Transfer of Extracellular Vesicle-Associated-RNAs Induces Drug Resistance in ALK-Translocated Lung Adenocarcinoma

 , ,
, ,
Abstract
:1. Introduction
2. Results
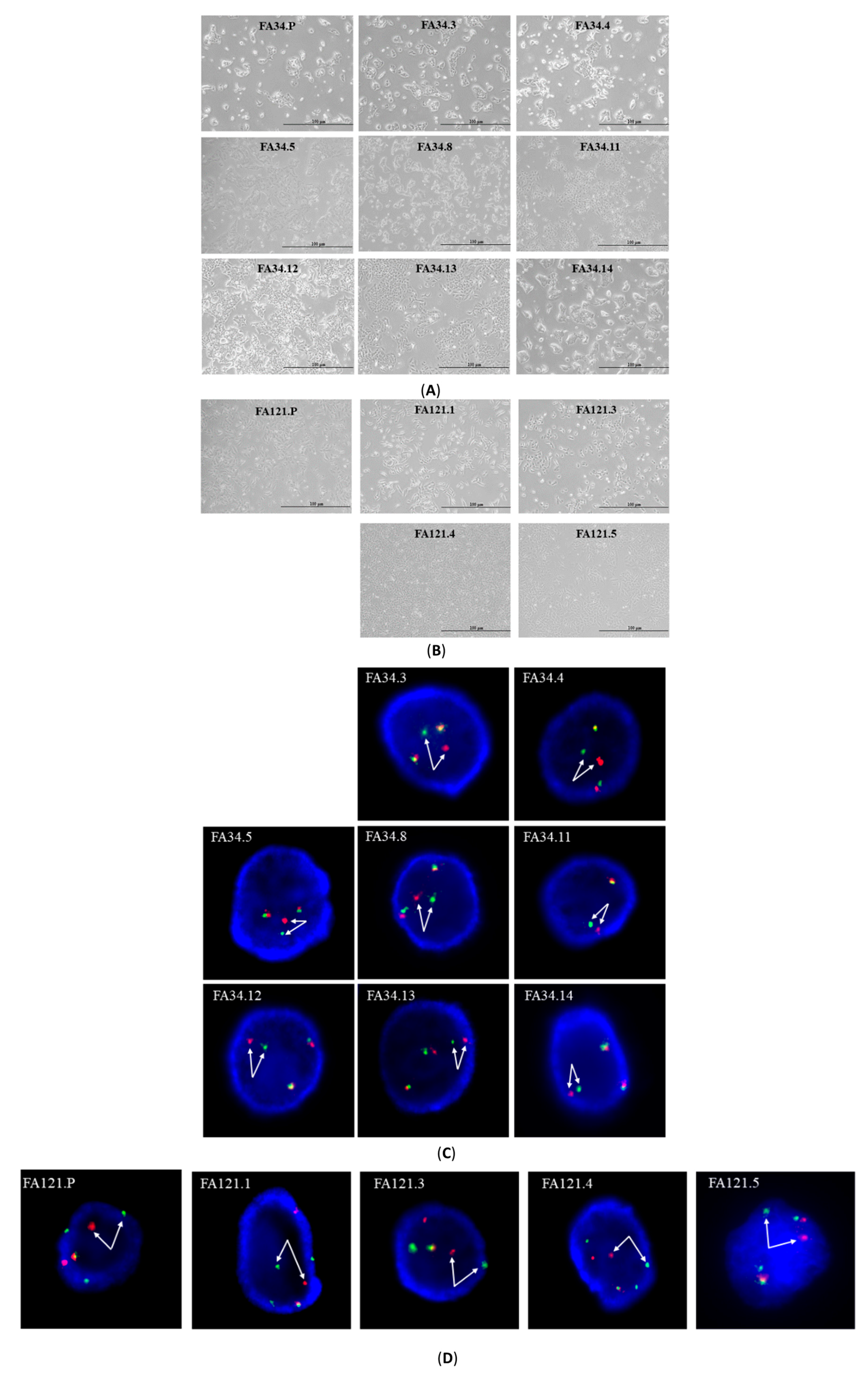
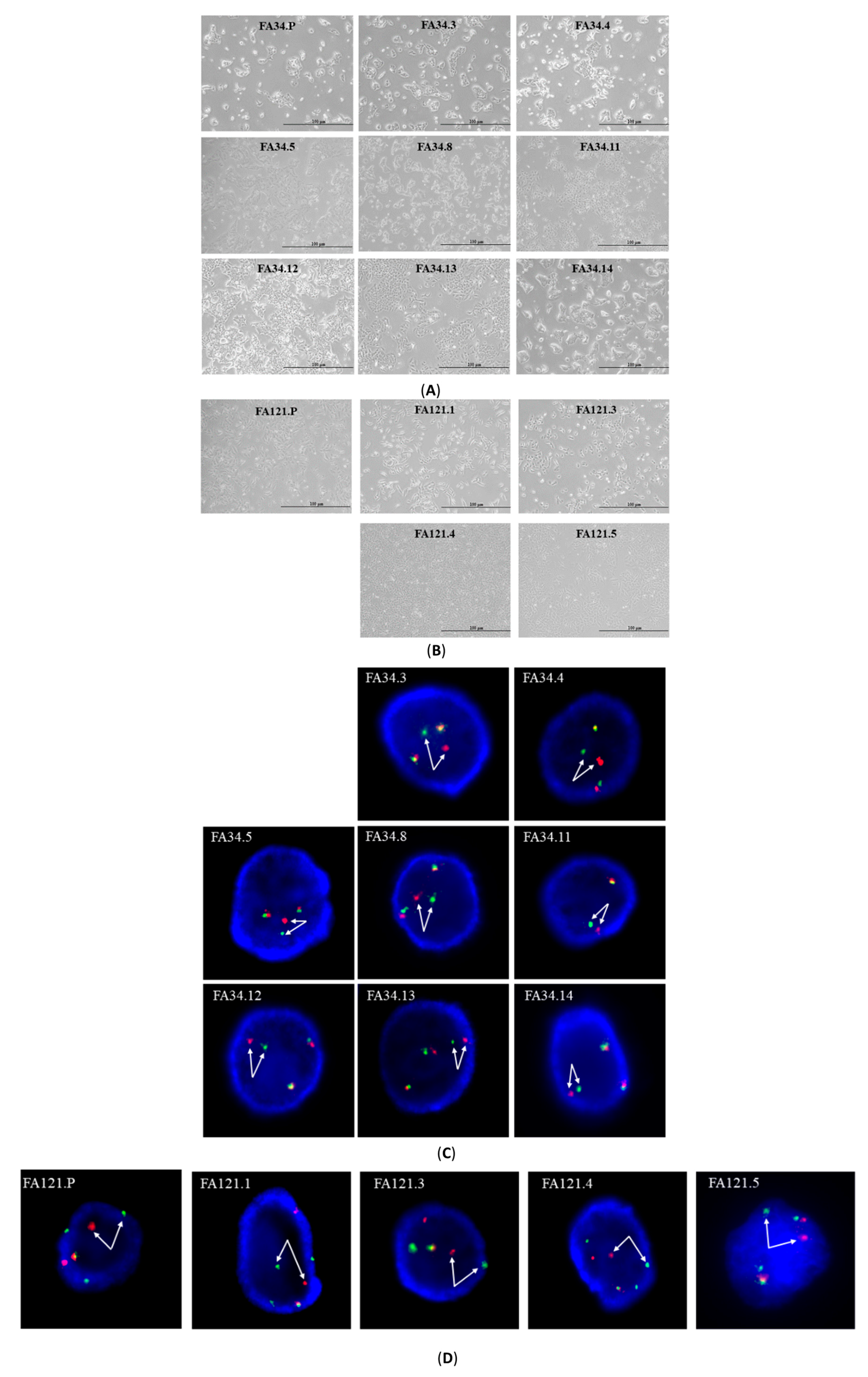
2.1. Characterization of EML4-ALK Lung Adenocarcinoma Cell Line Subclones
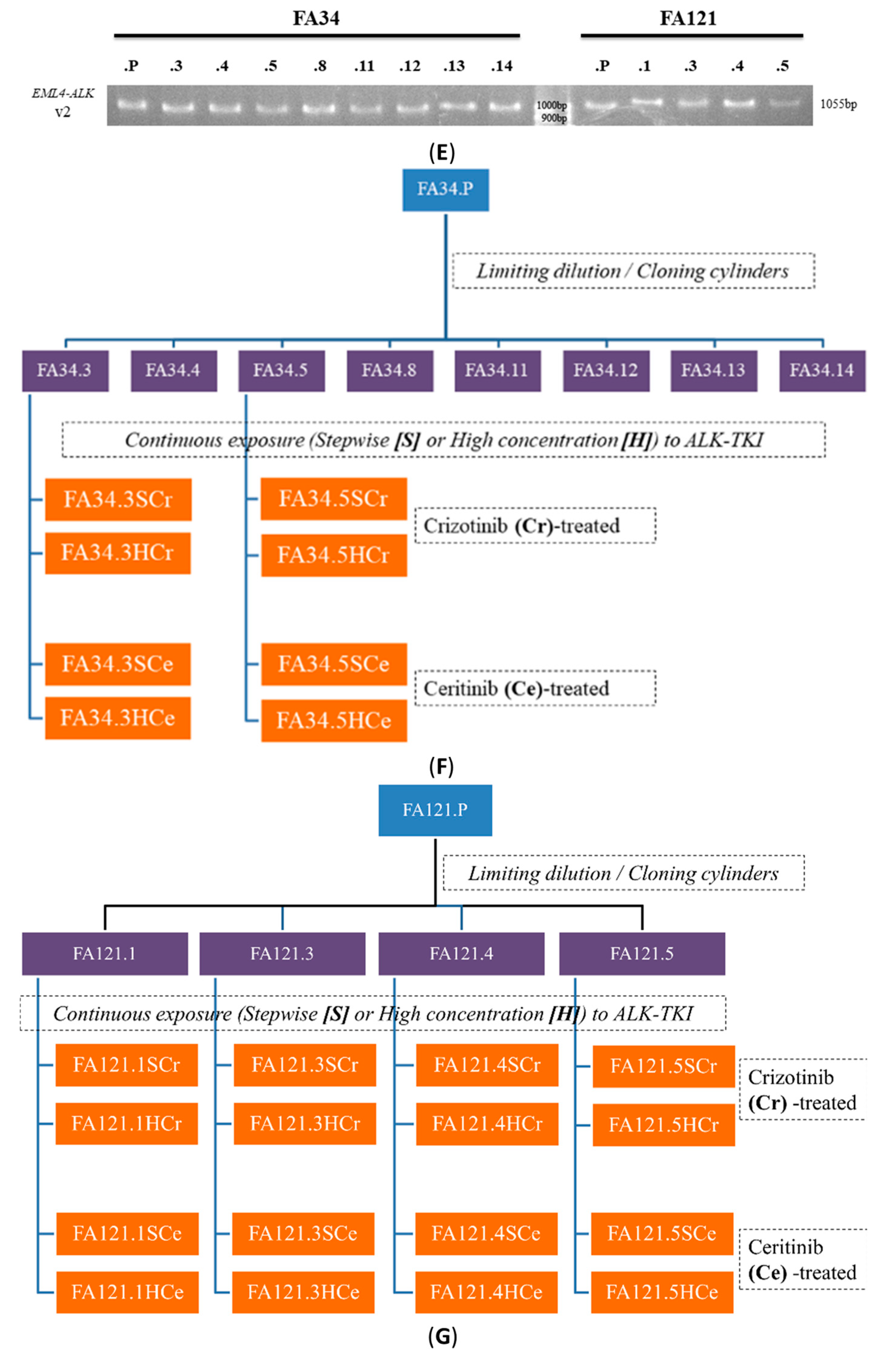
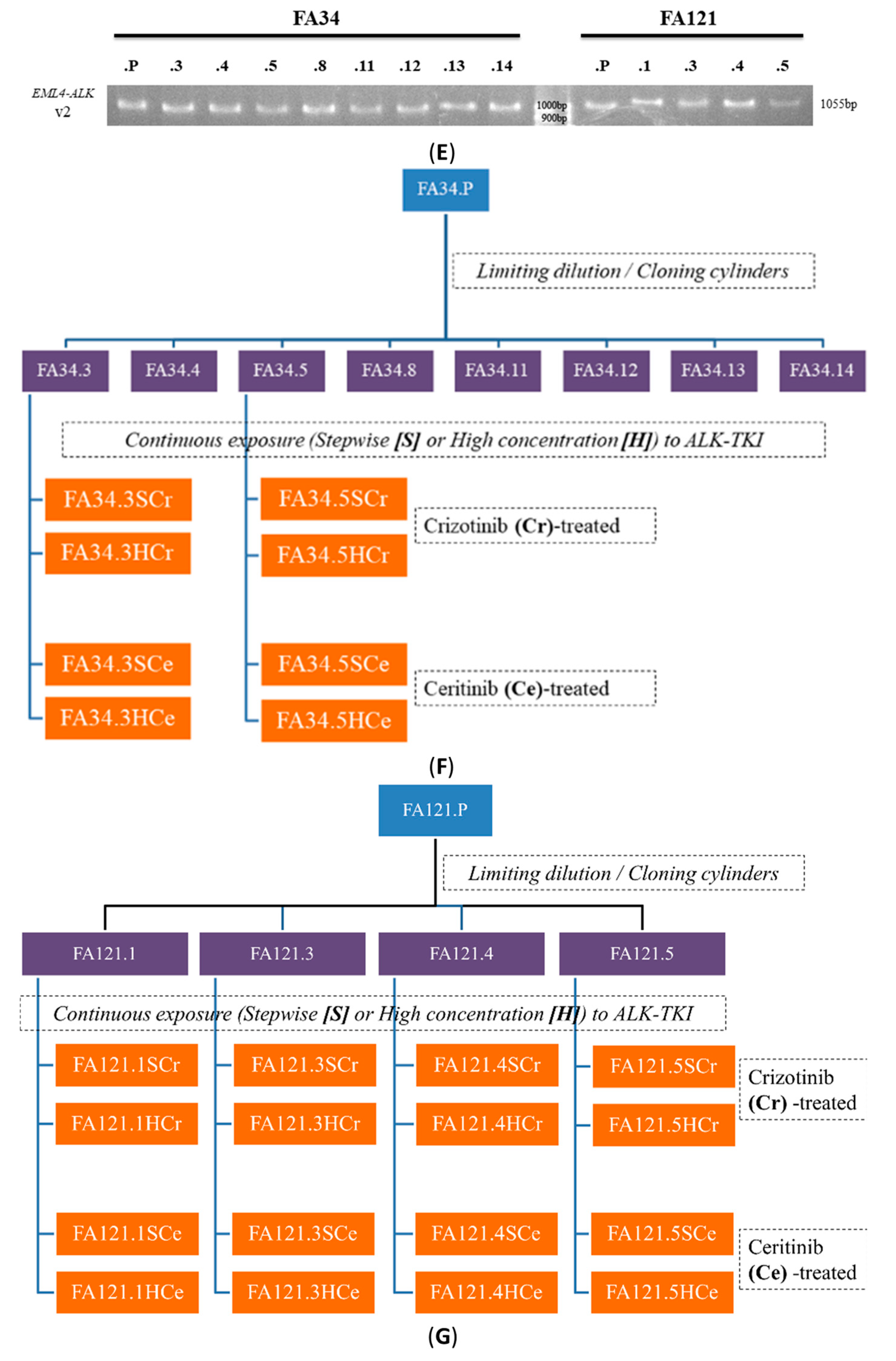
2.2. Establishment of Drug Resistant Lung Adenocarcinoma Cell Lines and Determination of Relevant Drug Resistant Mechanisms
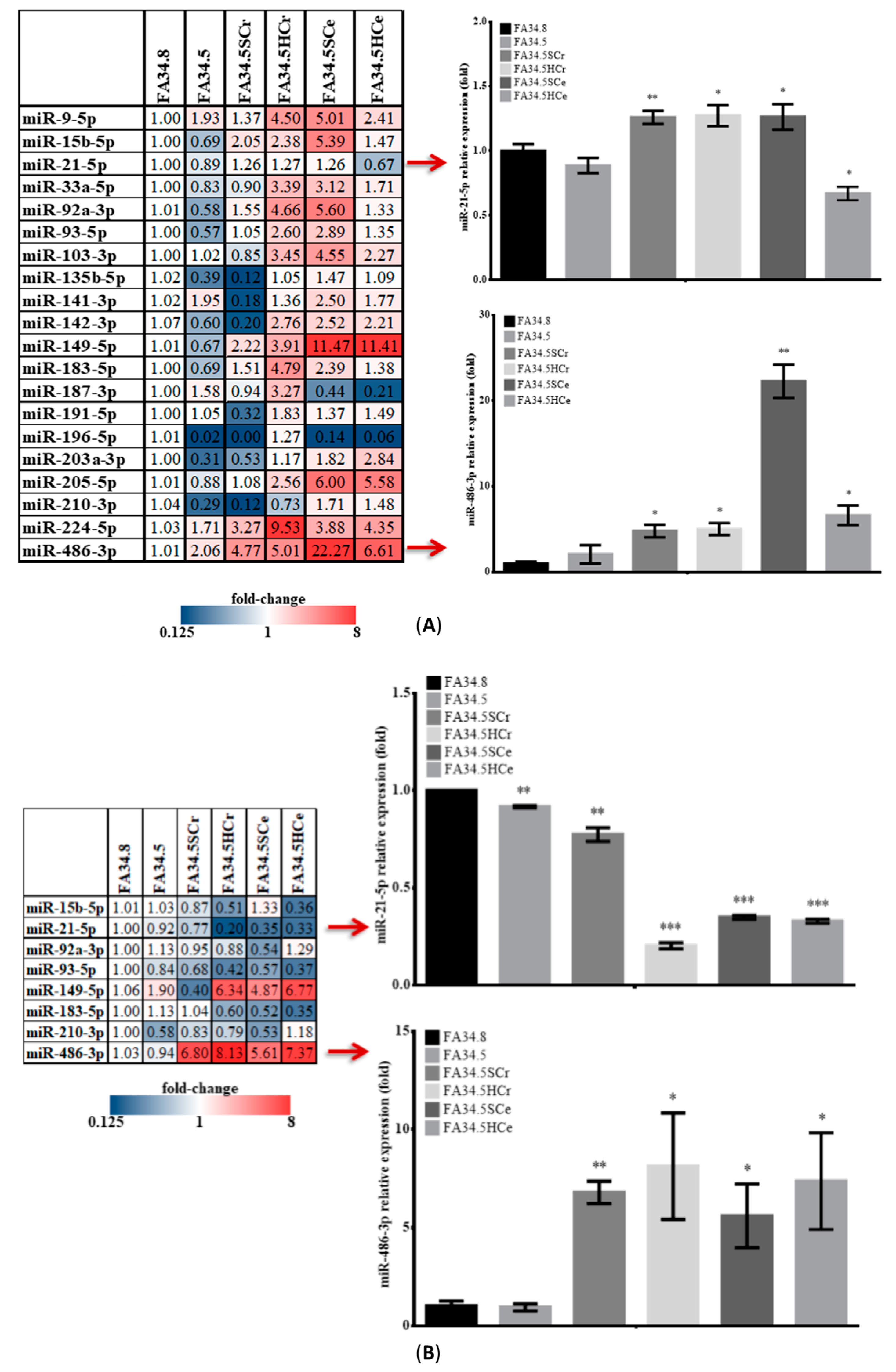
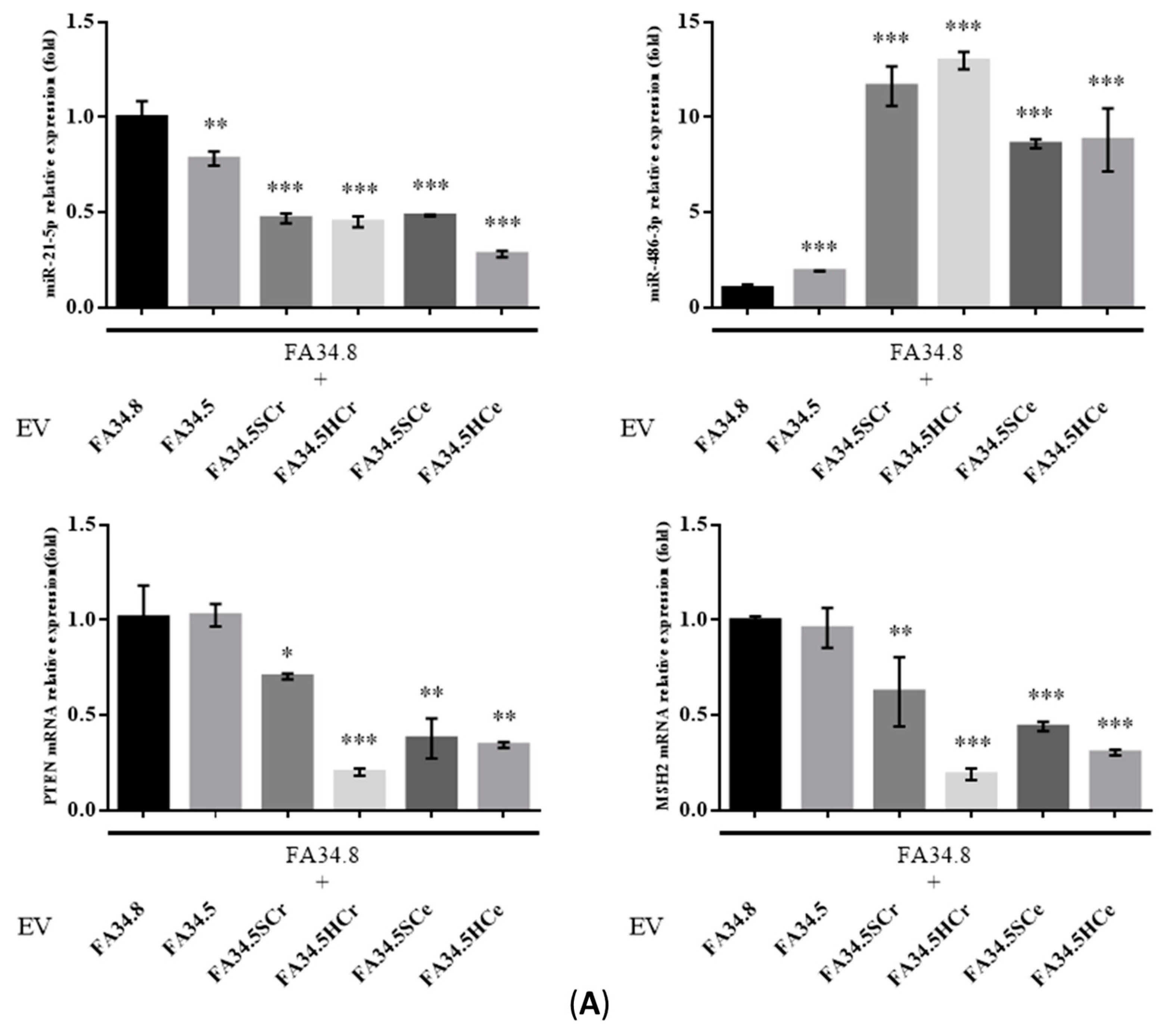
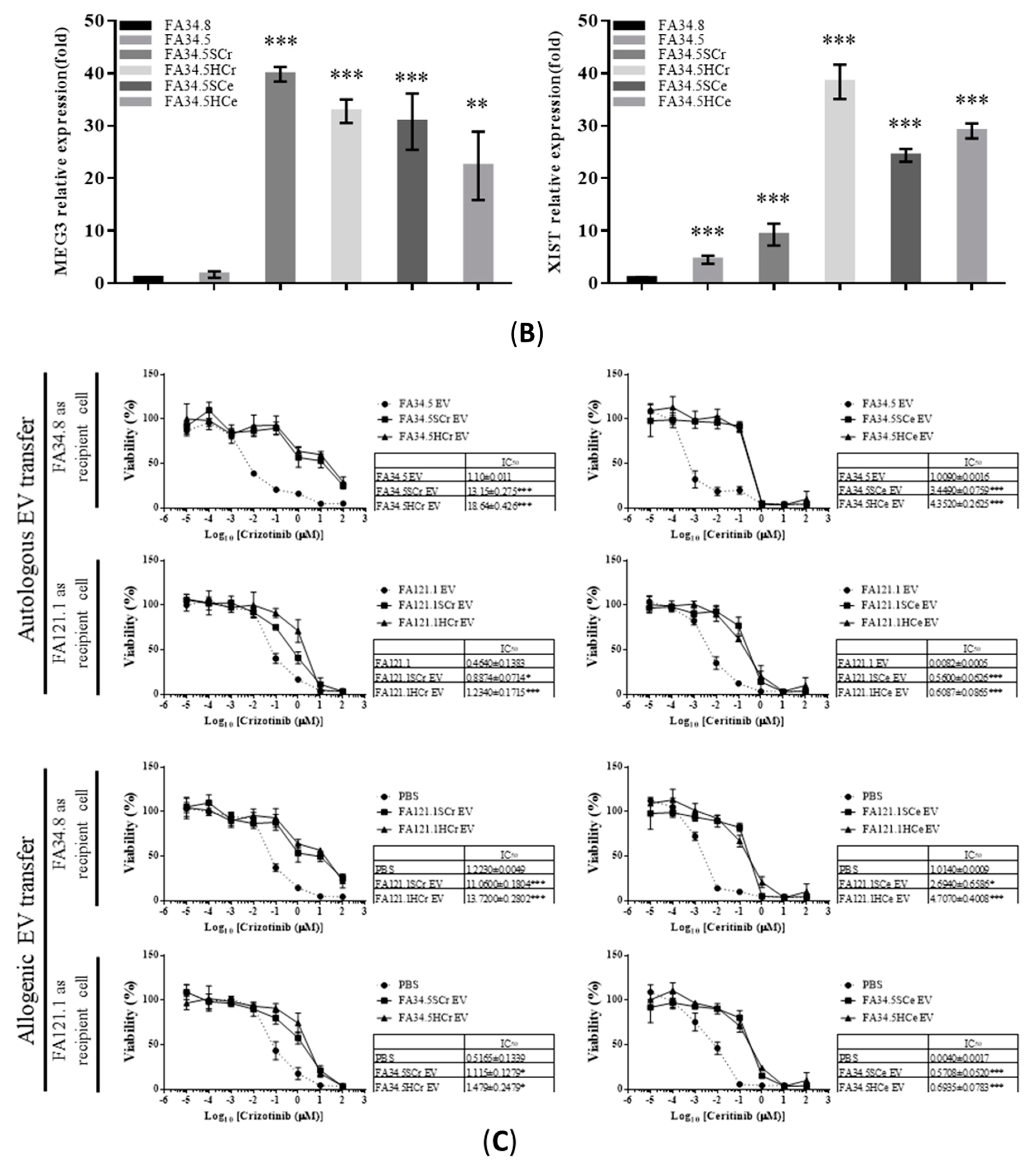
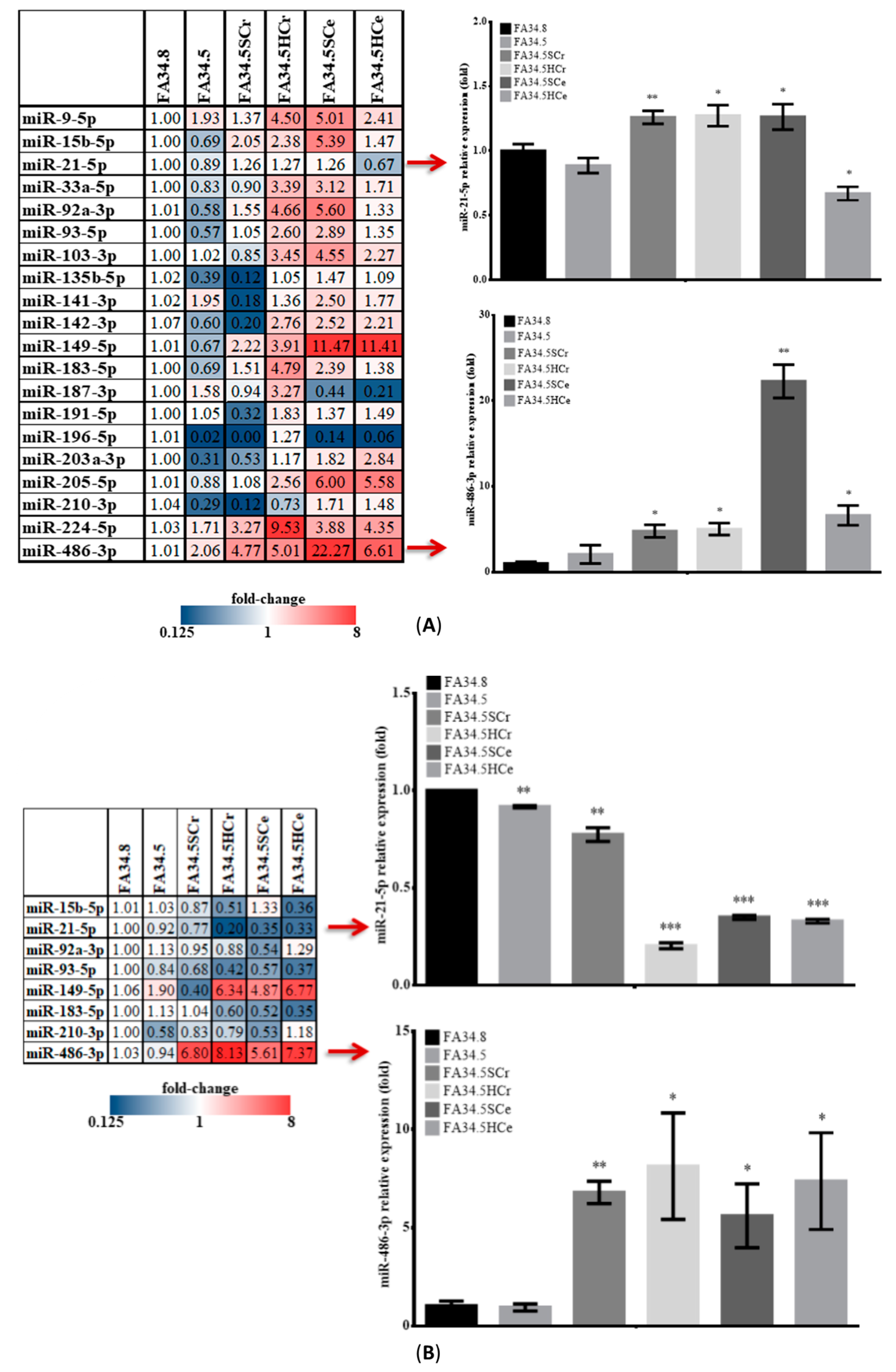
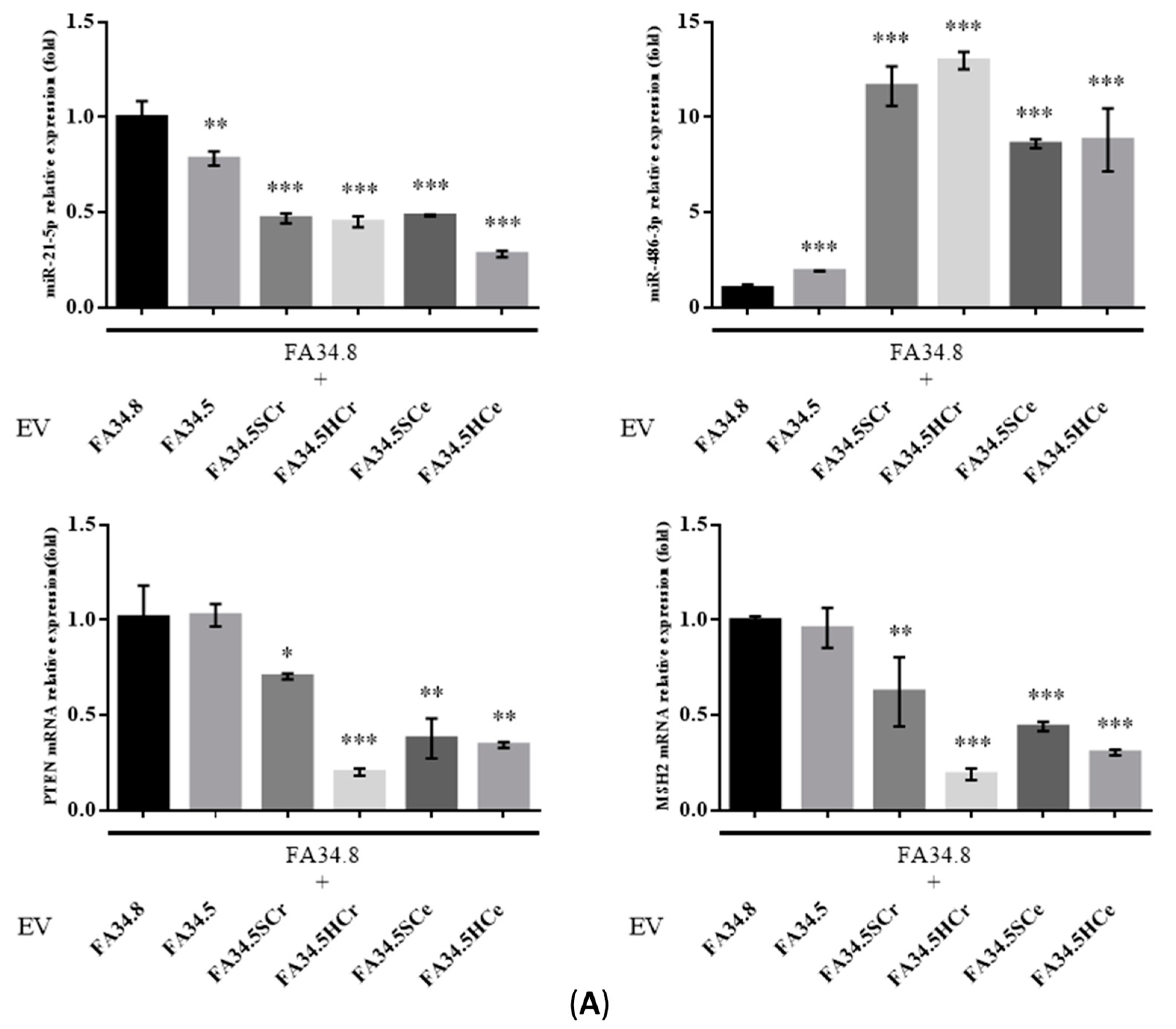
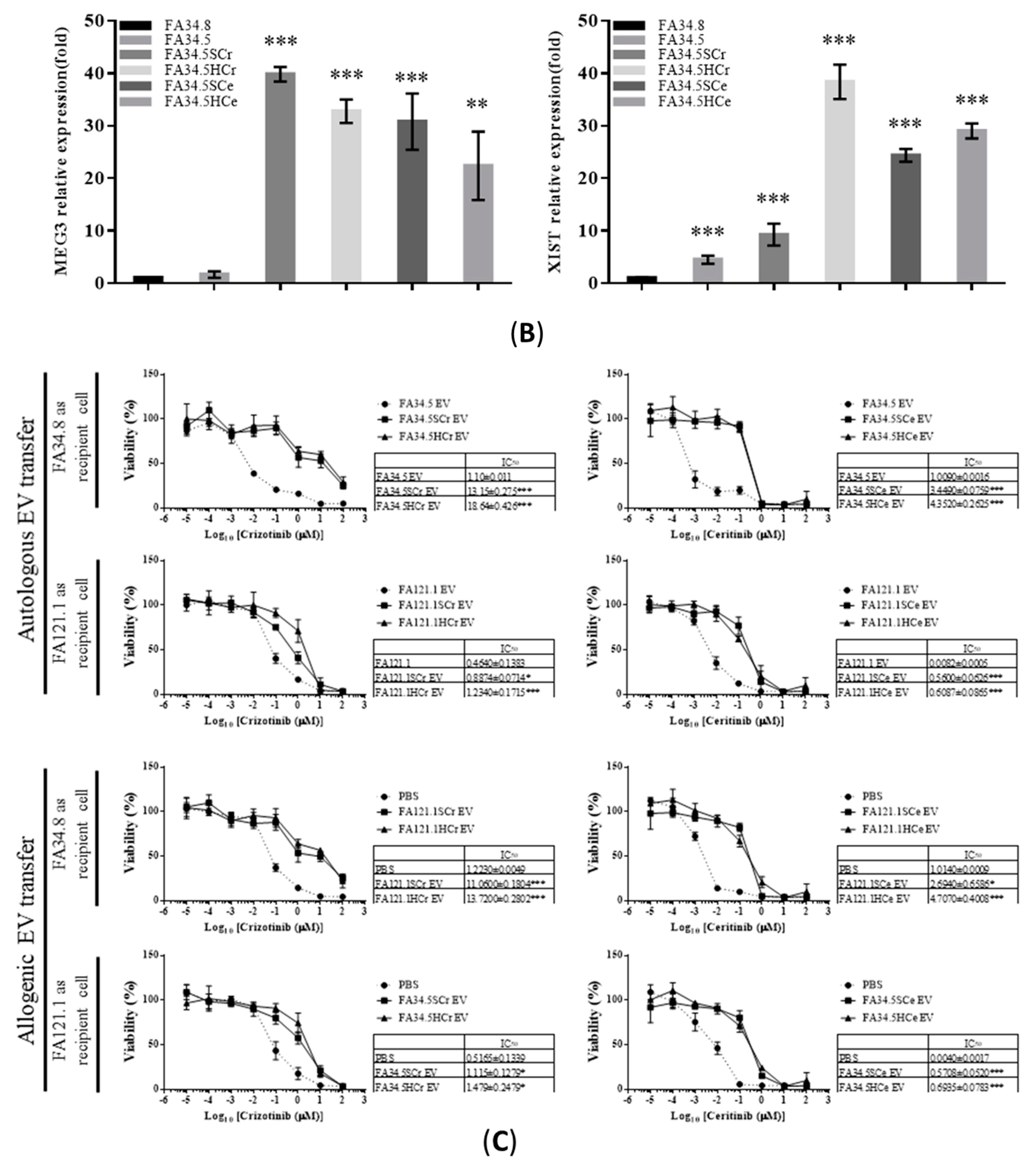
2.3. Identification of ALK-TKI Resistance-Related EV-RNAs in Lung Adenocarcinoma Subclones
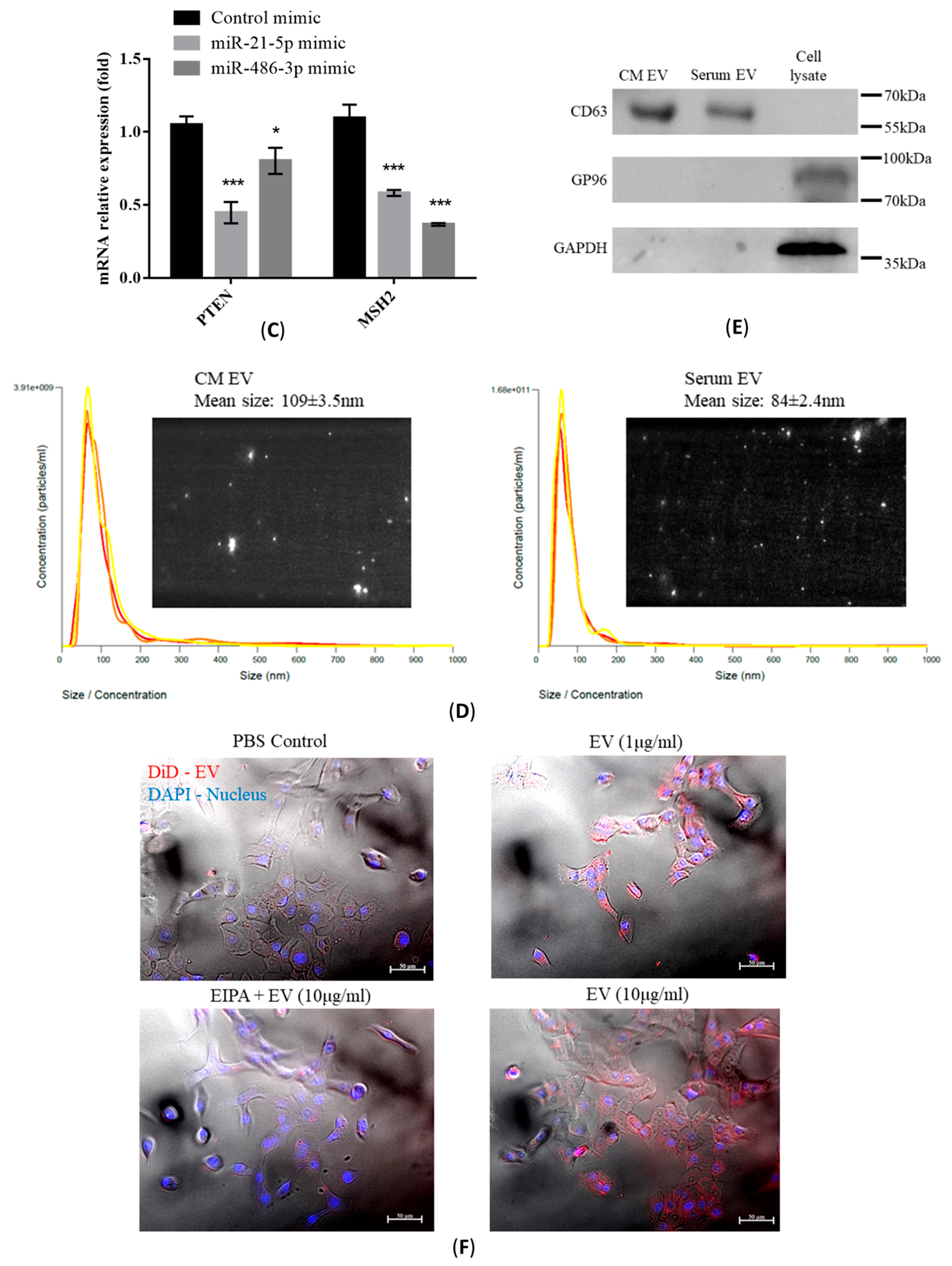
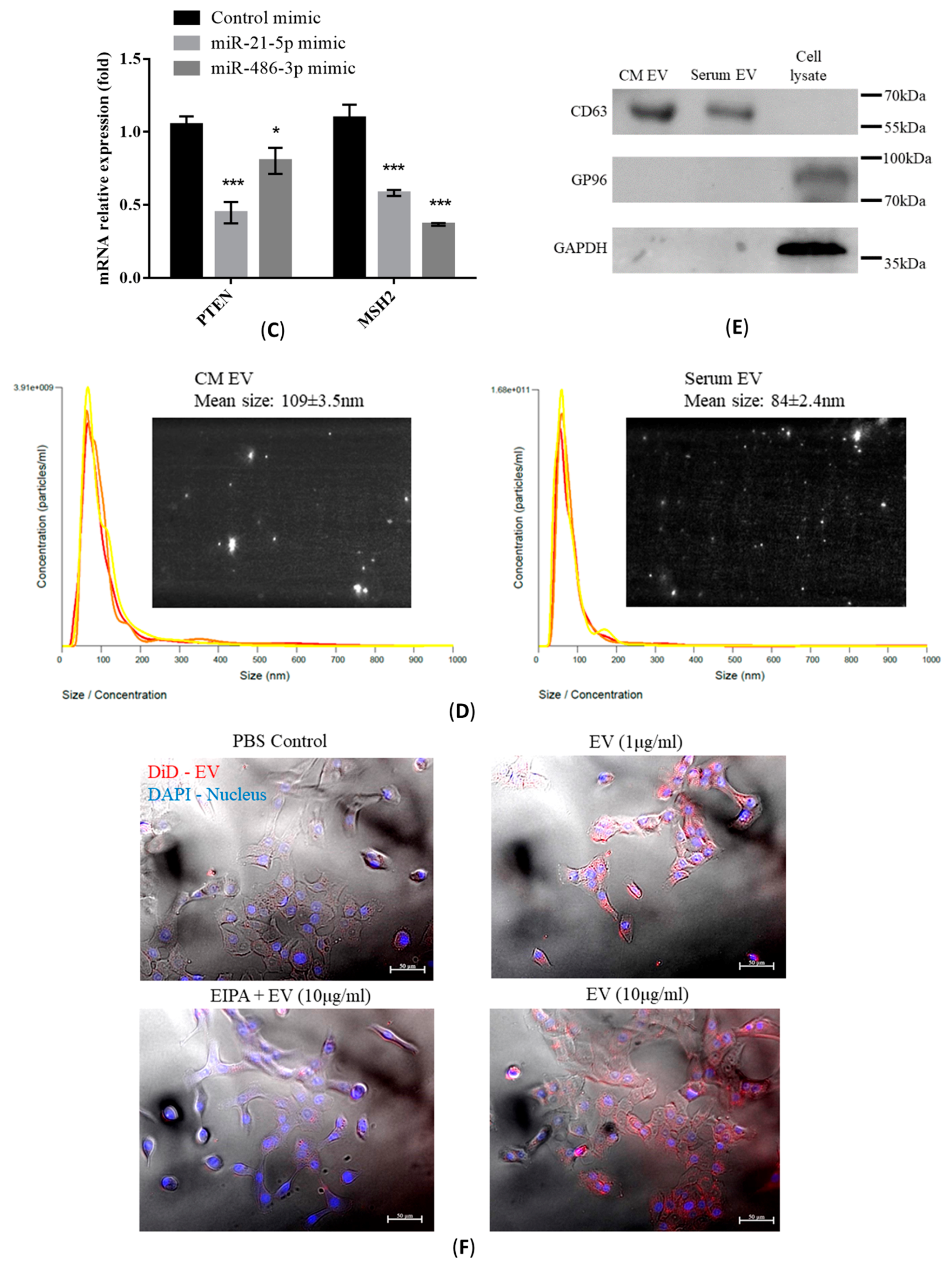
2.4. Characterization of Isolated EVs
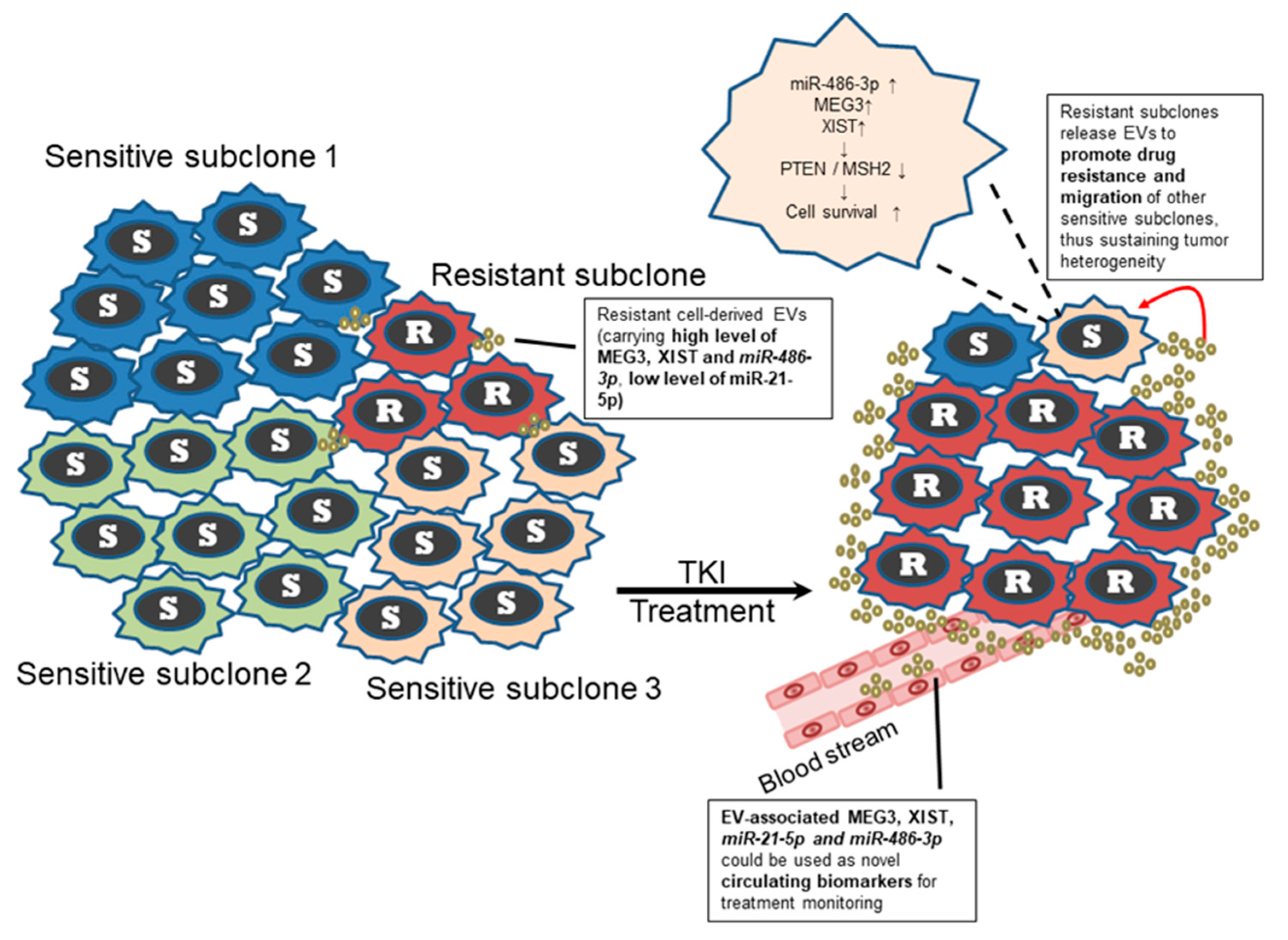
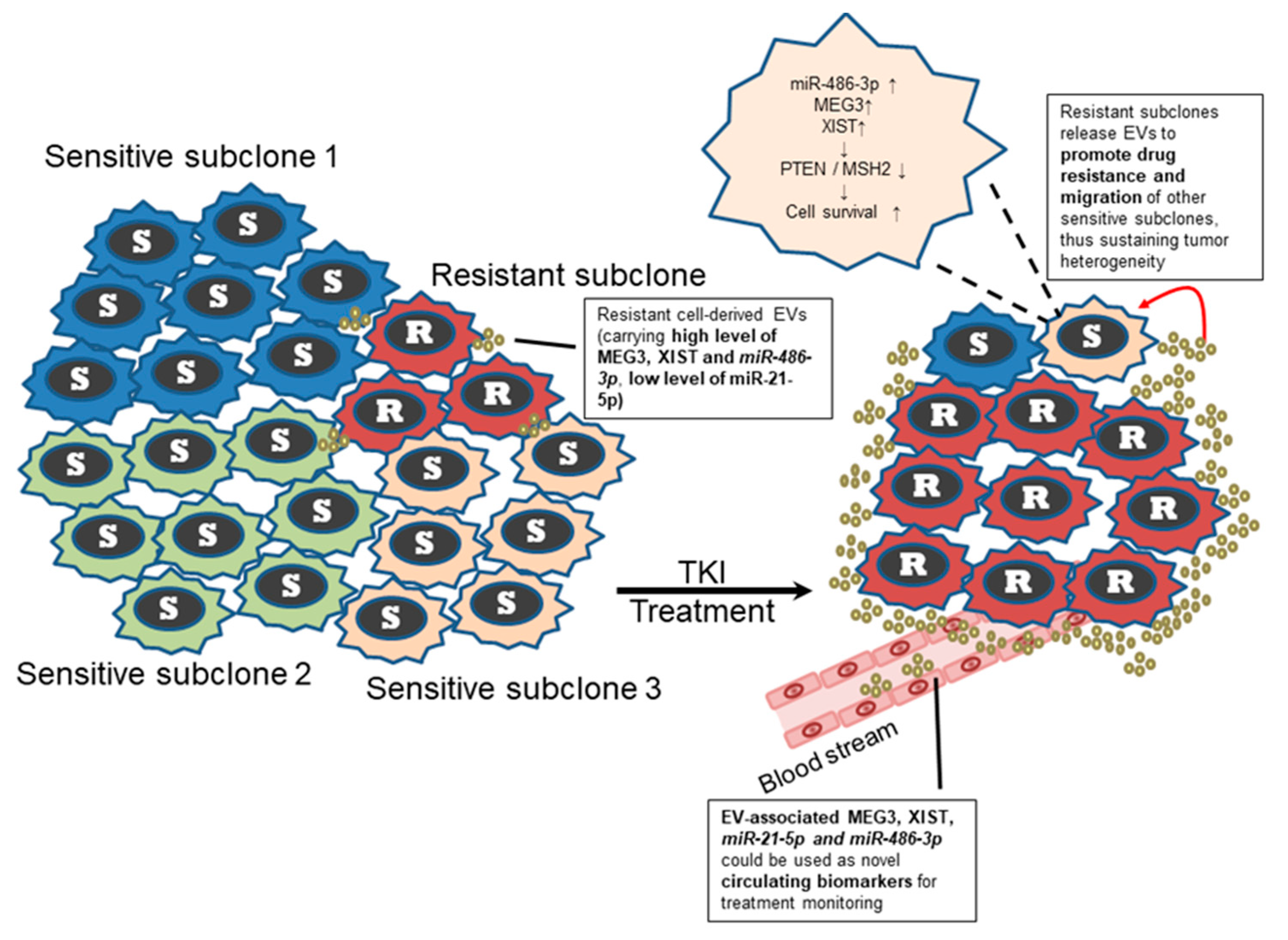
2.5. Transfer of EV-RNAs Induced Drug-Resistance and Cell Migration
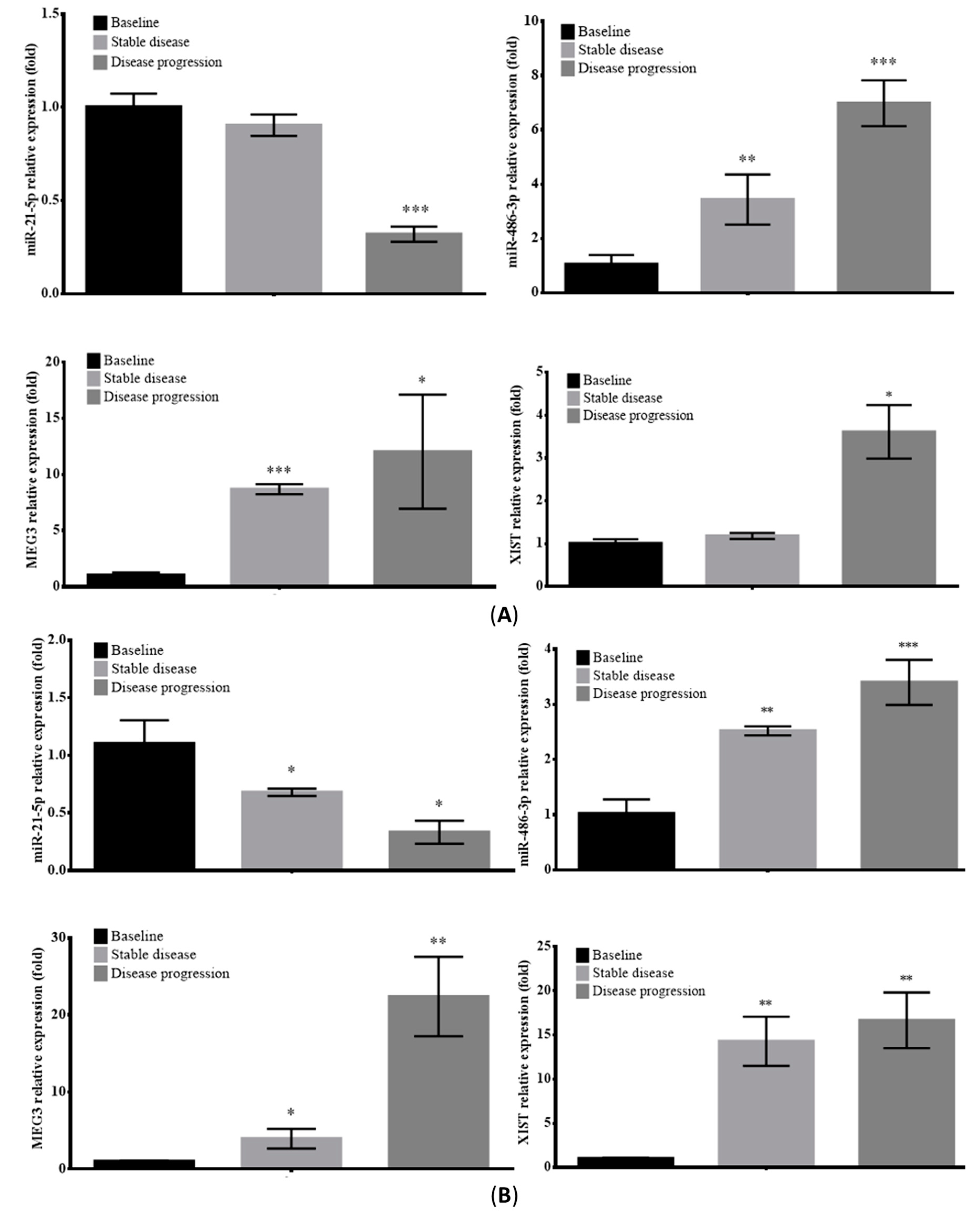
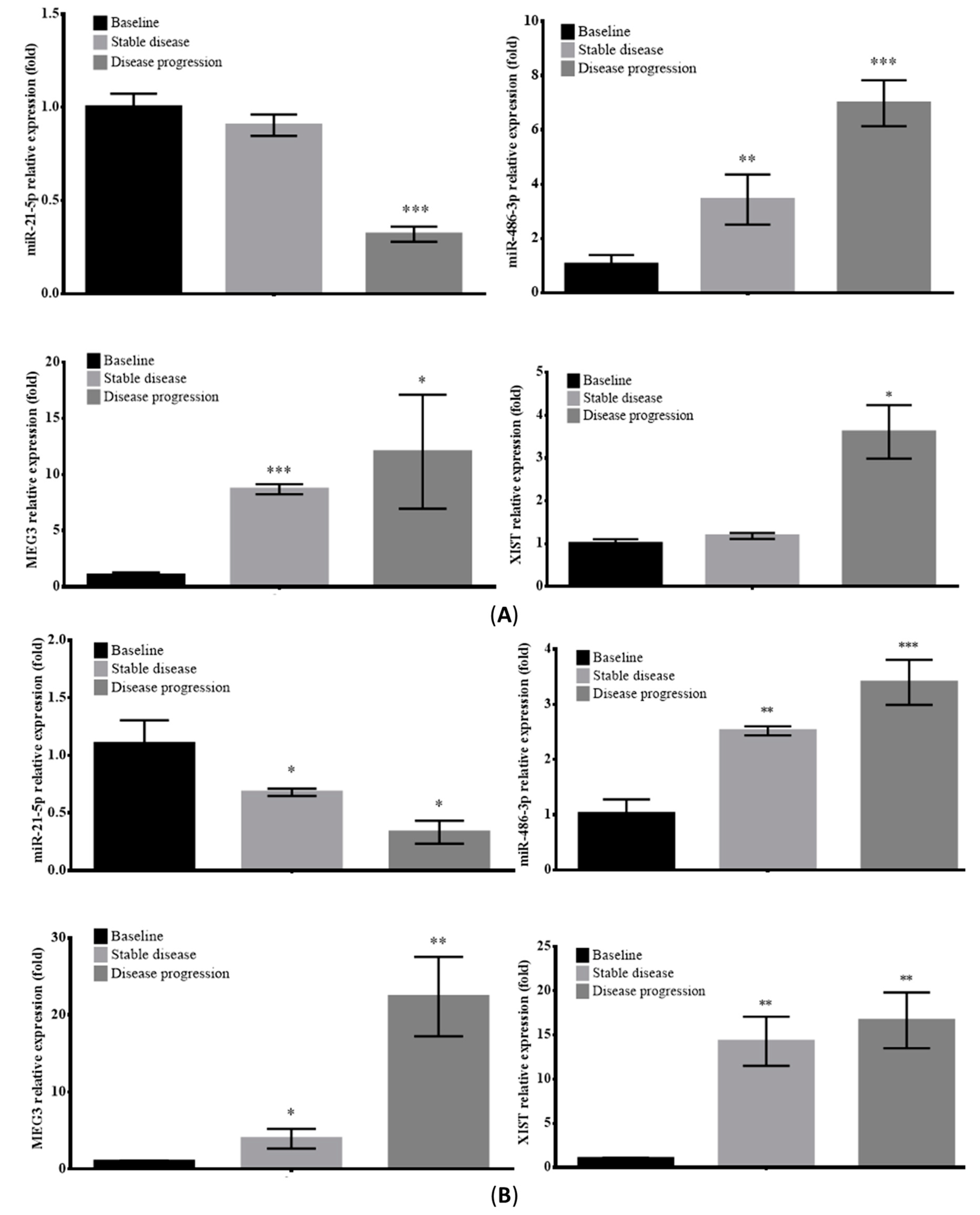
2.6. Prognostic Value of Circulating EV-RNAs in ALK-Translocated Lung Adenocarcinoma Patients
3. Discussion
4. Materials and Methods
4.1. ALK-Translocated Lung Adenocarcinoma Cell Lines
4.2. Fluorescence In Situ Hybridization (FISH)
4.3. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
4.4. Establishment of Lung Adenocarcinoma Cell Lines with Resistance to Crizotinib or Ceritinib
4.5. Cell Viability Assay
4.6. Detection of Secondary ALK Mutations or Amplifications
4.7. Extraction and Characterization of EVs Released from Lung Cancer Cells into Conditioned Medium and Serum
4.8. EV-RNA Extraction, TaqMan microRNA Assay and Quantitative PCR (qPCR)
4.9. EV Transfer
