Oxidative Phosphorylation: A Target for Novel Therapeutic Strategies Against Ovarian Cancer



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Oxphos As Target for Hgsoc Therapy
2.1. Relevance of OXPHOS in Solid Tumors
2.2. Reliance of Ovarian Cancer Stem Cells on OXPHOS
2.3. OXPHOS in Chemoresistant HGSOC
2.4. OXPHOS and the Tumor Microenvironment
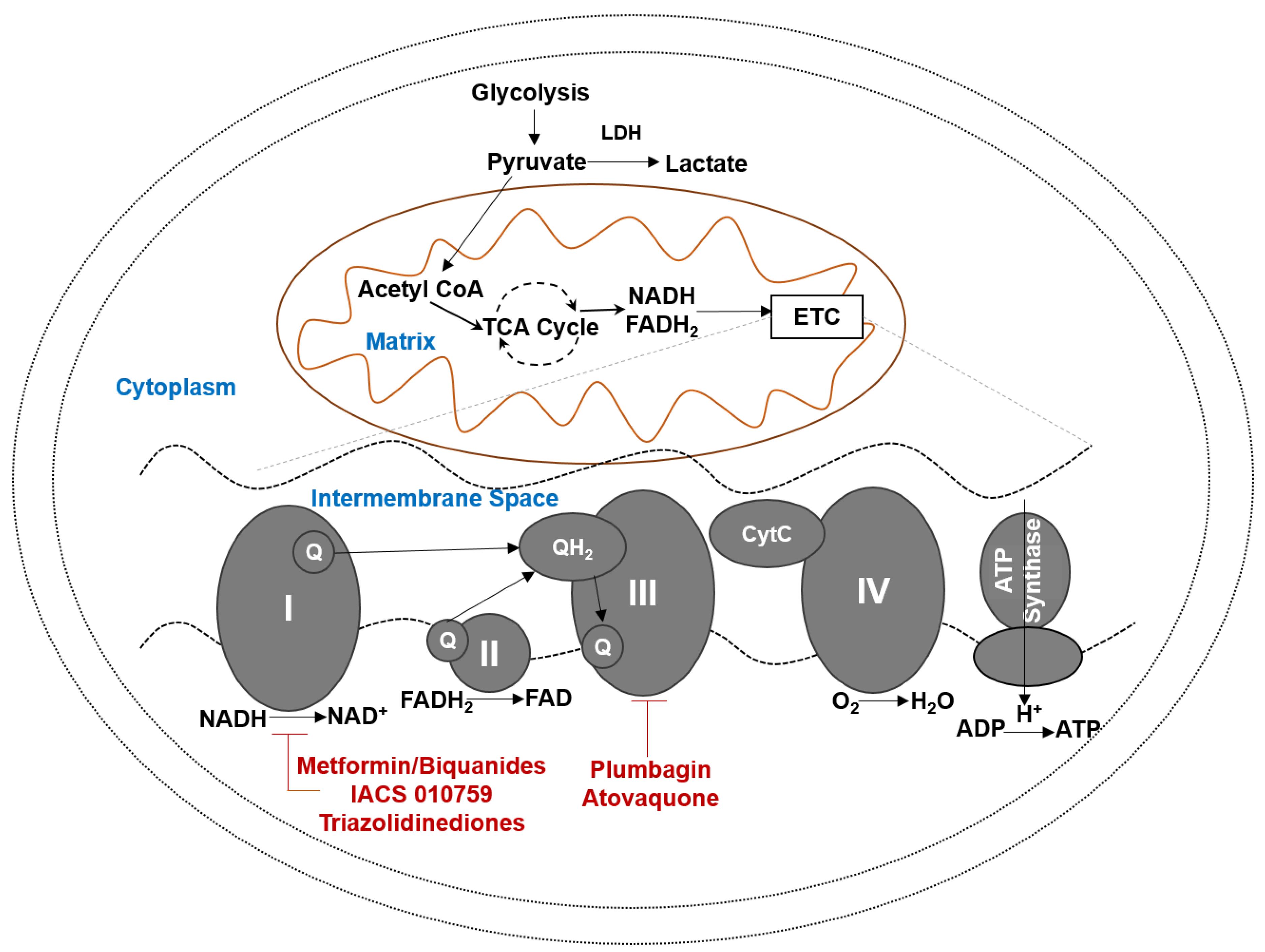
3. OXPHOS Provides Multiple Targets for Drug Development
4. OXPHOS Inhibitors
4.1. Complex I Inhibiting Biguanides
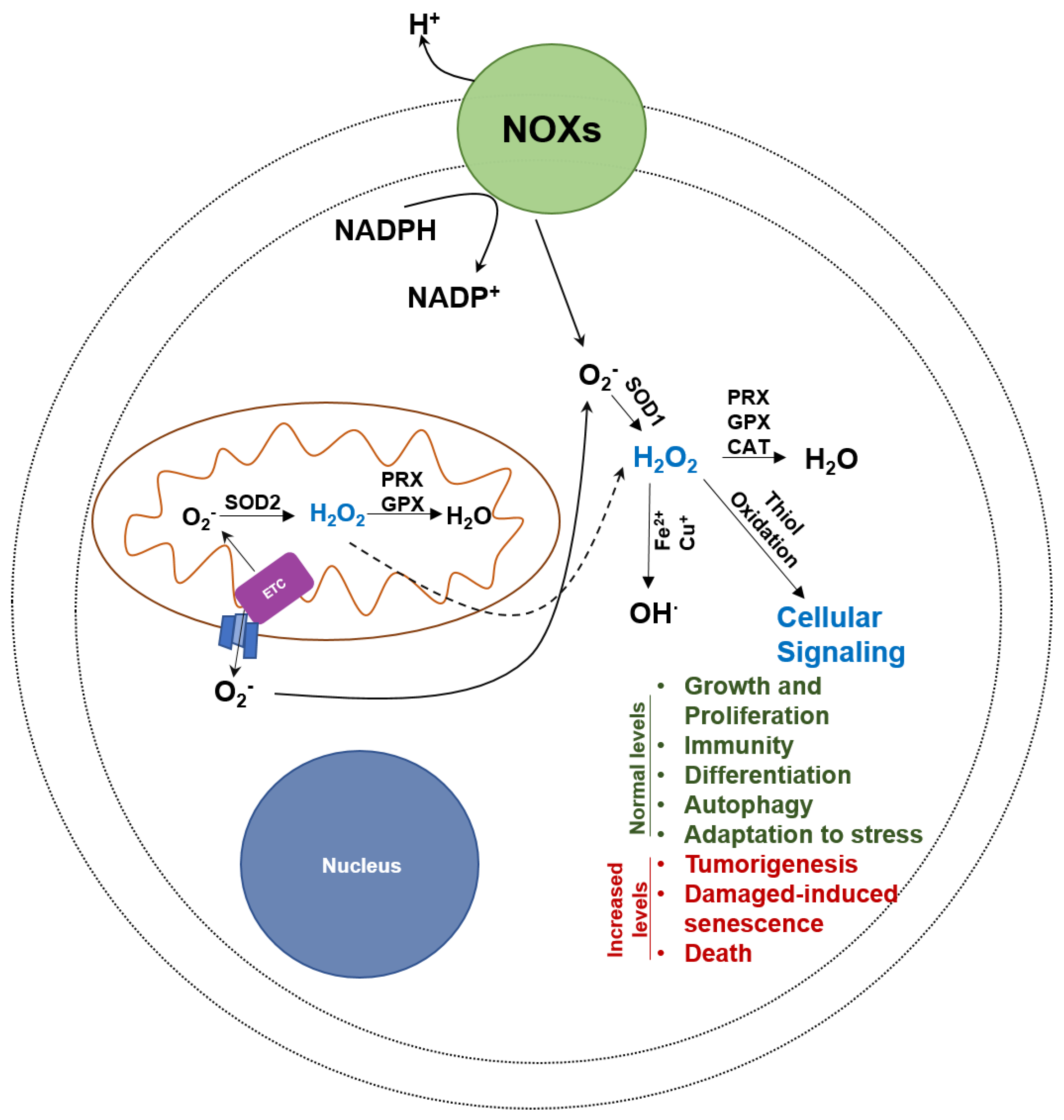
4.2. Oxidative Stress Inducers
4.3. Superoxide Anion (O2−)
4.4. Hydrogen Peroxide (H2O2)
4.5. Hydroxyl Radical (OH∙)
5. Barriers to Using OXPHOS Inhibitors for HGSOC Therapy
5.1. Potential Toxicity of OXPHOS Inhibitors
5.2. Anti-Oxidant Mechanisms and Chemoresistance
5.3. Mitochondrial Adaptations to Oxidative Stress
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Cori, C.F.; Cori, G.T. The Carbohydrate metabolism of tumors: I. The free sugar, lactic acid, and glycogen content of malignant tumors. J. Biol. Chem. 1925, 64, 11–22. [Google Scholar]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Hatzivassiliou, G.; Zhao, F.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005, 8, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Wu, N.; Asara, J.M.; Cantley, L.C. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 2008, 452, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Boxer, M.B.; Jiang, J.K.; Vander Heiden, M.G.; Shen, M.; Skoumbourdis, A.P.; Southall, N.; Veith, H.; Leister, W.; Austin, C.P.; Park, H.W.; et al. Evaluation of substituted N,N’diarylsulfonamides as activators of the tumor cell specific M2 isoform of pyruvate kinase. J. Med. Chem. 2010, 53, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Anastasiou, D.; Yu, Y.; Israelsen, W.J.; Jiang, J.K.; Boxer, M.B.; Hong, B.S.; Tempel, W.; Dimov, S.; Shen, M.; Jha, A.; et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat. Chem. Biol. 2012, 8, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Ning, X.; Qi, H.; Li, R.; Jin, Y.; McNutt, M.A.; Yin, Y. Synthesis and antitumor activity of novel 2, 3-didithiocarbamate substituted naphthoquinones as inhibitors of pyruvate kinase M2 isoform. J. Enzyme Inhib. Med. Chem. 2018, 33, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Sugden, M.C.; Holness, M.J. Therapeutic potential of the mammalian pyruvate dehydrogenase kinases in the prevention of hyperglycaemia. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2002, 2, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, S.L.; Hu, X.; Tam, K.Y. Phenyl butyrate inhibits pyruvate dehydrogenase kinase 1 and contributes to its anti-cancer effect. Eur. J. Pharm. Sci. 2017, 110, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; Hu, X.; Zhang, W.; Yao, H.; Tam, K.Y. Development of pyruvate dehydrogenase kinase inhibitors in medicinal chemistry with particular emphasis as anticancer agents. Drug Discov. Today 2015, 20, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Magid, A.F. Inhibitors of Mutant IDH for the Treatment of Cancer. ACS Med. Chem. Lett. 2014, 5, 1184–1185. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Huwaimel, B.; Verma, K.; Miller, J.; Germain, T.M.; Kinarivala, N.; Pappas, D.; Brookes, P.S.; Trippier, P.C. Synthesis and Antineoplastic Evaluation of Mitochondrial Complex II (Succinate Dehydrogenase) Inhibitors Derived from Atpenin A5. ChemMedChem 2017, 12, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Rai, G.; Brimacombe, K.R.; Mott, B.T.; Urban, D.J.; Hu, X.; Yang, S.M.; Lee, T.D.; Cheff, D.M.; Kouznetsova, J.; Benavides, G.A.; et al. Discovery and Optimization of Potent, Cell-Active Pyrazole-Based Inhibitors of Lactate Dehydrogenase (LDH). J. Med. Chem. 2017, 60, 9184–9204. [Google Scholar] [CrossRef] [PubMed]
- Pasto, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Fiorillo, M.; Peiris-Pages, M.; Ozsvari, B.; Smith, D.L.; Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Cappello, A.R.; Pezzi, V.; Lisanti, M.P.; et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget 2015, 6, 14777–14795. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.; Bonuccelli, G.; Ozsvari, B.; Peiris-Pages, M.; Fiorillo, M.; Smith, D.L.; Bevilacqua, G.; Mazzanti, C.M.; McDonnell, L.A.; Naccarato, A.G.; et al. Mitochondrial mass, a new metabolic biomarker for stem-like cancer cells: Understanding WNT/FGF-driven anabolic signaling. Oncotarget 2015, 6, 30453–30471. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih Ie, M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer--shifting the paradigm. Hum. Pathol. 2011, 42, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih Ie, M. The origin and pathogenesis of epithelial ovarian cancer: A proposed unifying theory. Am. J. Surg. Pathol. 2010, 34, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih Ie, M. Pathogenesis of ovarian cancer: lessons from morphology and molecular biology and their clinical implications. Int. J. Gynecol. Pathol. 2008, 27, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Network, C.G.A.R. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Kuhn, E.; Kurman, R.J.; Vang, R.; Sehdev, A.S.; Han, G.; Soslow, R.; Wang, T.L.; Shih Ie, M. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma-evidence supporting the clonal relationship of the two lesions. J. Pathol. 2012, 226, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Vang, R.; Levine, D.A.; Soslow, R.A.; Zaloudek, C.; Shih Ie, M.; Kurman, R.J. Molecular Alterations of TP53 are a Defining Feature of Ovarian High-Grade Serous Carcinoma: A Rereview of Cases Lacking TP53 Mutations in The Cancer Genome Atlas Ovarian Study. Int. J. Gynecol. Pathol. 