Alternating Electric Fields (TTFields) Activate Cav1.2 Channels in Human Glioblastoma Cells


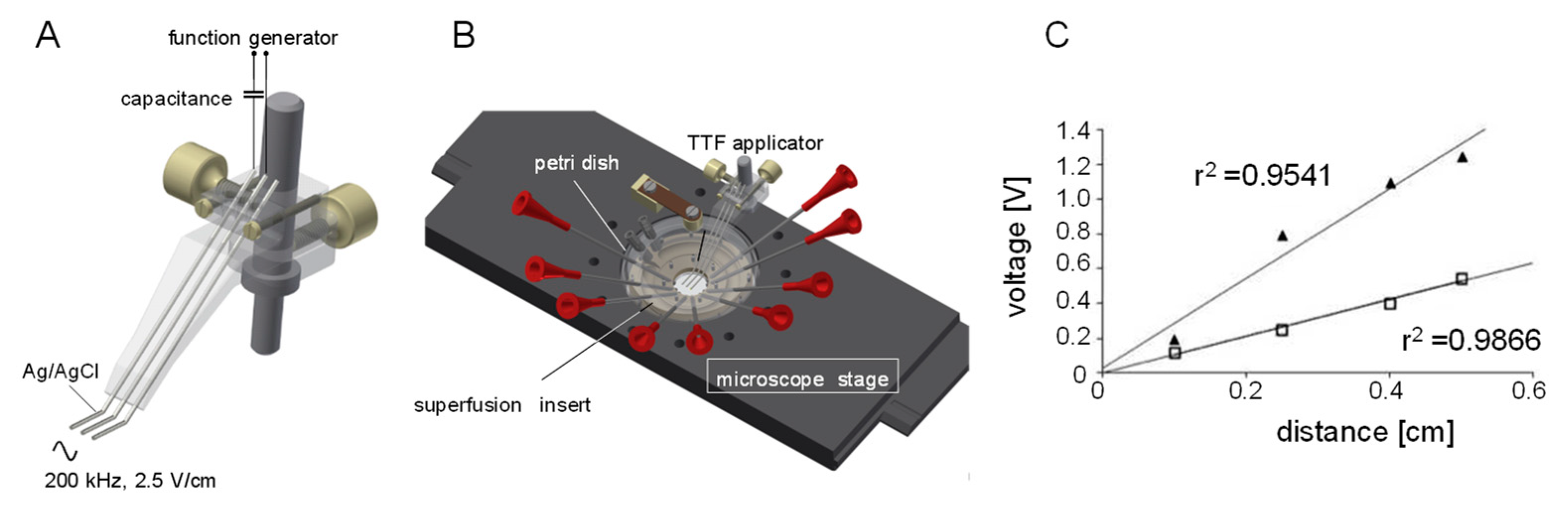{kind=link}
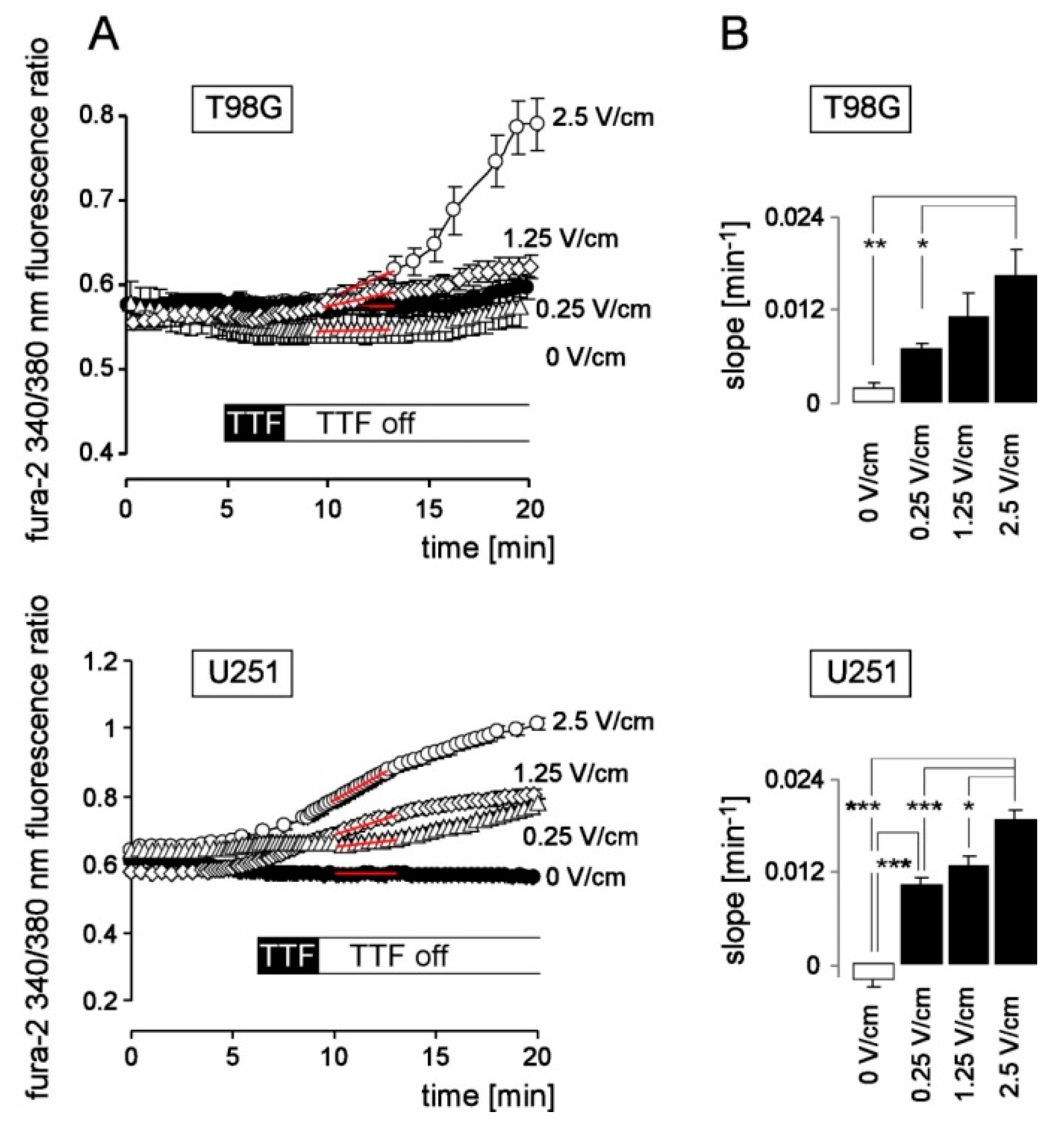{kind=link}
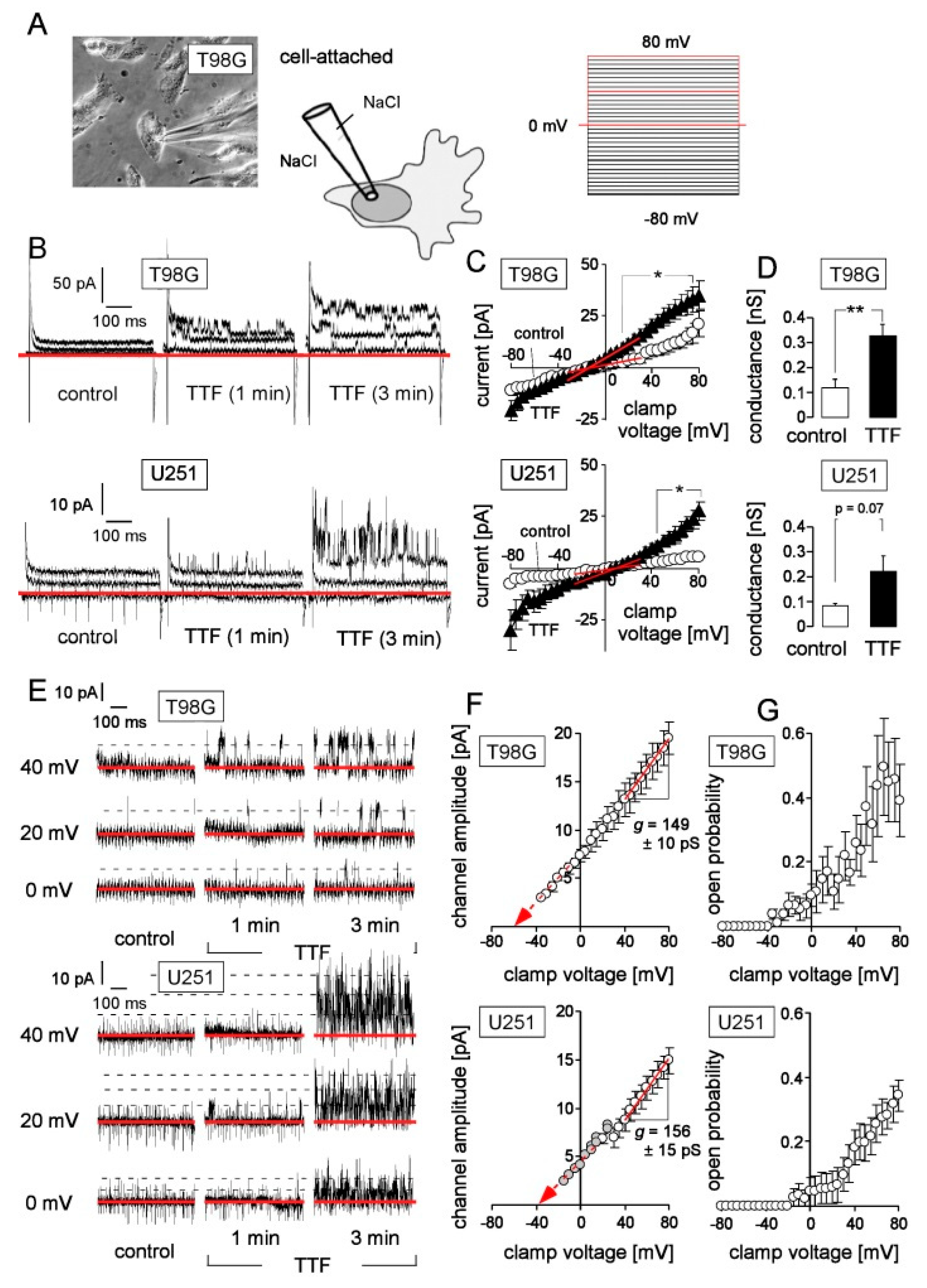{kind=link}
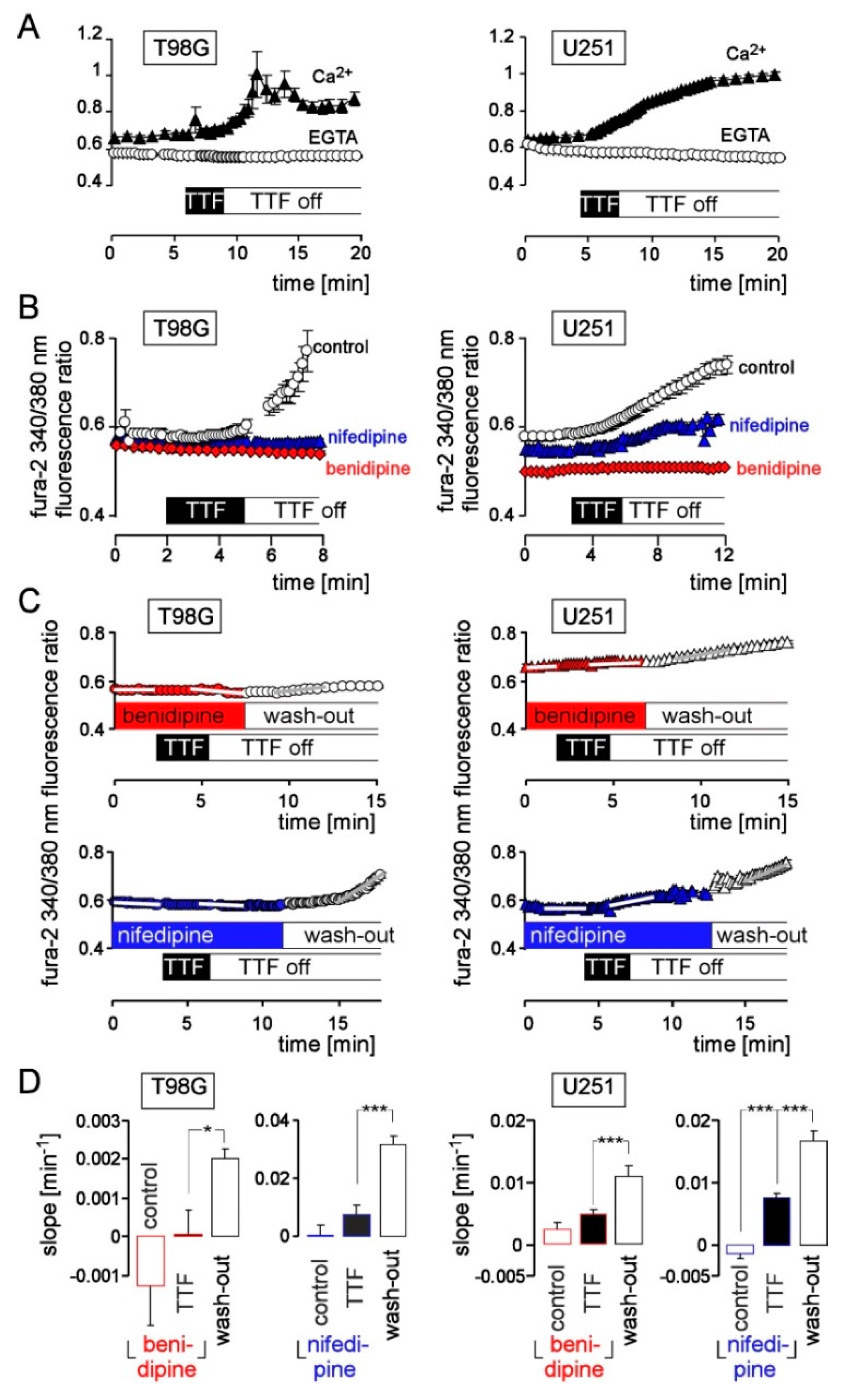{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
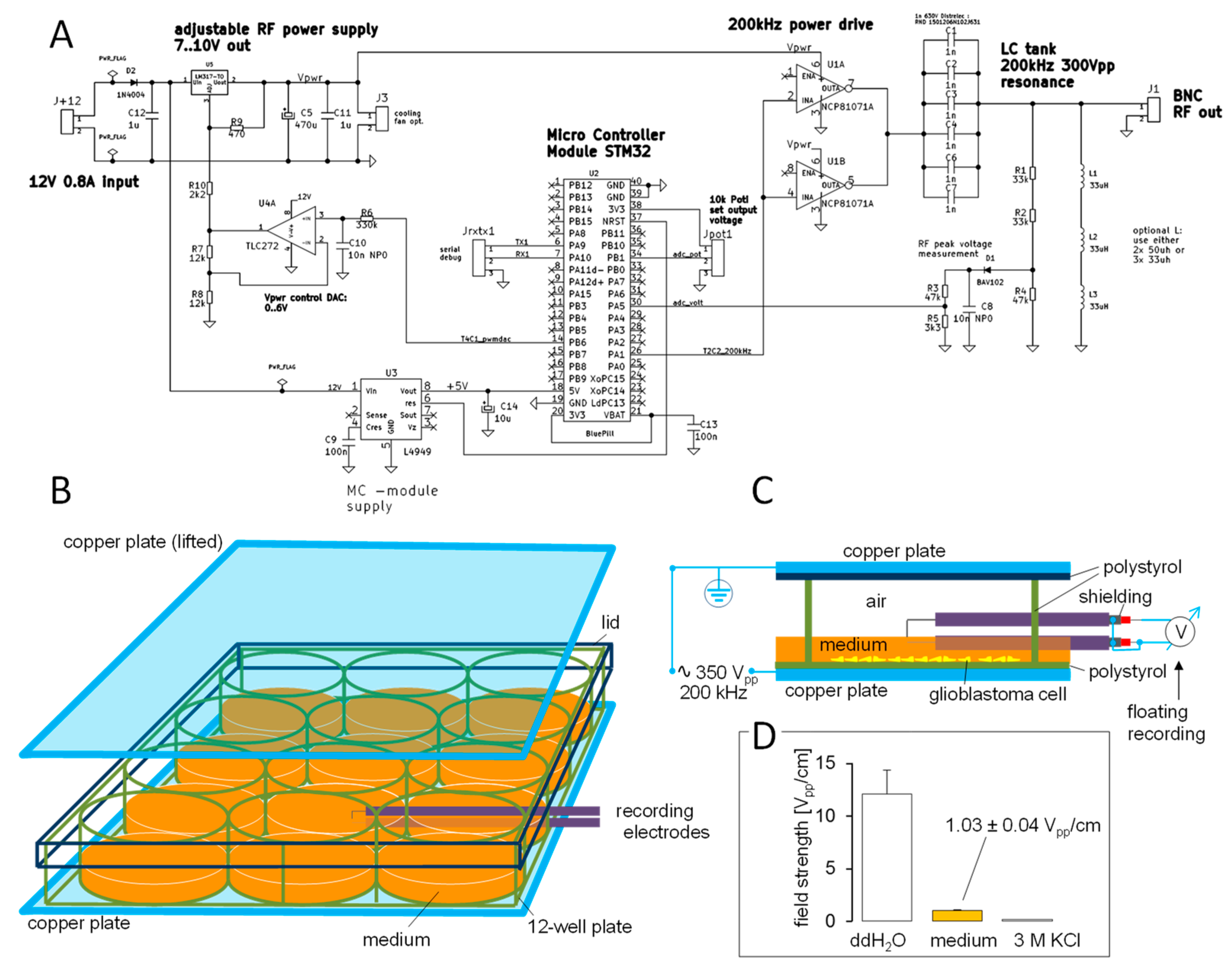
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Fura-2 Fluorescence Imaging of Cytosolic Free Ca2+ Concentration (free[Ca2+]i)
2.3. Patch Clamp Recording
2.4. Quantitative RT-PCR
2.5. Analysis of Cell Cycle, DNA Degradation and Aneuploidy
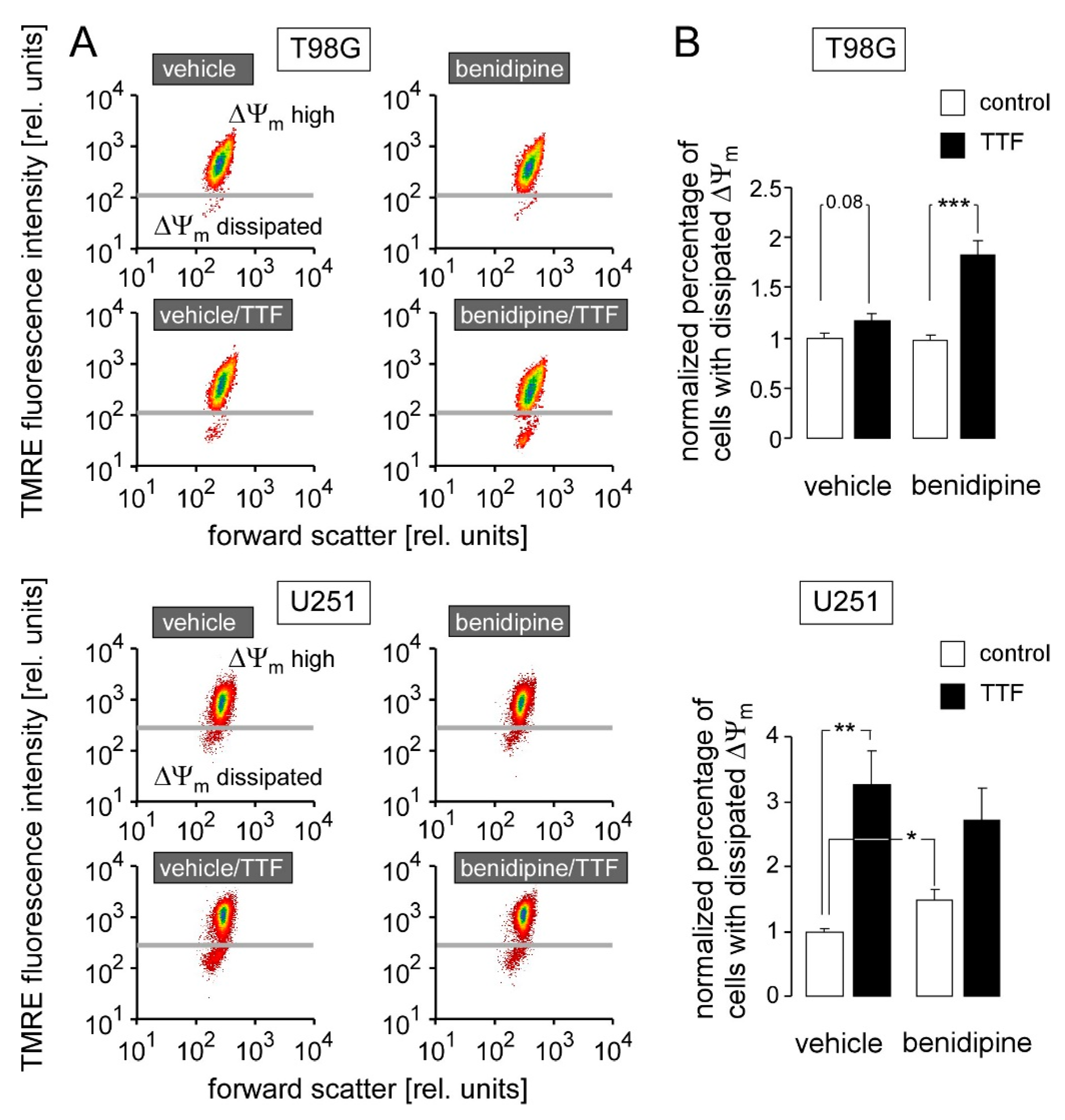
2.6. Determination of Inner Mitochondrial Membrane Potential (ΔΨm)
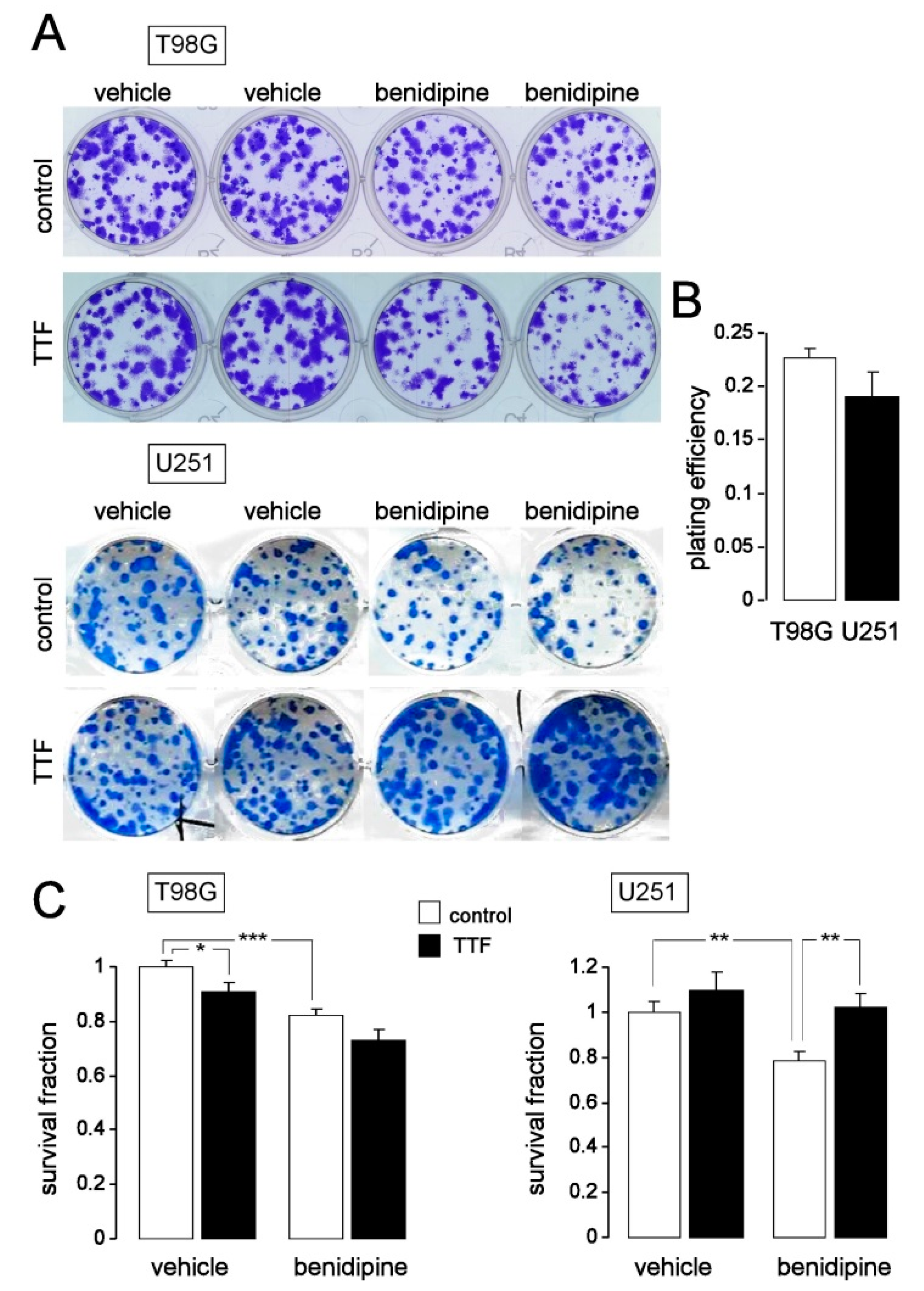
2.7. Colony Formation Assay
2.8. Statistics
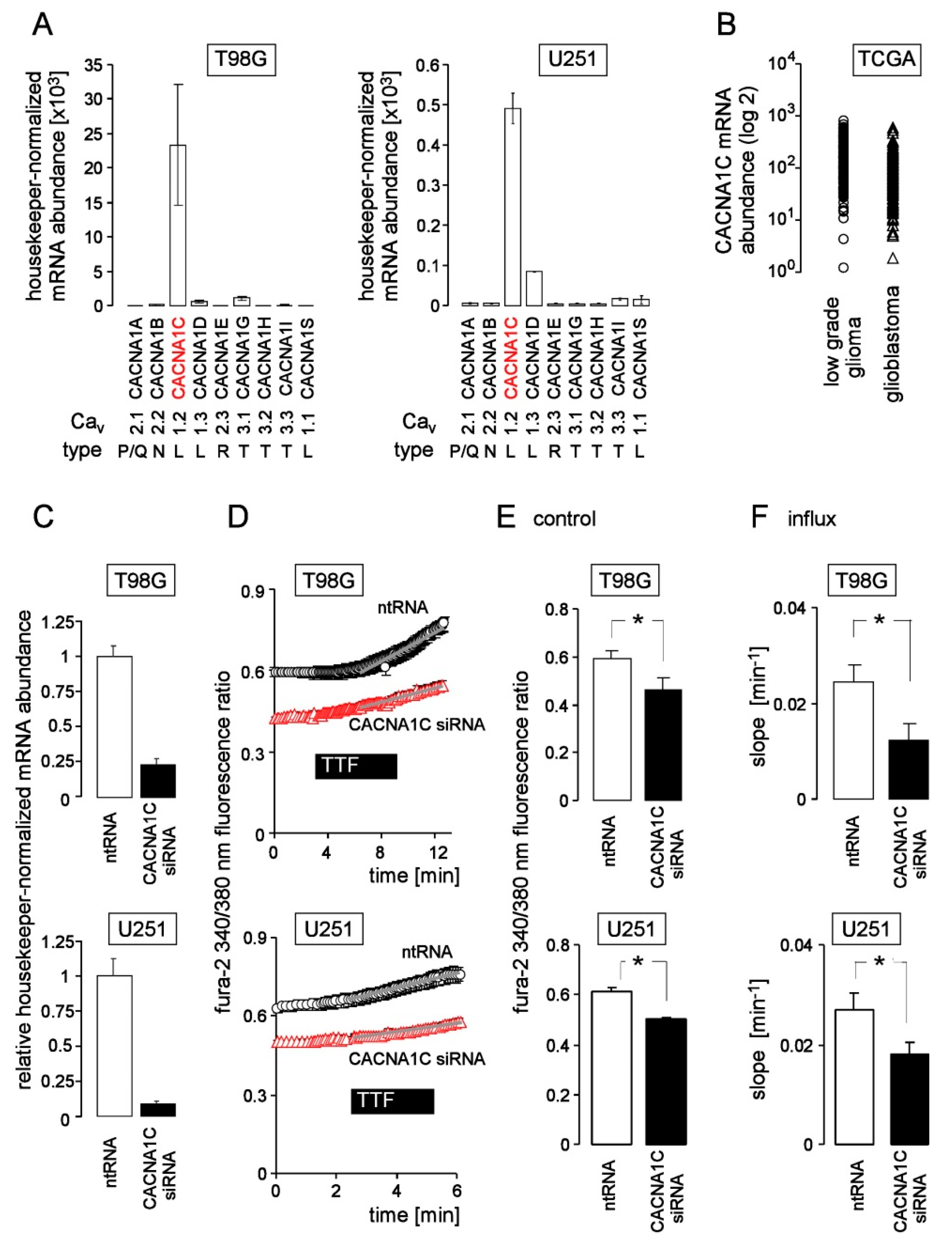
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Wong, E.T.; Kanner, A.A.; Steinberg, D.; Engelhard, H.; Heidecke, V.; Kirson, E.D.; Taillibert, S.; Liebermann, F.; Dbaly, V.; et al. NovoTTF-100A versus physician’s choice chemotherapy in recurrent glioblastoma: A randomised phase III trial of a novel treatment modality. Eur. J. Cancer (Oxford, England: 1990) 2012, 48, 2192–2202. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Lacouture, M.E.; Davis, M.E.; Elzinga, G.; Butowski, N.; Tran, D.; Villano, J.L.; DiMeglio, L.; Davies, A.M.; Wong, E.T. Characterization and management of dermatologic adverse events with the NovoTTF-100A System, a novel anti-mitotic electric field device for the treatment of recurrent glioblastoma. Semin. Oncol. 2014, 41 (Suppl. 4), S1–S14. [Google Scholar] [CrossRef]
- Kirson, E.D.; Gurvich, Z.; Schneiderman, R.; Dekel, E.; Itzhaki, A.; Wasserman, Y.; Schatzberger, R.; Palti, Y. Disruption of cancer cell replication by alternating electric fields. Cancer Res. 2004, 64, 3288–3295. [Google Scholar] [CrossRef] [PubMed]