4.10. Serial Circulating EV-RNA Levels in ALK-Translocated Lung Adenocarcinoma Patients on ALK-TKI Treatment
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tan, W.-L.; Jain, A.; Takano, A.; Newell, E.W.; Iyer, N.G.; Lim, W.-T.; Tan, E.-H.; Zhai, W.; Hillmer, A.M.; Tam, W.-L.; et al. Novel therapeutic targets on the horizon for lung cancer. Lancet Oncol. 2016, 17, e347–e362. [Google Scholar] [CrossRef]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [Green Version]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. New Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Silva, A.S.; Gillies, R.J.; Frieden, B.R. Adaptive therapy. Cancer Res. 2009, 69, 4894–4903. [Google Scholar] [CrossRef] [PubMed]
- Taverna, S.; Giallombardo, M.; Gil-Bazo, I.; Carreca, A.P.; Castiglia, M.; Chacartegui, J.; Araujo, A.; Alessandro, R.; Pauwels, P.; Peeters, M.; et al. Exosomes isolation and characterization in serum is feasible in non-small cell lung cancer patients: Critical analysis of evidence and potential role in clinical practice. Oncotarget 2016, 7, 28748–28760. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife 2016, 5, e10250. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Plebanek, M.P.; Angeloni, N.L.; Vinokour, E.; Li, J.; Henkin, A.; Martinez-Marin, D.; Filleur, S.; Bhowmick, R.; Henkin, J.; Miller, S.D.; et al. Pre-metastatic cancer exosomes induce immune surveillance by patrolling monocytes at the metastatic niche. Nat. Commun. 2017, 8, 1319. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, Z.; Lu, N. From pathogenesis to clinical application: Insights into exosomes as transfer vectors in cancer. J. Exp. Clin. Cancer Res. 2016, 35, 156. [Google Scholar] [CrossRef]
- Jin, Y.; Chen, K.; Wang, Z.; Wang, Y.; Liu, J.; Lin, L.; Shao, Y.; Gao, L.; Yin, H.; Cui, C.; et al. DNA in serum extracellular vesicles is stable under different storage conditions. BMC Cancer 2016, 16, 753. [Google Scholar] [CrossRef]
