T-type Calcium Channels in Cancer


Abstract
1. Introduction
2. Classification of Voltage-Activated Ca2+ Channels
3. Biophysical Properties of T-type Ca2+ Channels
4. Expression of T-type Ca2+ Channels in Prostate Cancer
5. Expression of T-type Ca2+ Channels in Breast Cancer
6. Expression of Voltage-Activated Ca2+ Channels in Ovarian and Other Cancers
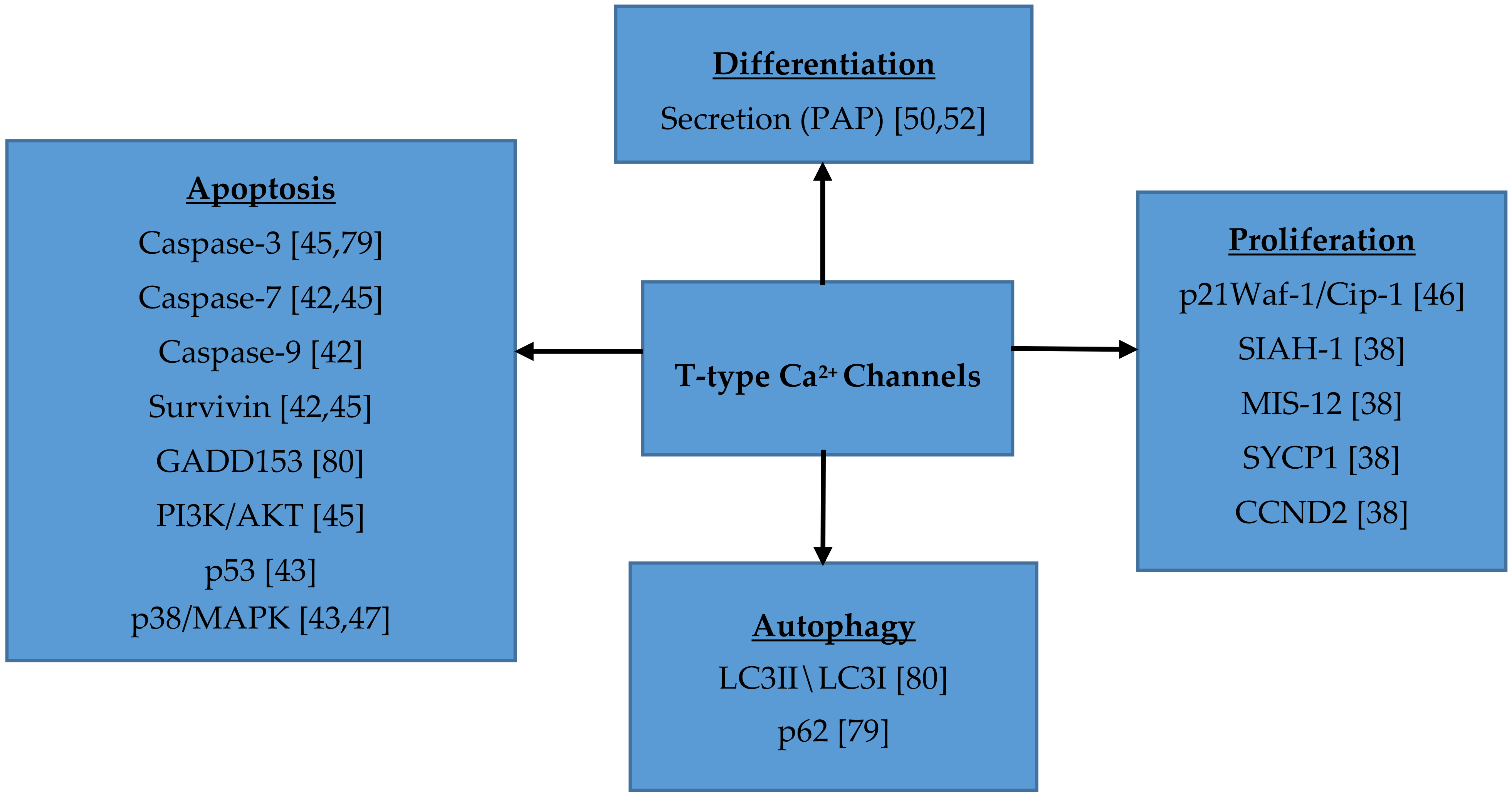
7. Role of T-type Ca2+ Channels in Cell Proliferation, Migration, Survival and Differentiation
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Catterall, W.A. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 2000, 16, 521–555. [Google Scholar] [CrossRef]
- Lambert, R.C.; Bessaih, T.; Leresche, N. Modulation of neuronal T-type calcium channels. CNS Neurol. Disord. Drug Targets 2006, 5, 611–627. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Seagar, M.J.; Jones, J.F.; Reber, B.F.X.; Catterall, W.A. Subunit structure of dihydropyridine-sensitive calcium channels from skeletal muscle. Proc. Natl. Acad. Sci. USA 1987, 84, 5478–5482. [Google Scholar] [CrossRef] [PubMed]
- Ertel, S.I.; Ertel, E.A.; Clozel, J.P. T-type Ca2+ channels and pharmacological blockade: Potential pathophysiological relevance. Cardiovasc. Drugs Ther. 1997, 11, 723–739. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, S.; Byerly, L. Calcium channel. Annu. Rev. Neurosci. 1981, 4, 69–125. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, S.; Ozawa, S.; Sand, O. Voltage clamp analysis of two inward current mechanisms in the egg cell membrane of a starfish. J. Gen. Physiol. 1975, 65, 617–644. [Google Scholar] [CrossRef] [PubMed]
- Ríos, E.; Pizarro, G. Voltage sensor of excitation-contraction coupling in skeletal muscle. Physiol. Rev. 1991, 71, 849–908. [Google Scholar] [CrossRef]
- Perez-Reyes, E. Molecular physiology of low-voltage-activated T-type calcium channels. Physiol. Rev. 2003, 83, 117–161. [Google Scholar] [CrossRef]
- Kang, M.G.; Chen, C.C.; Felix, R.; Letts, V.A.; Frankel, W.N.; Mori, Y.; Campbell, K.P. Biochemical and biophysical evidence for gamma 2 subunit association with neuronal voltage-activated Ca2+ channels. J. Biol. Chem. 2001, 276, 32917–32924. [Google Scholar] [CrossRef] [PubMed]
- Klugbauer, N.; Lacinová, L.; Marais, E.; Hobom, M.; Hofmann, F. Molecular diversity of the calcium channel α2δ subunit. J. Neurosci. 1999, 19, 684–691. [Google Scholar] [CrossRef]
- Gao, B.; Sekido, Y.; Maximov, A.; Saad, M.; Forgacs, E.; Latif, F.; Wei, M.H.; Lerman, M.; Lee, J.H.; Perez-Reyes, E.; et al. Functional properties of a new voltage-dependent calcium channel α2δ auxiliary subunit gene (CACNA2D2). J. Biol. Chem. 2000, 275, 12237–12242. [Google Scholar] [CrossRef]
- Qin, N.; Yagel, S.; Momplaisir, M.L.; Codd, E.E.; D’Andrea, M.R. Molecular cloning and characterization of the human voltage-gated calcium channel α(2)δ-4 subunit. Mol. Pharmacol. 2002, 62, 485–496. [Google Scholar] [CrossRef]
- Gurnett, C.A.; De Waard, M.; Campbell, K.P. Dual function of the voltage-dependent Ca2+ channel α2δ subunit in current stimulation and subunit interaction. Neuron 1996, 16, 431–440. [Google Scholar] [CrossRef]
- Gurnett, C.A.; Felix, R.; Campbell, K.P. Extracellular interaction of the voltage-dependent Ca2+ channel α2δ and α1 subunits. J. Biol. Chem. 1997, 272, 18508–18512. [Google Scholar] [CrossRef] [PubMed]