- Giladi, M.; Schneiderman, R.S.; Porat, Y.; Munster, M.; Itzhaki, A.; Mordechovich, D.; Cahal, S.; Kirson, E.D.; Weinberg, U.; Palti, Y. Mitotic disruption and reduced clonogenicity of pancreatic cancer cells in vitro and in vivo by tumor treating fields. Pancreatology 2014, 14, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Giladi, M.; Schneiderman, R.S.; Voloshin, T.; Porat, Y.; Munster, M.; Blat, R.; Sherbo, S.; Bomzon, Z.; Urman, N.; Itzhaki, A.; et al. Mitotic Spindle Disruption by Alternating Electric Fields Leads to Improper Chromosome Segregation and Mitotic Catastrophe in Cancer Cells. Sci. Rep. 2015, 5, 18046. [Google Scholar] [CrossRef] [PubMed]
- Gera, N.; Yang, A.; Holtzman, T.S.; Lee, S.X.; Wong, E.T.; Swanson, K.D. Tumor treating fields perturb the localization of septins and cause aberrant mitotic exit. PLoS ONE 2015, 10, e0125269. [Google Scholar] [CrossRef]
- Gutin, P.H.; Wong, E.T. Noninvasive application of alternating electric fields in glioblastoma: A fourth cancer treatment modality. Am. Soc. Clin. Oncol. Educ. Book. 2012, 32, 126–131. [Google Scholar] [CrossRef]
- Kirson, E.D.; Dbaly, V.; Tovarys, F.; Vymazal, J.; Soustiel, J.F.; Itzhaki, A.; Mordechovich, D.; Steinberg-Shapira, S.; Gurvich, Z.; Schneiderman, R.; et al. Alternating electric fields arrest cell proliferation in animal tumor models and human brain tumors. Proc. Natl. Acad. Sci. USA 2007, 104, 10152–10157. [Google Scholar] [CrossRef]
- Kirson, E.D.; Schneiderman, R.S.; Dbaly, V.; Tovarys, F.; Vymazal, J.; Itzhaki, A.; Mordechovich, D.; Gurvich, Z.; Shmueli, E.; Goldsher, D.; et al. Chemotherapeutic treatment efficacy and sensitivity are increased by adjuvant alternating electric fields (TTFields). BMC Med. Phys. 2009, 9, 1. [Google Scholar] [CrossRef]
- Schneiderman, R.S.; Shmueli, E.; Kirson, E.D.; Palti, Y. TTFields alone and in combination with chemotherapeutic agents effectively reduce the viability of MDR cell sub-lines that over-express ABC transporters. BMC Cancer 2010, 10, 229. [Google Scholar] [CrossRef] [PubMed]
- Giladi, M.; Weinberg, U.; Schneiderman, R.S.; Porat, Y.; Munster, M.; Voloshin, T.; Blatt, R.; Cahal, S.; Itzhaki, A.; Onn, A.; et al. Alternating electric fields (tumor-treating fields therapy) can improve chemotherapy treatment efficacy in non-small cell lung cancer both in vitro and in vivo. Semin. Oncol. 2014, 41 (Suppl. 6), S35–S41. [Google Scholar] [CrossRef]