- Jin, X.; Chen, Y.; Chen, H.; Fei, S.; Chen, D.; Cai, X.; Liu, L.; Lin, B.; Su, H.; Zhao, L.; et al. Evaluation of tumor-derived exosomal miRNA as potential diagnostic biomarkers for early stage non-small-cell lung cancer using next-generation sequencing. Clin. Cancer Res. 2017, 23, 5311–5319. [Google Scholar] [CrossRef] [PubMed]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Masliah-Planchon, J.; Pasmant, E.; Luscan, A.; Laurendeau, I.; Ortonne, N.; Hivelin, M.; Varin, J.; Valeyrie-Allanore, L.; Dumaine, V.; Lantieri, L.; et al. MicroRNAome profiling in benign and malignant neurofibromatosis type 1-associated nerve sheath tumors: Evidences of PTEN pathway alterations in early NF1 tumorigenesis. BMC Genom. 2013, 14, 473. [Google Scholar] [CrossRef] [PubMed]
- Balaguer, F.; Moreira, L.; Lozano, J.J.; Link, A.; Ramirez, G.; Shen, Y.; Cuatrecasas, M.; Arnold, M.; Meltzer, S.J.; Syngal, S.; et al. Colorectal cancers with microsatellite instability display unique miRNA profiles. Clin. Cancer Res. 2011, 17, 6239–6249. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Henson, R.; Wehbe-Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Valeri, N.; Gasparini, P.; Braconi, C.; Paone, A.; Lovat, F.; Fabbri, M.; Sumani, K.M.; Alder, H.; Amadori, D.; Patel, T.; et al. MicroRNA-21 induces resistance to 5-fluorouracil by down-regulating human DNA MutS homolog 2 (hMSH2). Proc. Natl. Acad. Sci. USA 2010, 107, 21098–21103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, L.; Ding, J.; Chen, C.; Wu, Z.J.; Liu, B.; Gao, Y.; Chen, W.; Liu, F.; Sun, W.; Li, X.F.; et al. Exosome-Transmitted lncARSR Promotes Sunitinib Resistance in Renal Cancer by Acting as a Competing Endogenous RNA. Cancer Cell 2016, 29, 653–668. [Google Scholar] [CrossRef]