- Dolphin, A.C.; Wyatt, C.N.; Richards, J.; Beattie, R.E.; Craig, P.; Lee, J.H.; Cribbs, L.L.; Volsen, S.G.; Perez-Reyes, E. The effect of α2δ and other accessory subunits on expression and properties of the calcium channel α1G. J. Physiol. 1999, 519, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Dubel, S.J.; Altier, C.; Chaumont, S.; Lory, P.; Bourinet, E.; Nargeot, J. Plasma membrane expression of T-type calcium channel α1 subunits is modulated by high voltage-activated auxiliary subunits. J. Biol. Chem. 2004, 279, 29263–29269. [Google Scholar] [CrossRef] [PubMed]
- Helton, T.D.; Horne, W.A. Alternative splicing of the β4 subunit has α1 subunit subtype-specific effects on Ca2+ channel gating. J. Neurosci. 2002, 22, 1573–1582. [Google Scholar] [CrossRef]
- Pragnell, M.; De Waard, M.; Mori, Y.; Tanabe, T.; Snutch, T.P.; Campbell, K.P. Calcium channel beta-subunit binds to a conserved motif in the I-II cytoplasmic linker of the α1-subunit. Nature 1994, 368, 67–70. [Google Scholar] [CrossRef]
- De Waard, M.; Pragnell, M.; Campbell, K.P. Ca2+ channel regulation by a conserved beta subunit domain. Neuron 1994, 13, 495–503. [Google Scholar] [CrossRef]
- Bichet, D.; Cornet, V.; Geib, S.; Carlier, E.; Volsen, S.; Hoshi, T.; Mori, Y.; De Waard, M. The I-II loop of the Ca2+ channel α1 subunit contains an endoplasmic reticulum retention signal antagonized by the beta subunit. Neuron 2000, 25, 177–190. [Google Scholar] [CrossRef]
- Leuranguer, V.; Bourinet, E.; Lory, P.; Nargeot, J. Antisense depletion of β-subunits fails to affect T-type calcium channels properties in a neuroblastoma cell line. Neuropharmacology 1998, 37, 701–708. [Google Scholar] [CrossRef]
- Chen, R.S.; Deng, T.C.; Garcia, T.; Sellers, Z.M.; Best, P.M. Calcium channel γ subunits: A functionally diverse protein family. Cell. Biochem. Biophys. 2007, 47, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Rousset, M.; Cens, T.; Restituito, S.; Barrere, C.; Black, J.L., 3rd; McEnery, M.W.; Charnet, P. Functional roles of γ2, γ3 and γ4, three new Ca2+ channel subunits, in P/Q-type Ca2+ channel expressed in Xenopus oocytes. J. Physiol. 2001, 532, 583–593. [Google Scholar] [CrossRef]
- Senatore, A.; Guan, W.; Spafford, J.D. Cav3 T-type channels: Regulators for gating, membrane expression, and cation selectivity. Pflugers Arch. 2014, 466, 645–660. [Google Scholar] [CrossRef]
- Fedulova, S.A.; Kostyuk, P.G.; Veselovsky, N.S. Two types of calcium channels in the somatic membrane of new-born rat dorsal root ganglion neurones. J. Physiol. 1985, 359, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Shcheglovitov, A.; Kostyuk, P.; Shuba, Y. Selectivity signatures of three isoforms of recombinant T-type Ca2+ channels. Biochim. Biophys. Acta 2007, 1768, 1406–1419. [Google Scholar] [CrossRef]
- Lee, J.H.; Gomora, J.C.; Cribbs, L.L.; Perez-Reyes, E. Nickel block of three cloned T-type calcium channels: Low concentrations selectively block α1H. Biophys J. 1999, 77, 3034–3042. [Google Scholar] [CrossRef]
- Narahashi, T.; Tsunoo, A.; Yoshii, M. Characterization of two types of calcium channels in mouse neuroblastoma cells. J. Physiol. 1987, 383, 231–249. [Google Scholar] [CrossRef]