- Voloshin, T.; Munster, M.; Blatt, R.; Shteingauz, A.; Roberts, P.C.; Schmelz, E.M.; Giladi, M.; Schneiderman, R.S.; Zeevi, E.; Porat, Y.; et al. Alternating electric fields (TTFields) in combination with paclitaxel are therapeutically effective against ovarian cancer cells in vitro and in vivo. Int. J. Cancer 2016, 139, 2850–2858. [Google Scholar] [CrossRef] [PubMed]
- Karanam, N.K.; Srinivasan, K.; Ding, L.; Sishc, B.; Saha, D.; Story, M.D. Tumor-treating fields elicit a conditional vulnerability to ionizing radiation via the downregulation of BRCA1 signaling and reduced DNA double-strand break repair capacity in non-small cell lung cancer cell lines. Cell Death Dis. 2017, 8, e2711. [Google Scholar] [CrossRef] [PubMed]
- Kirson, E.D.; Giladi, M.; Gurvich, Z.; Itzhaki, A.; Mordechovich, D.; Schneiderman, R.S.; Wasserman, Y.; Ryffel, B.; Goldsher, D.; Palti, Y. Alternating electric fields (TTFields) inhibit metastatic spread of solid tumors to the lungs. Clin. Exp. Metastasis 2009, 26, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Castellvi, Q.; Ginesta, M.M.; Capella, G.; Ivorra, A. Tumor growth delay by adjuvant alternating electric fields which appears non-thermally mediated. Bioelectrochemistry 2015, 105, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Guo, C.; Wang, Z.; Gao, K.; Shi, X.; Liu, J. Electrical stimulation towards melanoma therapy via liquid metal printed electronics on skin. Clin. Transl. Med. 2016, 5, 21. [Google Scholar] [CrossRef]
- Salzberg, M.; Kirson, E.; Palti, Y.; Rochlitz, C. A pilot study with very low-intensity, intermediate-frequency electric fields in patients with locally advanced and/or metastatic solid tumors. Onkologie 2008, 31, 362–365. [Google Scholar] [CrossRef]
- Pless, M.; Droege, C.; von Moos, R.; Salzberg, M.; Betticher, D. A phase I/II trial of Tumor Treating Fields (TTFields) therapy in combination with pemetrexed for advanced non-small cell lung cancer. Lung Cancer 2013, 81, 445–450. [Google Scholar] [CrossRef]
- Vergote, I.; von Moos, R.; Manso, L.; Van Nieuwenhuysen, E.; Concin, N.; Sessa, C. Tumor Treating Fields in combination with paclitaxel in recurrent ovarian carcinoma: Results of the INNOVATE pilot study. Gynecol. Oncol. 2018, 150, 471–477. [Google Scholar] [CrossRef]