- Zhang, J.; Yao, T.; Wang, Y.; Yu, J.; Liu, Y.; Lin, Z. Long noncoding RNA MEG3 is downregulated in cervical cancer and affects cell proliferation and apoptosis by regulating miR-21. Cancer Biol. Ther. 2016, 17, 104–113. [Google Scholar] [CrossRef]
- Zhang, R.; Xia, T. Long non-coding RNA XIST regulates PDCD4 expression by interacting with miR-21-5p and inhibits osteosarcoma cell growth and metastasis. Int. J. Oncol. 2017, 51, 1460–1470. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef]
- Wang, W.; Li, J.; Zhu, W.; Gao, C.; Jiang, R.; Li, W.; Hu, Q.; Zhang, B. MicroRNA-21 and the clinical outcomes of various carcinomas: A systematic review and meta-analysis. BMC Cancer 2014, 14, 819. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.; Chin, A.R.; et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gu, Y.; Han, Y.; Zhang, Q.; Jiang, Z.; Zhang, X.; Huang, B.; Xu, X.; Zheng, J.; Cao, X. Tumor Exosomal RNAs Promote Lung Pre-metastatic Niche Formation by Activating Alveolar Epithelial TLR3 to Recruit Neutrophils. Cancer Cell 2016, 30, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yu, Z.; Yuan, S.; Xie, W.; Li, C.; Hu, Z.; Xiang, Y.; Wu, N.; Wu, L.; Bai, L.; et al. Circulating exosomal microRNAs as prognostic biomarkers for non-small-cell lung cancer. Oncotarget 2017, 8, 13048–13058. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Lin, C.; Gong, H.; Wang, C.; Liu, L.; Wu, J.; Tao, S.; Hu, B.; Cheng, S.Y.; Li, M.; et al. miR-486 sustains NF-kappaB activity by disrupting multiple NF-κB-negative feedback loops. Cell Res. 2013, 23, 274–289. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tian, X.; Han, R.; Zhang, X.; Wang, X.; Shen, H.; Xue, L.; Liu, Y.; Yan, X.; Shen, J.; et al. Downregulation of miR-486-5p contributes to tumor progression and metastasis by targeting protumorigenic ARHGAP5 in lung cancer. Oncogene 2014, 33, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Wei, J.; Taron, M. Circulating MicroRNA Signatures of Tumor-Derived Exosomes for Early