- Tombaugh, G.C.; Somjen, G.G. Differential sensitivity to intracellular pH among high- and low-threshold Ca2+ currents in isolated rat CA1 neurons. J. Neurophysiol. 1997, 77, 639–653. [Google Scholar] [CrossRef]
- Shah, M.J.; Meis, S.; Munsch, T.; Pape, H.C. Modulation by extracellular pH of low- and high-voltage-activated calcium currents of rat thalamic relay neurons. J. Neurophysiol. 2001, 85, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Chemin, J.; Traboulsie, A.; Lory, P. Molecular pathways underlying the modulation of T-type calcium channels by neurotransmitters and hormones. Cell. Calcium 2006, 40, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Chemin, J.; Mezghrani, A.; Bidaud, I.; Dupasquier, S.; Marger, F.; Barrère, C.; Nargeot, J.; Lory, P. Temperature-dependent modulation of CaV3 T-type calcium channels by protein kinases C and A in mammalian cells. J. Biol. Chem. 2007, 282, 32710–32718. [Google Scholar] [CrossRef]
- Chemin, J.; Taiakina, V.; Monteil, A.; Piazza, M.; Guan, W.; Stephens, R.F.; Kitmitto, A.; Pang, Z.P.; Dolphin, A.C.; Perez-Reyes, E.; et al. Calmodulin regulates Cav3 T-type channels at their gating brake. J. Biol. Chem. 2017, 292, 20010–20031. [Google Scholar] [CrossRef] [PubMed]
- Huguenard, J.R.; Prince, D.A. A novel T-type current underlies prolonged Ca2+-dependent burst firing in GABAergic neurons of rat thalamic reticular nucleus. J. Neurosci. 1992, 12, 3804–3817. [Google Scholar] [CrossRef]
- Fry, C.H.; Sui, G.; Wu, C. T-type Ca2+ channels in non-vascular smooth muscles. Cell. Calcium 2006, 40, 231–239. [Google Scholar] [CrossRef]
- Iftinca, M.C.; Zamponi, G.W. Regulation of neuronal T-type calcium channels. Trends Pharmacol. Sci. 2009, 30, 32–40. [Google Scholar] [CrossRef]
- Díaz-Lezama, N.; Hernández-Elvira, M.; Sandoval, A.; Monroy, A.; Felix, R.; Monjaraz, E. Ghrelin inhibits proliferation and increases T-type Ca2+ channel expression in PC-3 human prostate carcinoma cells. Biochem. Biophys. Res. Commun. 2010, 403, 24–29. [Google Scholar] [CrossRef]
- Ohkubo, T.; Yamazaki, J. T-type voltage-activated calcium channel Cav3.1, but not Cav3.2, is involved in the inhibition of proliferation and apoptosis in MCF-7 human breast cancer cells. Int. J. Oncol. 2012, 41, 267–275. [Google Scholar] [CrossRef]
- Bertolesi, G.E.; Shi, C.; Elbaum, L.; Jollimore, C.; Rozenberg, G.; Barnes, S.; Kelly, M.E. The Ca2+ channel antagonists mibefradil and pimozide inhibit cell growth via different cytotoxic mechanisms. Mol. Pharmacol. 2002, 62, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.T.; Huang, L.; Pottle, J.E.; Liu, K.; Yang, Y.; Zeng, X.; Keyser, B.M.; Agrawal, K.C.; Hansen, J.B.; Li, M. Selective blockade of T-type Ca2+ channels suppresses human breast cancer cell proliferation. Cancer Lett. 2008, 267, 116–124. [Google Scholar] [CrossRef]
- Li, W.; Zhang, S.L.; Wang, N.; Zhang, B.B.; Li, M. Blockade of T-type Ca2+ channels inhibits human ovarian cancer cell proliferation. Cancer Investig. 2011, 29, 339–346. [Google Scholar] [CrossRef]
- Dziegielewska, B.; Casarez, E.V.; Yang, W.Z.; Gray, L.S.; Dziegielewski, J.; Slack-Davis, J.K. T-Type Ca2+ Channel Inhibition Sensitizes Ovarian Cancer to Carboplatin. Mol. Cancer Ther. 2016, 15, 460–470. [Google Scholar] [CrossRef]