- Porat, Y.; Giladi, M.; Schneiderman, R.S.; Blat, R.; Shteingauz, A.; Zeevi, E.; Munster, M.; Voloshin, T.; Kaynan, N.; Tal, O.; et al. Determining the Optimal Inhibitory Frequency for Cancerous Cells Using Tumor Treating Fields (TTFields). J. Vis. Exp. 2017. [Google Scholar] [CrossRef] [PubMed]
- Meletath, S.K.; Pavlick, D.; Brennan, T.; Hamilton, R.; Chmielecki, J.; Elvin, J.A.; Palma, N.; Ross, J.S.; Miller, V.A.; Stephens, P.J.; et al. Personalized Treatment for a Patient With a BRAF V600E Mutation Using Dabrafenib and a Tumor Treatment Fields Device in a High-Grade Glioma Arising From Ganglioglioma. J. Natl. Compr. Cancer Netw. 2016, 14, 1345–1350. [Google Scholar] [CrossRef]
- Giladi, M.; Munster, M.; Schneiderman, R.S.; Voloshin, T.; Porat, Y.; Blat, R.; Zielinska-Chomej, K.; Haag, P.; Bomzon, Z.; Kirson, E.D.; et al. Tumor treating fields (TTFields) delay DNA damage repair following radiation treatment of glioma cells. Radiat. Oncol. 2017, 12, 206. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.T.; Lok, E.; Gautam, S.; Swanson, K.D. Dexamethasone exerts profound immunologic interference on treatment efficacy for recurrent glioblastoma. Br. J. Cancer 2015, 113, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Bellono, N.W.; Leitch, D.B.; Julius, D. Molecular basis of ancestral vertebrate electroreception. Nature 2017, 543, 391–396. [Google Scholar] [CrossRef]
- Morgado-Valle, C.; Verdugo-Diaz, L.; Garcia, D.E.; Morales-Orozco, C.; Drucker-Colin, R. The role of voltage-gated Ca2+ channels in neurite growth of cultured chromaffin cells induced by extremely low frequency (ELF) magnetic field stimulation. Cell Tissue Res. 1998, 291, 217–230. [Google Scholar] [CrossRef]
- Grassi, C.; D’Ascenzo, M.; Torsello, A.; Martinotti, G.; Wolf, F.; Cittadini, A.; Azzena, G.B. Effects of 50 Hz electromagnetic fields on voltage-gated Ca2+ channels and their role in modulation of neuroendocrine cell proliferation and death. Cell Calcium 2004, 35, 307–315. [Google Scholar] [CrossRef]
- Piacentini, R.; Ripoli, C.; Mezzogori, D.; Azzena, G.B.; Grassi, C. Extremely low-frequency electromagnetic fields promote in vitro neurogenesis via upregulation of Ca(v)1-channel activity. J. Cell. Physiol. 2008, 215, 129–139. [Google Scholar] [CrossRef]