2016, 35, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Kindelberger, D.W.; Lee, Y.; Miron, A.; Hirsch, M.S.; Feltmate, C.; Medeiros, F.; Callahan, M.J.; Garner, E.O.; Gordon, R.W.; Birch, C.; et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: Evidence for a causal relationship. Am. J. Surg. Pathol. 2007, 31, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Perets, R.; Wyant, G.A.; Muto, K.W.; Bijron, J.G.; Poole, B.B.; Chin, K.T.; Chen, J.Y.; Ohman, A.W.; Stepule, C.D.; Kwak, S.; et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell 2013, 24, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Przybycin, C.G.; Kurman, R.J.; Ronnett, B.M.; Shih Ie, M.; Vang, R. Are all pelvic (nonuterine) serous carcinomas of tubal origin? Am. J. Surg. Pathol. 2010, 34, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive, relapsed serous ovarian cancer and a BRCA mutation: Overall survival adjusted for postprogression poly(adenosine diphosphate ribose) polymerase inhibitor therapy. Cancer 2016, 122, 1844–1852. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.A.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: An updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol. 2016, 17, 1579–1589. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Dar, S.; Chhina, J.; Mert, I.; Chitale, D.; Buekers, T.; Kaur, H.; Giri, S.; Munkarah, A.; Rattan, R. Bioenergetic Adaptations in Chemoresistant Ovarian Cancer Cells. Sci. Rep. 2017, 7, 8760. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.S.; Roberts, P.C.; Frisard, M.I.; McMillan, R.P.; Brown, T.J.; Lawless, M.H.; Hulver, M.W.; Schmelz, E.M. Metabolic changes during ovarian cancer progression as targets for sphingosine treatment. Exp. Cell Res. 2013, 319, 1431–1442. [Google Scholar] [CrossRef] [PubMed]
- Dier, U.; Shin, D.H.; Hemachandra, L.P.; Uusitalo, L.M.; Hempel, N. Bioenergetic analysis of ovarian cancer cell lines: Profiling of histological subtypes and identification of a mitochondria-defective cell line. PLoS ONE 2014, 9, e98479. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.K.; Huang, T.S.; Liao, Y.P.; Huang, R.L.; Su, P.H.; Shen, H.Y.; Lai, H.C.; Wang, Y.C. Pyruvate kinase M2 is a poor prognostic marker of and a therapeutic target in ovarian cancer. PLoS ONE 2017, 12, e0182166. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Gu, J.; Xin, X.; Li, Y.; Wang, H. Expression of hexokinase 2 in epithelial ovarian tumors and its clinical significance in serous ovarian cancer. Eur. J. Gynaecol. Oncol. 2014, 35, 519–524. [Google Scholar] [PubMed]
- Xintaropoulou, C.; Ward, C.; Wise, A.; Queckborner, S.; Turnbull, A.; Michie, C.O.; Williams, A.R.W.; Rye, T.; Gourley, C.; Langdon, S.P. Expression of glycolytic enzymes in ovarian cancers and evaluation of the glycolytic pathway as a strategy for ovarian cancer treatment. BMC Cancer 2018, 18, 636. [Google Scholar] [CrossRef] [PubMed]