- Dziegielewska, B.; Brautigan, D.L.; Larner, J.M.; Dziegielewski, J. T-type Ca2+ channel inhibition induces p53-dependent cell growth arrest and apoptosis through activation of p38-MAPK in colon cancer cells. Mol. Cancer Res. 2014, 12, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Latour, I.; Louw, D.F.; Beedle, A.M.; Hamid, J.; Sutherland, G.R.; Zamponi, G.W. Expression of T-type calcium channel splice variants in human glioma. Glia 2004, 48, 112–119. [Google Scholar] [CrossRef]
- Valerie, N.C.; Dziegielewska, B.; Hosing, A.S.; Augustin, E.; Gray, L.S.; Brautigan, D.L.; Larner, J.M.; Dziegielewski, J. Inhibition of T-type calcium channels disrupts Akt signaling and promotes apoptosis in glioblastoma cells. Biochem. Pharmacol. 2013, 85, 888–897. [Google Scholar] [CrossRef]
- Lu, F.; Chen, H.; Zhou, C.; Liu, S.; Guo, M.; Chen, P.; Zhuang, H.; Xie, D.; Wu, S. T-type Ca2+ channel expression in human esophageal carcinomas: A functional role in proliferation. Cell. Calcium 2008, 43, 49–58. [Google Scholar] [CrossRef]
- Li, Y.; Liu, S.; Lu, F.; Zhang, T.; Chen, H.; Wu, S.; Zhuang, H. A role of functional T-type Ca2+ channel in hepatocellular carcinoma cell proliferation. Oncol. Rep. 2009, 22, 1229–1235. [Google Scholar] [PubMed]
- Das, A.; Pushparaj, C.; Bahí, N.; Sorolla, A.; Herreros, J.; Pamplona, R.; Vilella, R.; Matias-Guiu, X.; Martí, R.M.; Cantí, C. Functional expression of voltage-gated calcium channels in human melanoma. Pigment. Cell. Melanoma Res. 2012, 25, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Mariot, P.; Vanoverberghe, K.; Lalevee, N.; Rossier, M.F.; Prevarskaya, N. Overexpression of an alpha 1H (Cav3.2) T-type calcium channel during neuroendocrine differentiation of human prostate cancer cells. J. Biol. Chem. 2002, 277, 10824–10833. [Google Scholar] [CrossRef]
- Gackière, F.; Bidaux, G.; Delcourt, P.; Van Coppenolle, F.; Katsogiannou, M.; Dewailly, E.; Bavencoffe, A.; Van Chuoï-Mariot, M.T.; Mauroy, B.; Prevarskaya, N.; et al. CaV3.2 T-type calcium channels are involved in calcium-dependent secretion of neuroendocrine prostate cancer cells. J. Biol. Chem. 2008, 283, 10162–10173. [Google Scholar] [CrossRef] [PubMed]
- Gackière, F.; Warnier, M.; Katsogiannou, M.; Derouiche, S.; Delcourt, P.; Dewailly, E.; Slomianny, C.; Humez, S.; Prevarskaya, N.; Roudbaraki, M.; et al. Functional coupling between large-conductance potassium channels and Cav3.2 voltage-dependent calcium channels participates in prostate cancer cell growth. Biol. Open 2013, 2, 941–951. [Google Scholar] [CrossRef]
- Fukami, K.; Sekiguchi, F.; Yasukawa, M.; Asano, E.; Kasamatsu, R.; Ueda, M.; Yoshida, S.; Kawabata, A. Functional upregulation of the H2S/Cav3.2 channel pathway accelerates secretory function in neuroendocrine-differentiated human prostate cancer cells. Biochem. Pharmacol. 2015, 97, 300–309. [Google Scholar] [CrossRef]
- Weaver, E.M.; Zamora, F.J.; Hearne, J.L.; Martin-Caraballo, M. Posttranscriptional regulation of T-type Ca2+ channel expression by interleukin-6 in prostate cancer cells. Cytokine 2015, 76, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Weaver, E.M.; Zamora, F.J.; Puplampu-Dove, Y.A.; Kiessu, E.; Hearne, J.L.; Martin-Caraballo, M. Regulation of T-type calcium channel expression by sodium butyrate in prostate cancer cells. Eur. J. Pharmacol. 2015, 749, 20–31. [Google Scholar] [CrossRef]