- Li, Y.; Yan, X.; Liu, J.; Li, L.; Hu, X.; Sun, H.; Tian, J. Pulsed electromagnetic field enhances brain-derived neurotrophic factor expression through L-type voltage-gated calcium channel- and Erk-dependent signaling pathways in neonatal rat dorsal root ganglion neurons. Neurochem. Int. 2014, 75, 96–104. [Google Scholar] [CrossRef]
- Luo, F.L.; Yang, N.; He, C.; Li, H.L.; Li, C.; Chen, F.; Xiong, J.X.; Hu, Z.A.; Zhang, J. Exposure to extremely low frequency electromagnetic fields alters the calcium dynamics of cultured entorhinal cortex neurons. Environ. Res. 2014, 135, 236–246. [Google Scholar] [CrossRef]
- Burke, R.C.; Bardet, S.M.; Carr, L.; Romanenko, S.; Arnaud-Cormos, D.; Leveque, P.; O’Connor, R.P. Nanosecond pulsed electric fields depolarize transmembrane potential via voltage-gated K+, Ca2+ and TRPM8 channels in U87 glioblastoma cells. Biochim. Biophys. Acta Biomembr. 2017, 1859, 2040–2050. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Sohn, U.D.; Kim, H.G.; Kim, H.R. Exposure to 835 MHz RF-EMF decreases the expression of calcium channels, inhibits apoptosis, but induces autophagy in the mouse hippocampus. Korean J. Physiol. Pharm. 2018, 22, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Pall, M.L. Electromagnetic fields act via activation of voltage-gated calcium channels to produce beneficial or adverse effects. J. Cell Mol. Med. 2013, 17, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Pall, M.L. Scientific evidence contradicts findings and assumptions of Canadian Safety Panel 6: Microwaves act through voltage-gated calcium channel activation to induce biological impacts at non-thermal levels, supporting a paradigm shift for microwave/lower frequency electromagnetic field action. Rev. Environ. Health 2015, 30, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Pall, M.L. Microwave frequency electromagnetic fields (EMFs) produce widespread neuropsychiatric effects including depression. J. Chem. Neuroanat. 2016, 75, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, J.; Jiang, D.; Zhang, D.; Qian, Z.; Liu, C.; Tao, J. Inhibition of T-type Ca2+ channels by endostatin attenuates human glioblastoma cell proliferation and migration. Br. J. Pharmacol. 2012, 166, 1247–1260. [Google Scholar] [CrossRef]