- Suh, D.H.; Kim, M.A.; Kim, H.; Kim, M.K.; Kim, H.S.; Chung, H.H.; Kim, Y.B.; Song, Y.S. Association of overexpression of hexokinase II with chemoresistance in epithelial ovarian cancer. Clin. Exp. Med. 2014, 14, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Tsukioka, M.; Matsumoto, Y.; Noriyuki, M.; Yoshida, C.; Nobeyama, H.; Yoshida, H.; Yasui, T.; Sumi, T.; Honda, K.; Ishiko, O. Expression of glucose transporters in epithelial ovarian carcinoma: Correlation with clinical characteristics and tumor angiogenesis. Oncol. Rep. 2007, 18, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Kalir, T.; Wang, B.Y.; Goldfischer, M.; Haber, R.S.; Reder, I.; Demopoulos, R.; Cohen, C.J.; Burstein, D.E. Immunohistochemical staining of GLUT1 in benign, borderline, and malignant ovarian epithelia. Cancer 2002, 94, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Lee, Y.S.; Kim, J.; Chung, J.Y.; Kim, J.H. Overexpression of glucose transporter-1 (GLUT-1) predicts poor prognosis in epithelial ovarian cancer. Cancer Investig. 2013, 31, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, T.; Yoshida, Y.; Kawahara, K.; Tsuchida, T.; Okazawa, H.; Fujibayashi, Y.; Yonekura, Y.; Kotsuji, F. Expression of GLUT-1 glucose transfer, cellular proliferation activity and grade of tumor correlate with [F-18]-fluorodeoxyglucose uptake by positron emission tomography in epithelial tumors of the ovary. Int. J. Cancer 2004, 109, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Hitosugi, T.; Kang, S.; Vander Heiden, M.G.; Chung, T.-W.; Elf, S.; Lythgoe, K.; Dong, S.; Lonial, S.; Wang, X.; Chen, G.Z.; et al. Tyrosine Phosphorylation Inhibits PKM2 to Promote the Warburg Effect and Tumor Growth. Sci. Signal. 2009, 2, ra73. [Google Scholar] [CrossRef] [PubMed]
- Privat, M.; Radosevic-Robin, N.; Aubel, C.; Cayre, A.; Penault-Llorca, F.; Marceau, G.; Sapin, V.; Bignon, Y.J.; Morvan, D. BRCA1 induces major energetic metabolism reprogramming in breast cancer cells. PLoS ONE 2014, 9, e102438. [Google Scholar] [CrossRef] [PubMed]
- Simabuco, F.M.; Morale, M.G.; Pavan, I.C.B.; Morelli, A.P.; Silva, F.R.; Tamura, R.E. p53 and metabolism: From mechanism to therapeutics. Oncotarget 2018, 9, 23780–23823. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Chien, J.; Staub, J.; Avula, R.; Greene, E.L.; Matthews, T.A.; Smith, D.I.; Kaufmann, S.H.; Roberts, L.R.; Shridhar, V. Loss of HSulf-1 up-regulates heparin-binding growth factor signaling in cancer. J. Biol. Chem. 2003, 278, 23107–23117. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Roy, D.; Camacho-Pereira, J.; Khurana, A.; Chini, E.; Yang, L.; Baddour, J.; Stilles, K.; Padmabandu, S.; Leung, S.; et al. HSulf-1 deficiency dictates a metabolic reprograming of glycolysis and TCA cycle in ovarian cancer. Oncotarget 2015, 6, 33705–33719. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, J.; Takemitsu, M.; Nonaka, I. Recovery of the missing tumorigenicity in mitochondrial DNA-less HeLa cells by introduction of mitochondrial DNA from normal human cells. Somat. Cell Mol. Genet. 1992, 18, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, L.R.; Varella-Garcia, M.; Liang, B.C. Diminished tumorigenic phenotype after depletion of mitochondrial DNA. Cell Growth Differ. 1997, 8, 1189–1198. [Google Scholar] [PubMed]