- Hall, M.; Todd, B.; Allen, E.D., Jr.; Nguyen, N.; Kwon, Y.J.; Nguyen, V.; Hearne, J.L.; Martin-Caraballo, M. Androgen receptor signaling regulates T-type Ca2+ channel expression and neuroendocrine differentiation in prostate cancer cells. Am. J. Cancer Res. 2018, 8, 732–747. [Google Scholar] [PubMed]
- Marker, P.C.; Donjacour, A.A.; Dahiya, R.; Cunha, G.R. Hormonal, cellular, and molecular control of prostatic development. Dev. Biol. 2003, 253, 165–174. [Google Scholar] [CrossRef]
- Abrahamsson, P.A. Neuroendocrine cells in tumour growth of the prostate. Endocr. Relat. Cancer 1999, 6, 503–519. [Google Scholar] [CrossRef]
- Vashchenko, N.; Abrahamsson, P.A. Neuroendocrine differentiation in prostate cancer: Implications for new treatment modalities. Eur. Urol. 2005, 47, 147–155. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Farach, A.; Ding, Y.; Lee, M.; Creighton, C.; Delk, N.A.; Ittmann, M.; Miles, B.; Rowley, D.; Farach-Carson, M.C.; Ayala, G.E. Neuronal Trans-Differentiation in Prostate Cancer Cells. Prostate 2016, 76, 1312–1325. [Google Scholar] [CrossRef]
- Yuan, T.C.; Veeramani, S.; Lin, M.F. Neuroendocrine-like prostate cancer cells: Neuroendocrine transdifferentiation of prostate adenocarcinoma cells. Endocr. Relat. Cancer 2007, 14, 531–547. [Google Scholar] [CrossRef]
- Van Bokhoven, A.; Varella-Garcia, M.; Korch, C.; Johannes, W.U.; Smith, E.E.; Miller, H.L.; Nordeen, S.K.; Miller, G.J.; Lucia, M.S. Molecular characterization of human prostate carcinoma cell lines. Prostate 2003, 57, 205–225. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; You, Z. In vitro and in vivo model systems used in prostate cancer research. J. Biol. Methods 2015, 2, e17. [Google Scholar] [CrossRef] [PubMed]
- Dziegielewska, B.; Gray, L.S.; Dziegielewski, J. T-type calcium channels blockers as new tools in cancer therapies. Pflugers Arch. 2014, 466, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Toyota, M.; Ho, C.; Ohe-Toyota, M.; Baylin, S.B.; Issa, J.P. Inactivation of CACNA1G, a T-type calcium channel gene, by aberrant methylation of its 5′ CpG island in human tumors. Cancer Res. 1999, 59, 4535–4541. [Google Scholar] [PubMed]
- Van Loo, K.M.; Schaub, C.; Pernhorst, K.; Yaari, Y.; Beck, H.; Schoch, S.; Becker, A.J. Transcriptional regulation of T-type calcium channel CaV3.2: Bi-directionality by early growth response 1 (Egr1) and repressor element 1 (RE-1) protein-silencing transcription factor (REST). J. Biol. Chem. 2012, 287, 15489–15501. [Google Scholar] [CrossRef] [PubMed]
- Warnier, M.; Roudbaraki, M.; Derouiche, S.; Delcourt, P.; Bokhobza, A.; Prevarskaya, N.; Mariot, P. CACNA2D2 promotes tumorigenesis by stimulating cell proliferation and angiogenesis. Oncogene 2015, 34, 5383–5394. [Google Scholar] [CrossRef] [PubMed]
- Brinton, L.A.; Schairer, C.; Hoover, R.N.; Fraumeni, J.F., Jr. Menstrual factors and risk of breast cancer. Cancer Investig. 1988, 6, 245–254. [Google Scholar] [CrossRef]
- Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and breastfeeding: Collaborative reanalysis of individual data from 47 epidemiological studies in 30 countries, including 50302 women with breast cancer and 96973 women without the disease. Lancet 2002, 360, 187–195. [Google Scholar] [CrossRef]
- Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and hormone replacement therapy: Collaborative reanalysis of data from 51 epidemiological studies of 52,705 women with breast cancer and 108,411 women without breast cancer. Lancet 1997, 350, 1047–1059. [Google Scholar] [CrossRef]
- Cuzick, J.; Sestak, I.; Bonanni, B.; Costantino, J.P.; Cummings, S.; DeCensi, A.; Dowsett, M.; Forbes, J.F.; Ford, L.; LaCroix, A.Z.; et al. Selective oestrogen receptor modulators in prevention of breast cancer: An updated meta-analysis of individual participant data. Lancet 2013, 381, 1827–1834. [Google Scholar] [CrossRef]
- Lee, A.V.; Oesterreich, S.; Davidson, N.E. MCF-7 cells—Changing the course of breast cancer research and care for 45 years. J. Natl. Cancer Inst. 2015, 107, djv073. [Google Scholar] [CrossRef] [PubMed]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.P.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006, 10, 515–527. [Google Scholar] [CrossRef]
- Horwitz, K.B.; Costlow, M.E.; McGuire, W.L. MCF-7; a human breast cancer cell line with estrogen, androgen, progesterone, and glucocorticoid receptors. Steroids 1975, 26, 785–795. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, M.; Vest, P.A.; Bhattacharjee, A.; Liu, L.; Li, M. A mibefradil metabolite is a potent intracellular blocker of L-type Ca2+ currents in pancreatic β-cells. J. Pharmacol. Exp. Ther. 2000, 292, 939–943. [Google Scholar]
- Kang, J.; Chen, X.L.; Rampe, D. The antipsychotic drugs sertindole and pimozide block erg3, a human brain K+ channel. Biochem. Biophys. Res. Commun. 2001, 286, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Wang, L.; Cai, F.; Rampe, D. High affinity blockade of the HERG cardiac K+ channel by the neuroleptic pimozide. Eur. J. Pharmacol. 2000, 392, 137–140. [Google Scholar] [CrossRef]
- Huang, J.B.; Kindzelskii, A.L.; Clark, A.J.; Petty, H.R. Identification of channels promoting calcium spikes and waves in HT1080 tumor cells: Their apparent roles in cell motility and invasion. Cancer Res. 2004, 64, 2482–2489. [Google Scholar] [CrossRef] [PubMed]
- Krouse, A.J.; Gray, L.; Macdonald, T.; McCray, J. Repurposing and Rescuing of Mibefradil, an Antihypertensive, for Cancer: A Case Study. Assay Drug Dev. Technol. 2015, 13, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, C.; Rudraraju, B.; Monteverde, M.; Lattanzio, L.; Gojis, O.; Brizio, R.; Garrone, O.; Merlano, M.; Syed, N.; Lo Nigro, C.; et al. Methylation of the calcium channel regulatory subunit α2δ-3 (CACNA2D3) predicts site-specific relapse in oestrogen receptor-positive primary breast carcinomas. Br. J. Cancer 2012, 107, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Ha, S.E.; Lee, M.Y.; Kurahashi, M.; Wei, L.; Jorgensen, B.G.; Park, C.; Park, P.J.; Redelman, D.; Sasse, K.C.; Becker, L.S.; et al. Transcriptome analysis of PDGFRα+ cells identifies T-type Ca2+ channel CACNA1G as a new pathological marker for PDGFRα+ cell hyperplasia. PLoS ONE 2017, 12, e0182265. [Google Scholar] [CrossRef]
- Brain Tumors: Types of Brain Tumors. Available online: https://www.aans.org/Patients/Neurosurgical-Conditions-and-Treatments/Brain-Tumors# (accessed on 2 October 2018).