Diagnosis of Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2009, 10, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Khatun, Z.; Shiras, A. Tumor exosomes: Cellular postmen of cancer diagnosis and personalized therapy. Nanomedicine (Lond. Engl.) 2016, 11, 421–437. [Google Scholar] [CrossRef]
- Hida, T.; Nokihara, H.; Kondo, M.; Kim, Y.H.; Azuma, K.; Seto, T.; Takiguchi, Y.; Nishio, M.; Yoshioka, H.; Imamura, F.; et al. Alectinib versus crizotinib in patients with ALK-positive non-small-cell lung cancer (J-ALEX): An open-label, randomised phase 3 trial. Lancet 2017, 390, 29–39. [Google Scholar] [CrossRef]
- Soria, J.C.; Tan, D.S.; Chiari, R.; Wu, Y.L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.J.; et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): A randomised, open-label, phase 3 study. Lancet 2017, 389, 917–929. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, S.; Yao, J.; Lowery, F.J.; Zhang, Q.; Huang, W.C.; Li, P.; Li, M.; Wang, X.; Zhang, C.; et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 2015, 527, 100–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Au Yeung, C.L.; Co, N.N.; Tsuruga, T.; Yeung, T.L.; Kwan, S.Y.; Leung, C.S.; Li, Y.; Lu, E.S.; Kwan, K.; Wong, K.K.; et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat. Commun. 2016, 7, 11150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarroya-Beltri, C.; Gutierrez-Vazquez, C.; Sanchez-Cabo, F.; Perez-Hernandez, D.; Vazquez, J.; Martin-Cofreces, N.; Martinez-Herrera, D.J.; Pascual-Montano, A.; Mittelbrunn, M.; Sanchez-Madrid, F. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 2013, 4, 2980. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A.; et al. Cancer Exosomes Perform Cell-Independent MicroRNA Biogenesis and Promote Tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach, D.H.; Hong, J.Y.; Pak, H.J.; Lee, S.K. The role of exosomes and miRNAs in drug-resistance of cancer cells. Int. J. Cancer 2017, 141, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Lam, D.C.; Luo, S.Y.; Deng, W.; Kwan, J.; Rodriguez-Canales, J.; Cheung, A.L.; Cheng, G.H.; Lin, C.H.; Wistuba, I.I.; Sham, P.C.; et al. Oncogenic mutation profiling in new lung cancer and mesothelioma cell lines. Onco Targets Ther. 2015, 8, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F.; Linnoila, R.I.; Kurita, Y.; Oie, H.K.; Mulshine, J.L.; Clark, J.C.; Whitsett, J.A. Peripheral airway cell differentiation in human lung cancer cell lines. Cancer Res. 1990, 50, 5481–5487. [Google Scholar]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Bavi, P.; Jehan, Z.; Bu, R.; Prabhakaran, S.; Al-Sanea, N.; Al-Dayel, F.; Al-Assiri, M.; Al-Halouly, T.; Sairafi, R.; Uddin, S.; et al. ALK gene amplification is associated with poor prognosis in colorectal carcinoma. Br. J. Cancer 2013, 109, 2735–2743. [Google Scholar] [CrossRef] [Green Version]
- Consortium, E.-T.; Van Deun, J.; Mestdagh, P.; Agostinis, P.; Akay, Ö.; Anand, S.; Anckaert, J.; Martinez, Z.A.; Baetens, T.; Beghein, E.; et al. EV-TRACK: Transparent reporting and centralizing knowledge in extracellular vesicle research. Nat. Methods 2017, 14, 228–232. [Google Scholar]
- Kasinski, A.L.; Slack, F.J. Epigenetics and genetics. MicroRNAs en route to the clinic: Progress in validating and targeting microRNAs for cancer therapy. Nat. Rev. Cancer 2011, 11, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Croce, C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 2009, 10, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Mine, N.; Kufe, D.W.; Von Hoff, D.D.; Kawabe, T. CBP501 inhibits EGF-dependent cell migration, invasion and epithelial-to–mesenchymal transition of non-small cell lung cancer cells by blocking KRas to calmodulin binding. Oncotarget 2017, 8, 74006–74018. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A. IC50 values of different FA34 and FA121 subclones against the three ALK-TKIs tested | |||||