- Morais, R.; Zinkewich-Peotti, K.; Parent, M.; Wang, H.; Babai, F.; Zollinger, M. Tumor-forming ability in athymic nude mice of human cell lines devoid of mitochondrial DNA. Cancer Res. 1994, 54, 3889–3896. [Google Scholar] [PubMed]
- Lim, H.Y.; Ho, Q.S.; Low, J.; Choolani, M.; Wong, K.P. Respiratory competent mitochondria in human ovarian and peritoneal cancer. Mitochondrion 2011, 11, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Corti, D.; Draetta, G.F. Tumors and mitochondrial respiration: A neglected connection. Cancer Res. 2015, 75, 3685–3686. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Lu, M.; Jia, D.; Ma, J.; Ben-Jacob, E.; Levine, H.; Kaipparettu, B.A.; Onuchic, J.N. Modeling the Genetic Regulation of Cancer Metabolism: Interplay between Glycolysis and Oxidative Phosphorylation. Cancer Res. 2017, 77, 1564–1574. [Google Scholar] [CrossRef] [PubMed]
- Cuezva, J.M.; Krajewska, M.; de Heredia, M.L.; Krajewski, S.; Santamaria, G.; Kim, H.; Zapata, J.M.; Marusawa, H.; Chamorro, M.; Reed, J.C. The bioenergetic signature of cancer: A marker of tumor progression. Cancer Res. 2002, 62, 6674–6681. [Google Scholar] [PubMed]
- Hjerpe, E.; Egyhazi Brage, S.; Carlson, J.; Frostvik Stolt, M.; Schedvins, K.; Johansson, H.; Shoshan, M.; Avall-Lundqvist, E. Metabolic markers GAPDH, PKM2, ATP5B and BEC-index in advanced serous ovarian cancer. BMC Clin. Pathol. 2013, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.S.; Roberts, P.C.; Frisard, M.I.; Hulver, M.W.; Schmelz, E.M. Ovarian tumor-initiating cells display a flexible metabolism. Exp. Cell Res. 2014, 328, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.C.; Mottillo, E.P.; Baxa, A.C.; Heng, H.H.; Doyon-Reale, N.; Gregoire, L.; Lancaster, W.D.; Rabah, R.; Schmelz, E.M. Sequential molecular and cellular events during neoplastic progression: A mouse syngeneic ovarian cancer model. Neoplasia 2005, 7, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Song, H.Y.; Dunbar, J.D.; Zhang, Y.X.; Guo, D.; Donner, D.B. Identification of a protein with homology to hsp90 that binds the type 1 tumor necrosis factor receptor. J. Biol. Chem. 1995, 270, 3574–3581. [Google Scholar] [CrossRef] [PubMed]
- Matassa, D.S.; Amoroso, M.R.; Lu, H.; Avolio, R.; Arzeni, D.; Procaccini, C.; Faicchia, D.; Maddalena, F.; Simeon, V.; Agliarulo, I.; et al. Oxidative metabolism drives inflammation-induced platinum resistance in human ovarian cancer. Cell Death Differ. 2016, 23, 1542–1554. [Google Scholar] [CrossRef] [PubMed]
