth, T.; Nakada, S.; Reavie, L.B.; Drake, K.L.; Hoelzinger, D.B.; Berens, M.E. The phosphorylation of ephrin-B2 ligand promotes glioma cell migration and invasion. Int. J. Cancer 2010, 126, 1155–1165. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bensalma, S.; Turpault, S.; Balandre, A.-C.; De Boisvilliers, M.; Gaillard, A.; Chadéneau, C.; Muller, J.-M. PKA at a Cross-Road of Signaling Pathways Involved in the Regulation of Glioblastoma Migration and Invasion by the Neuropeptides VIP and PACAP. Cancers 2019, 11, 123. https://doi.org/10.3390/cancers11010123
Bensalma S, Turpault S, Balandre A-C, De Boisvilliers M, Gaillard A, Chadéneau C, Muller J-M. PKA at a Cross-Road of Signaling Pathways Involved in the Regulation of Glioblastoma Migration and Invasion by the Neuropeptides VIP and PACAP. Cancers. 2019; 11(1):123. https://doi.org/10.3390/cancers11010123
Chicago/Turabian StyleBensalma, Souheyla, Soumaya Turpault, Annie-Claire Balandre, Madryssa De Boisvilliers, Afsaneh Gaillard, Corinne Chadéneau, and Jean-Marc Muller. 2019. "PKA at a Cross-Road of Signaling Pathways Involved in the Regulation of Glioblastoma Migration and Invasion by the Neuropeptides VIP and PACAP" Cancers 11, no. 1: 123. https://doi.org/10.3390/cancers11010123
APA StyleBensalma, S., Turpault, S., Balandre, A.-C., De Boisvilliers, M., Gaillard, A., Chadéneau, C., & Muller, J.-M. (2019). PKA at a Cross-Road of Signaling Pathways Involved in the Regulation of Glioblastoma Migration and Invasion by the Neuropeptides VIP and PACAP. Cancers, 11(1), 123. https://doi.org/10.3390/cancers11010123