- Ernest, N.J.; Logsdon, N.J.; McFerrin, M.B.; Sontheimer, H.; Spiller, S.E. Biophysical properties of human medulloblastoma cells. J. Membr. Biol. 2010, 237, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Kahl, C.R.; Means, A.R. Regulation of cell cycle progression by calcium/calmodulin-dependent pathways. Endocr. Rev. 2003, 24, 719–736. [Google Scholar] [CrossRef] [PubMed]
- Becchetti, A. Ion channels and transporters in cancer. 1. Ion channels and cell proliferation in cancer. Am. J. Physiol. Cell. Physiol. 2011, 301, C255–C265. [Google Scholar] [CrossRef] [PubMed]
- Holdhoff, M.; Ye, X.; Supko, J.G.; Nabors, L.B.; Desai, A.S.; Walbert, T.; Lesser, G.J.; Read, W.L.; Lieberman, F.S.; Lodge, M.A.; et al. Timed sequential therapy of the selective T-type calcium channel blocker mibefradil and temozolomide in patients with recurrent high-grade gliomas. Neuro Oncol. 2017, 19, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, J.; Jiang, D.; Zhang, D.; Qian, Z.; Liu, C.; Tao, J. Inhibition of T-type Ca2+ channels by endostatin attenuates human glioblastoma cell proliferation and migration. Br. J. Pharmacol. 2012, 166, 1247–1460. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Kondratskyi, A.; Yassine, M.; Kondratska, K.; Skryma, R.; Slomianny, C.; Prevarskaya, N. Calcium-permeable ion channels in control of autophagy and cancer. Front. Physiol 2013, 4, 272. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Pushparaj, C.; Herreros, J.; Nager, M.; Vilella, R.; Portero, M.; Pamplona, R.; Matias-Guiu, X.; Martí, R.M.; Cantí, C. T-type calcium channel blockers inhibit autophagy and promote apoptosis of malignant melanoma cells. Pigment. Cell. Melanoma Res. 2013, 26, 874–885. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Channel | Sample | mRNA | Protein | Functional Channels | Molecular/Functional Regulation | Cellular Function |
|---|---|---|---|---|---|---|
| Cav3.1 | Prostate cancer (PC-3) | + | + | + | Ghrelin ↑ Cav3.1 mRNA, ↑ protein expression | Pharmacological inhibition of channel function promotes apoptosis and decreases proliferation [37] |
| Breast cancer (MCF-7) | + | + | ND | Molecular knockdown of Cav3.1 expression decreases proliferation without any effect on apoptosis Overexpression of Cav3.1 promotes apoptosis and decreases proliferation [38] | ||
| Breast cancer (MCF-7) | + | ND | + | Pharmacological inhibition of channel function decreases proliferation [39] | ||
| Breast cancer (MCF-7, MDA-MB-231) | + | + | + | Cell confluency ↓ Cav3.1 mRNA, ↓ protein expression | Pharmacological inhibition of Cav3.1 decreases proliferation [40] | |
| Retinoblastoma (Y79) | + | ND | + | Pharmacological inhibition of channel function or molecular knockdown of Cav3.1 expression decreases proliferation [39] | ||
| Ovarian cancer (HO8910, A2780) | ND | + | ND | Pharmacological inhibition of channel function or molecular knockdown of Cav3.1 expression decreases proliferation and arrests cells in G0/G1 phase [41] | ||
| Ovarian cancer (A2780, A2780Cis, IGROV-1) (1) | + | ND | ND | Pharmacological inhibition of channel function or molecular knockdown of Cav3.1 expression decreases cell viability, increases apoptosis, arrests cells in G1 and/or G2 phase, decreases survivin and BIRC5 expression, increases sensitivity to carboplatin [42] | ||
| Colon cancer (HCT116 p53wt, HCT116 p53-/-) | + | + | ND | Pharmacological inhibition of channel function or molecular knockdown of Cav3.1 expression decreases proliferation and increases apoptosis [43] | ||
| Glioma (U251N, U563, U87, biopsies) | + | + | + | ND [44] | ||
| Glioblastoma (U251, U87, T98G) | + | ND | ND | Pharmacological inhibition of channel function or molecular knockdown of Cav3.1 expression decreases cell viability and clonogenic potential and increases apoptosis [45] | ||
| Esophageal cancer (TE8) | + | ND | + | Pharmacological inhibition of channel function or molecular knockdown of Cav3.1 expression decreases proliferation without any effect on apoptosis and increases p21CIP1 expression [46] | ||
| Hepatocellular carcinoma (SNU449) | + | ND | + | Pharmacological inhibition of channel function, but not molecular knockdown of Cav3.1 expression, decreases proliferation, increases phosphorylated ERK 1/2, and downregulates certain genes [47] | ||
| Melanoma (M28, JG, M16, M29, M9, melanoma tissue samples) (1) | + | ND | + | Hypoxia ↑ Cav3.1 mRNA expression (M16, JG, M28) | Pharmacological inhibition of channel function or molecular knockdown of Cav3.1 expression arrests cells in G1 phase and decreases cell viability [48] | |