- Matassa, D.S.; Agliarulo, I.; Avolio, R.; Landriscina, M.; Esposito, F. TRAP1 regulation of cancer metabolism: Dual role as oncogene or tumor suppressor. Genes 2018, 9, 195. [Google Scholar] [CrossRef] [PubMed]
- Morrot, A.; da Fonseca, L.M.; Salustiano, E.J.; Gentile, L.B.; Conde, L.; Filardy, A.A.; Franklim, T.N.; da Costa, K.M.; Freire-de-Lima, C.G.; Freire-de-Lima, L. Metabolic Symbiosis and Immunomodulation: How Tumor Cell-Derived Lactate May Disturb Innate and Adaptive Immune Responses. Front. Oncol. 2018, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Curry, J.M.; Ko, Y.H.; Lin, Z.; Tuluc, M.; Cognetti, D.; Birbe, R.C.; Pribitkin, E.; Bombonati, A.; Pestell, R.G.; et al. Oncogenes and inflammation rewire host energy metabolism in the tumor microenvironment: RAS and NFkappaB target stromal MCT4. Cell Cycle 2013, 12, 2580–2597. [Google Scholar] [CrossRef] [PubMed]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Lin, Z.; Ertel, A.; Flomenberg, N.; Witkiewicz, A.K.; Birbe, R.C.; Howell, A.; Pavlides, S.; Gandara, R.; et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011, 10, 1772–1783. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; Dasgupta, A.; Nguyen, K.H.; Liu, C.; Kovatich, A.J.; Schwartz, G.F.; Pestell, R.G.; Sotgia, F.; Rui, H.; Lisanti, M.P. Stromal caveolin-1 levels predict early DCIS progression to invasive breast cancer. Cancer Biol. Ther. 2009, 8, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, F.; Martinez-Outschoorn, U.E.; Lisanti, M.P. Cancer metabolism: New validated targets for drug discovery. Oncotarget 2013, 4, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Monti, D.; Sotgia, F.; Whitaker-Menezes, D.; Tuluc, M.; Birbe, R.; Berger, A.; Lazar, M.; Cotzia, P.; Draganova-Tacheva, R.; Lin, Z.; et al. Pilot study demonstrating metabolic and anti-proliferative effects of in vivo anti-oxidant supplementation with N-Acetylcysteine in Breast Cancer. Semin. Oncol. 2017, 44, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Constance, J.E.; Lim, C.S. Targeting malignant mitochondria with therapeutic peptides. Ther. Deliv. 2012, 3, 961–979. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, M.; Becker, C.; Meier, C.; Jick, S.S.; Meier, C.R. Use of metformin and the risk of ovarian cancer: A case-control analysis. Gynecol. Oncol. 2011, 123, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Meuter, A.; Thapa, P.; Langstraat, C.; Giri, S.; Chien, J.; Rattan, R.; Cliby, W.; Shridhar, V. Metformin intake is associated with better survival in ovarian cancer: A case-control study. Cancer 2013, 119, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Romero, I.L.; McCormick, A.; McEwen, K.A.; Park, S.; Karrison, T.; Yamada, S.D.; Pannain, S.; Lengyel, E. Relationship of type II diabetes and metformin use to ovarian cancer progression, survival, and chemosensitivity. Obstet. Gynecol. 2012, 119, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Bakhru, A.; Buckanovich, R.J.; Griggs, J.J. The impact of diabetes on survival in women with ovarian cancer. Gynecol. Oncol. 