- Steinle, M.; Palme, D.; Misovic, M.; Rudner, J.; Dittmann, K.; Lukowski, R.; Ruth, P.; Huber, S.M. Ionizing radiation induces migration of glioblastoma cells by activating BK K+ channels. Radiother. Oncol. 2011, 101, 122–126. [Google Scholar] [CrossRef]
- Catacuzzeno, L.; Caramia, M.; Sforna, L.; Belia, S.; Guglielmi, L.; D’Adamo, M.C.; Pessia, M.; Franciolini, F. Reconciling the discrepancies on the involvement of large-conductance Ca2+-activated K channels in glioblastoma cell migration. Front. Cell. Neurosci. 2015, 9, 152. [Google Scholar] [CrossRef]
- Edalat, L.; Stegen, B.; Klumpp, L.; Haehl, E.; Schilbach, K.; Lukowski, R.; Kühnle, M.; Bernhardt, G.; Buschauer, A.; Zips, D.; et al. BK K+ channel blockade inhibits radiation-induced migration/brain infiltration of glioblastoma cells. Oncotarget 2016, 7, 14259–14278. [Google Scholar] [CrossRef]
- Rosa, P.; Catacuzzeno, L.; Sforna, L.; Mangino, G.; Carlomagno, S.; Mincione, G.; Petrozza, V.; Ragona, G.; Franciolini, F.; Calogero, A. BK channels blockage inhibits hypoxia-induced migration and chemoresistance to cisplatin in human glioblastoma cells. J. Cell. Physiol. 2018, 233, 6866–6877. [Google Scholar] [CrossRef] [PubMed]
- Stegen, B.; Butz, L.; Klumpp, L.; Zips, D.; Dittmann, K.; Ruth, P.; Huber, S.M. Ca2+-Activated IK K+ Channel Blockade Radiosensitizes Glioblastoma Cells. Mol. Cancer Res. 2015, 13, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, L.; Sezgin, E.C.; Skardelly, M.; Eckert, F.; Huber, S.M. KCa3.1 channels and glioblastoma: In vitro studies. Curr. Neuropharmacol. 2018, 16, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Catacuzzeno, L.; Franciolini, F. Role of KCa3.1 Channels in Modulating Ca2+ Oscillations during Glioblastoma Cell Migration and Invasion. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Eckert, M.; Klumpp, L.; Huber, S.M. Cellular Effects of the Antiepileptic Drug Valproic Acid in Glioblastoma. Cell. Physiol. Biochem. 2017, 44, 1591–1605. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Wolf, M.; Gulich, R.; Lunkenheimer, P.; Loidl, A. Broadband dielectric spectroscopy on human blood. Biochim. Biophys. Acta 2011, 1810, 727–740. [Google Scholar] [CrossRef]