| Subclones/IC50 (μM) | Crizotinib | Ceritinib | Alectinib | ||
| FA34.P | 0.0416 | 0.0535 | 0.0004 | ||
| FA34.3 | 0.4289 | 0.2769 | 0.3142 | ||
| FA34.4 | 0.9166 | 0.2484 | 0.0868 | ||
| FA34.5 | 2.306 | 0.4287 | 1.336 | ||
| FA34.8 | 0.2966 | 0.0107 | 0.0059 | ||
| FA34.11 | 0.2075 | 0.0212 | 0.0061 | ||
| FA34.12 | 0.42 | 0.3371 | 0.0257 | ||
| FA34.13 | 0.1015 | 0.0124 | 0.0004 | ||
| FA34.14 | 0.3062 | 0.0204 | 0.0224 | ||
| FA121.P | 0.03 | 0.04 | 0.01 | ||
| FA121.1 | 0.3625 | 0.0058 | 0.0165 | ||
| FA121.3 | 0.096 | 0.0004 | 0.0009 | ||
| FA121.4 | 0.7736 | 0.7354 | 0.6937 | ||
| FA121.5 | 0.0874 | 0.0029 | 0.0067 | ||
| B. IC50 values and the resistant mechanisms of different crizotinib-resistant subclones against the three ALK-TKIs tested. | |||||
| Subclones/IC50 (μM) | Crizotinib | Ceritinib | Alectinib | Secondary mutation | ALK amplification |
| FA34.3SCr | 19.6000 (471.2) | 2.2790 | 61.8400 | WT | YES |
| FA34.5SCr | 20.1200 (8.7) | 0.5629 | 4.9030 | WT | YES |
| FA34.3HCr | 16.8900 (39.4) | 2.2560 | 57.0300 | WT | YES |
| FA34.5HCr | 22.9800 (10.0) | 1.5690 | 31.4300 | WT | YES |
| FA121.1SCr | 1.2560 (3.5) | 1.2760 | 3.0690 | WT | NO |
| FA121.3SCr | 1.0960 (11.4) | 0.3091 | 2.0700 | WT | NO |
| FA121.4SCr | 1.9450 (2.5) | 0.8955 | 1.8940 | WT | YES |
| FA121.5SCr | 0.1884 (2.2) | 1.9920 | 0.3204 | WT | YES |
| FA121.1HCr | 1.6370 (4.5) | 0.0293 | 2.0180 | WT | YES |
| FA121.3HCr | 12.860 (134.0) | 1.9000 | 3.7180 | ALK, C1156S | NO |
| FA121.4HCr | 1.7230 (2.2) | 0.0544 | 0.0399 | WT | YES |
| FA121.5HCr | 2.7550 (31.5) | 0.0555 | 0.6398 | WT | YES |
| C. IC50 values and the resistant mechanisms of different ceritinib-resistant subclones against the three ALK-TKIs tested. | |||||
| Subclones/IC50 (μM) | Crizotinib | Ceritinib | Alectinib | Secondary mutation | ALK amplification |