2011, 121, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Bridges, H.R.; Sirvio, V.A.; Agip, A.N.; Hirst, J. Molecular features of biguanides required for targeting of mitochondrial respiratory complex I and activation of AMP-kinase. BMC Biol. 2016, 14, 65. [Google Scholar] [CrossRef] [PubMed]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Baran, N.; Han, L.; Herbrich, S.; Sweeney, S.R.; Lodi, A.; Gay, J.; Feng, N.; Molina, J.R.; Kaminski, M.; Guzman, M.L.; et al. Novel Complex I inhibitor IACS-010759 Targets Leukemia Initiating Cells (LICs) in AML Patients. Clin. Lymphoma Myeloma Leuk. 2017, 17, S297. [Google Scholar] [CrossRef]
- Baran, N.; Molina, J.; Cavazos, A.; Harutyunyan, K.; Feng, N.; Gay, J.; Piya, S.; Shanmuga Velandy, S.; Jabbour, E.J.; Andreeff, M.; et al. Mitochondrial Complex I Inhibition with Iacs-010759 in T-ALL Preclinical Models. Blood 2016, 128, 4028. [Google Scholar]
- Lissanu Deribe, Y.; Sun, Y.; Terranova, C.; Khan, F.; Martinez-Ledesma, J.; Gay, J.; Gao, G.; Mullinax, R.A.; Khor, T.; Feng, N.; et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nat. Med. 2018, 24, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Vangapandu, H.V.; Alston, B.; Morse, J.; Ayres, M.L.; Wierda, W.G.; Keating, M.J.; Marszalek, J.R.; Gandhi, V. Biological and metabolic effects of IACS-010759, an OxPhos inhibitor, on chronic lymphocytic leukemia cells. Oncotarget 2018, 9, 24980–24991. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative Stress: Introductory Remarks; Academic Press: London, UK, 1985. [Google Scholar]
- Lv, Z.; Yan, X.; Lu, L.; Su, C.; He, Y. Atovaquone enhances doxorubicin’s efficacy via inhibiting mitochondrial respiration and STAT3 in aggressive thyroid cancer. J. Bioenerg. Biomembr. 2018, 50, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Mutti, L.; Krstic-Demonacos, M.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Repurposing atovaquone: Targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget 2016, 7, 34084–34099. [Google Scholar] [CrossRef] [PubMed]
- Ashton, T.M.; Fokas, E.; Kunz-Schughart, L.A.; Folkes, L.K.; Anbalagan, S.; Huether, M.; Kelly, C.J.; Pirovano, G.; Buffa, F.M.; Hammond, E.M.; et al. The anti-malarial atovaquone increases radiosensitivity by alleviating tumour hypoxia. Nat. Commun. 2016, 7, 12308. [Google Scholar] [CrossRef] [PubMed]
- Kapur, A.; Beres, T.; Rathi, K.; Nayak, A.P.; Czarnecki, A.; Felder, M.; Gillette, A.; Ericksen, S.S.; Sampene, E.; Skala, M.C.; et al. Oxidative stress via inhibition of the mitochondrial electron transport and Nrf-2-mediated anti-oxidative response regulate the cytotoxic activity of plumbagin. Sci. Rep. 2018, 8, 1073. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, R.L.; Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Brand, M.D. Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J. Biol. Chem. 2015, 290, 209–227. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Hampton, M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 2008, 45, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M.; Jaruga, P. Mechanisms of free radical-induced damage to DNA. Free Radic. Res. 2012, 46, 382–419. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, B.B.; Zhong, W.; Mustafa, A.; Fischer, J.W.; Witkowsky, O.; Verma, A.K. Plumbagin inhibits prostate cancer development in TRAMP mice via targeting PKCepsilon, Stat3 and neuroendocrine markers. Carcinogenesis 2012, 33, 2586–2592. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, B.B.; Fischer, J.W.; Singh, A.; Zhong, W.; Mustafa, A.; Meske, L.; Sheikhani, M.O.; Verma, A.K. Plumbagin Inhibits Prostate Carcinogenesis in Intact and Castrated PTEN Knockout Mice via Targeting PKCepsilon, Stat3, and Epithelial-to-Mesenchymal Transition Markers. Cancer Prev. Res. 2015, 8, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, I.K.; Morrisey, J.M.; Darrouzet, E.; Daldal, F.; Vaidya, A.B. Resistance mutations reveal the atovaquone-binding domain of cytochrome b in malaria parasites. Mol. Microbiol. 1999, 33, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Plucinski, M.M.; Huber, C.S.; Akinyi, S.; Dalton, W.; Eschete, M.; Grady, K.; Silva-Flannery, L.; Mathison, B.A.; Udhayakumar, V.; Arguin, P.M.; et al. Novel Mutation in Cytochrome B of Plasmodium falciparum in One of Two Atovaquone-Proguanil Treatment Failures in Travelers Returning From Same Site in Nigeria. Open Forum Infect. Dis. 2014, 1, ofu059. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.P.; Nogueira, F.; Stromberg-Norklit, J.; Lindberg, J.; Carrolo, M.; Casimiro, C.; Lopes, D.; Arez, A.P.; Cravo, P.V.; Rosario, V.E. Detection of atovaquone and Malarone resistance conferring mutations in Plasmodium falciparum cytochrome b gene (cytb). Mol. Cell. Probes 2003, 17, 85–89. [Google Scholar] [CrossRef]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nayak, A.P.; Kapur, A.; Barroilhet, L.; Patankar, M.S. Oxidative Phosphorylation: A Target for Novel Therapeutic Strategies Against Ovarian Cancer. Cancers 2018, 10, 337. https://doi.org/10.3390/cancers10090337
Nayak AP, Kapur A, Barroilhet L, Patankar MS. Oxidative Phosphorylation: A Target for Novel Therapeutic Strategies Against Ovarian Cancer. Cancers. 2018; 10(9):337. https://doi.org/10.3390/cancers10090337
Chicago/Turabian StyleNayak, Amruta P., Arvinder Kapur, Lisa Barroilhet, and Manish S. Patankar. 2018. "Oxidative Phosphorylation: A Target for Novel Therapeutic Strategies Against Ovarian Cancer" Cancers 10, no. 9: 337. https://doi.org/10.3390/cancers10090337
APA StyleNayak, A. P., Kapur, A., Barroilhet, L., & Patankar, M. S. (2018). Oxidative Phosphorylation: A Target for Novel Therapeutic Strategies Against Ovarian Cancer. Cancers, 10(9), 337. https://doi.org/10.3390/cancers10090337