- Huang, H.; Lin, H.; Zhang, X.; Li, J. Resveratrol reverses temozolomide resistance by downregulation of MGMT in T98G glioblastoma cells by the NF-kappaB-dependent pathway. Oncol. Rep. 2012, 27, 2050–2056. [Google Scholar] [CrossRef]
- Qiu, Z.K.; Shen, D.; Chen, Y.S.; Yang, Q.Y.; Guo, C.C.; Feng, B.H.; Chen, Z.P. Enhanced MGMT expression contributes to temozolomide resistance in glioma stem-like cells. Chin. J. Cancer 2014, 33, 115–122. [Google Scholar] [CrossRef]
- Silginer, M.; Weller, M.; Stupp, R.; Roth, P. Biological activity of tumor-treating fields in preclinical glioma models. Cell Death Dis. 2017, 8, e2753. [Google Scholar] [CrossRef]
- Clark, P.A.; Gaal, J.T.; Strebe, J.K.; Pasch, C.A.; Deming, D.A.; Kuo, J.S.; Robins, H.I. The effects of tumor treating fields and temozolomide in MGMT expressing and non-expressing patient-derived glioblastoma cells. J. Clin. Neurosci. 2017, 36, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, J. The current and potential role of hyperthermia in radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 1989, 16, 535–549. [Google Scholar] [CrossRef]
- Kessler, A.F.; Frombling, G.E.; Gross, F.; Hahn, M.; Dzokou, W.; Ernestus, R.I.; Lohr, M.; Hagemann, C. Effects of tumor treating fields (TTFields) on glioblastoma cells are augmented by mitotic checkpoint inhibition. Cell Death Discov. 2018, 4, 12. [Google Scholar] [CrossRef]
- Navedo, M.F.; Amberg, G.C.; Votaw, V.S.; Santana, L.F. Constitutively active L-type Ca2+ channels. Proc. Natl. Acad. Sci. USA 2005, 102, 11112–11117. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Shim, J.H.; Kim, M.; Sun, Y.H.; Kwak, H.S.; Yan, X.; Choi, B.C.; Im, C.; Sim, S.S.; Jeong, J.H.; et al. MLCK and PKC Involvements via Gi and Rho A Protein in Contraction by the Electrical Field Stimulation in Feline Esophageal Smooth Muscle. Korean J. Physiol. Pharm. 2010, 14, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, D.; Frank, S.C.; Klumpp, L.; Sezgin, E.C.; Eckert, M.; Edalat, L.; Bastmeyer, M.; Zips, D.; Ruth, P.; Huber, S.M. TRPM8 is required for survival and radioresistance of glioblastoma cells. Oncotarget 2017, 8, 95896–95913. [Google Scholar] [CrossRef]