| FA34.5SCe | 17.6900 | 5.5000 (12.8) | 18.3600 | WT | YES |
| FA345.HCe | 24.2400 | 2.1280 (5.0) | 11.2600 | WT | YES |
| FA121.1SCe | 0.7165 | 0.4356 (75.1) | 0.2278 | WT | NO |
| FA121.3SCe | 0.3272 | 0.0557 (139.3) | 0.0965 | ALK, T1151M | YES |
| FA121.4SCe | 1.4610 | 0.9672 (1.3) | 0.1533 | ALK, T1151M | NO |
| FA121.5SCe | 1.9690 | 1.2190 (1.7) | 0.3851 | WT | NO |
| FA121.1HCe | 1.5330 | 0.5631 (97.1) | 10.8500 | WT | YES |
| FA121.3HCe | 7.6780 | 2.5650 (6412.5) | 9.0510 | WT | YES |
| FA121.4HCe | 0.8824 | 1.1534 (1.6) | 0.0845 | ALK, T1151M | NO |
| FA121.5HCe | 2.5500 | 0.4636 (159.8) | 7.5000 | WT | YES |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwok, H.-H.; Ning, Z.; Chong, P.W.-C.; Wan, T.S.-K.; Ng, M.H.-L.; Ho, G.Y.F.; Ip, M.S.-M.; Lam, D.C.-L. Transfer of Extracellular Vesicle-Associated-RNAs Induces Drug Resistance in ALK-Translocated Lung Adenocarcinoma. Cancers 2019, 11, 104. https://doi.org/10.3390/cancers11010104
Kwok H-H, Ning Z, Chong PW-C, Wan TS-K, Ng MH-L, Ho GYF, Ip MS-M, Lam DC-L. Transfer of Extracellular Vesicle-Associated-RNAs Induces Drug Resistance in ALK-Translocated Lung Adenocarcinoma. Cancers. 2019; 11(1):104. https://doi.org/10.3390/cancers11010104
Chicago/Turabian StyleKwok, Hoi-Hin, Ziyu Ning, Peony Wing-Chi Chong, Thomas Shek-Kong Wan, Margaret Heung-Ling Ng, Gloria Y.F. Ho, Mary Sau-Man Ip, and David Chi-Leung Lam. 2019. "Transfer of Extracellular Vesicle-Associated-RNAs Induces Drug Resistance in ALK-Translocated Lung Adenocarcinoma" Cancers 11, no. 1: 104. https://doi.org/10.3390/cancers11010104
APA StyleKwok, H.-H., Ning, Z., Chong, P. W.-C., Wan, T. S.-K., Ng, M. H.-L., Ho, G. Y. F., Ip, M. S.-M., & Lam, D. C.-L. (2019). Transfer of Extracellular Vesicle-Associated-RNAs Induces Drug Resistance in ALK-Translocated Lung Adenocarcinoma. Cancers, 11(1), 104. https://doi.org/10.3390/cancers11010104