| Cav3.2 | Prostate cancer (LNCaP) | + | ND | + | Induction of NED with Bt2cAMP and IBMX ↑ Cav3.2 mRNA, ↑ functional expression | Pharmacological inhibition of channel function reduces neurite outgrowth [49] Molecular knockdown of Cav3.2 expression inhibits secretion of PAP-prostate acidic phosphatase [50] Molecular knockdown of Cav3.2 expression decreases proliferation [51] |
| Prostate cancer (LNCaP) | + | + | + | Induction of NED with Bt2cAMP and IBMX or IL-6 increase ↑ Cav3.2 mRNA/protein and functional expressionChannel function can be modulated by H2S | Pharmacological inhibition of channel function decreases secretion of PAP-prostate acidic phosphatase [52] | |
| Prostate cancer (LNCaP) | + | + | + | Induction of NED with IL-6 or sodium butyrate increase ↑ Cav3.2 mRNA/protein and functional expression | Pharmacological inhibition of channel function reduces neurite outgrowth and decreases cell viability [53,54] | |
| Prostate cancer (LNCaP) | + | + | + | Induction of NED with androgen-depleted media or androgen receptor blocker bicalutamide increase ↑ Cav3.2 mRNA/protein and functional expression | Pharmacological inhibition of channel function decreases cell viability and increases sensitivity to anti-mitotic agents [55] | |
| Breast cancer (MCF-7) | + | + | ND | Overexpression or molecular knockdown of Cav3.2 expression have no effect on cellular proliferation [38] | ||
| Breast cancer (MCF-7) | + | ND | + | Pharmacological inhibition of channel function decreases proliferation [39] | ||
| Breast cancer (MCF-7, MDA-MB-231) | + | + | + | Cell confluency ↓ Cav3.2 mRNA, ↓ protein expression | Pharmacological inhibition of channel function or molecular knockdown of Cav3.2 expression decreases proliferation [40] | |
| Retinoblastoma (Y79) | + | ND | + | Differentiation ↓ Cav3.2 mRNA expression | Pharmacological inhibition of channel function or molecular knockdown of Cav3.2 expression decreases proliferation [39] | |
| Ovarian cancer (HO8910, A2780) | ND | + | ND | Pharmacological inhibition of channel function and molecular knockdown of Cav3.2 expression decreases proliferation and arrests cells in G0/G1 phase [41] | ||
| Ovarian cancer (A2780, A2780Cis, IGROV-1) (1) | + | ND | ND | Pharmacological inhibition of channel function or molecular knockdown of Cav3.2 expression decreases cell viability, increases apoptosis, arrests cells in G1 and/or G2 phase, decreases survivin and BIRC5 expression, increases sensitivity to carboplatin [42] | ||
| Colon cancer (HCT116 p53wt, HCT116 p53-/-) | + | + | ND | Pharmacological inhibition of channel function decreases proliferation and increases apoptosis [43] | ||
| Glioblastoma (U251, U87, T98G) | + | ND | ND | Pharmacological inhibition of channel function or molecular knockdown of Cav3.2 expression decreases cell viability and clonogenic potential, and increases apoptosis [45] | ||
| Esophageal cancer (TE8) | + | ND | + | Pharmacological inhibition of channel function decreases proliferation without any effect on apoptosis and increases p21CIP1 expression [46] | ||
| Hepatocellular carcinoma (SNU449) | + | ND | + | Pharmacological inhibition of channel function decreases proliferation, increases phosphorylated ERK 1/2, and downregulates certain genes [47] | ||
| Melanoma (M28, JG) (1) | + | ND | + | Hypoxia ↑ Cav3.2 mRNA expression | Pharmacological inhibition of channel function or molecular knockdown of Cav3.2 expression decreases cell viability and arrests cells in G1 phase [48] | |
| Cav3.3 | Ovarian cancer (A2780, A2780Cis, IGROV-1) (1) | + | ND | ND | Pharmacological inhibition of channel function decreases cell viability, increases apoptosis, and arrests cells in the G1 and/or G2 phase, decreases survivin and BIRC5 expression [42] | |
| Esophageal cancer (TE8) (1) | + | ND | + | Pharmacological inhibition of channel function decreases proliferation without any effect on apoptosis [46] | ||
| Hepatocellular carcinoma (SNU449) (1) | + | ND | + | Pharmacological inhibition of channel function decreases proliferation [47] | ||
| Melanoma (M28, JG) (1) | + | ND | + | Pharmacological inhibition of channel function decreases cell viability and arrests cells in the G1 phase [48] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antal, L.; Martin-Caraballo, M. T-type Calcium Channels in Cancer. Cancers 2019, 11, 134. https://doi.org/10.3390/cancers11020134
Antal L, Martin-Caraballo M. T-type Calcium Channels in Cancer. Cancers. 2019; 11(2):134. https://doi.org/10.3390/cancers11020134
Chicago/Turabian StyleAntal, Lauren, and Miguel Martin-Caraballo. 2019. "T-type Calcium Channels in Cancer" Cancers 11, no. 2: 134. https://doi.org/10.3390/cancers11020134
APA StyleAntal, L., & Martin-Caraballo, M. (2019). T-type Calcium Channels in Cancer. Cancers, 11(2), 134. https://doi.org/10.3390/cancers11020134