- Stegen, B.; Klumpp, L.; Misovic, M.; Edalat, L.; Eckert, M.; Klumpp, D.; Ruth, P.; Huber, S.M. K+ channel signaling in irradiated tumor cells. Eur. Biophys. J. 2016, 45, 585–598. [Google Scholar] [CrossRef]
- Yao, K.; Nagashima, K.; Miki, H. Pharmacological, pharmacokinetic, and clinical properties of benidipine hydrochloride, a novel, long-acting calcium channel blocker. J. Pharmacol. Sci. 2006, 100, 243–261. [Google Scholar] [CrossRef]
- Kobayashi, H.; Ohishi, T.; Nishiie, H.; Kobayashi, S.; Inoue, A.; Oka, T.; Nakamizo, N. Absorption, distribution and excretion after oral administration of 14C-benidipine hydrochloride in rats and dogs. Arzneim. Forsch. 1988, 38, 1742–1746. [Google Scholar]
- Larkin, J.G.; Thompson, G.G.; Scobie, G.; Forrest, G.; Drennan, J.E.; Brodie, M.J. Dihydropyridine calcium antagonists in mice: Blood and brain pharmacokinetics and efficacy against pentylenetetrazol seizures. Epilepsia 1992, 33, 760–769. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neuhaus, E.; Zirjacks, L.; Ganser, K.; Klumpp, L.; Schüler, U.; Zips, D.; Eckert, F.; Huber, S.M. Alternating Electric Fields (TTFields) Activate Cav1.2 Channels in Human Glioblastoma Cells. Cancers 2019, 11, 110. https://doi.org/10.3390/cancers11010110
Neuhaus E, Zirjacks L, Ganser K, Klumpp L, Schüler U, Zips D, Eckert F, Huber SM. Alternating Electric Fields (TTFields) Activate Cav1.2 Channels in Human Glioblastoma Cells. Cancers. 2019; 11(1):110. https://doi.org/10.3390/cancers11010110
Chicago/Turabian StyleNeuhaus, Eric, Lisa Zirjacks, Katrin Ganser, Lukas Klumpp, Uwe Schüler, Daniel Zips, Franziska Eckert, and Stephan M. Huber. 2019. "Alternating Electric Fields (TTFields) Activate Cav1.2 Channels in Human Glioblastoma Cells" Cancers 11, no. 1: 110. https://doi.org/10.3390/cancers11010110
APA StyleNeuhaus, E., Zirjacks, L., Ganser, K., Klumpp, L., Schüler, U., Zips, D., Eckert, F., & Huber, S. M. (2019). Alternating Electric Fields (TTFields) Activate Cav1.2 Channels in Human Glioblastoma Cells. Cancers, 11(1), 110. https://doi.org/10.3390/cancers11010110