Factors Secreted by Cancer-Associated Fibroblasts that Sustain Cancer Stem Properties in Head and Neck Squamous Carcinoma Cells as Potential Therapeutic Targets

 , , ,
, , ,  ,
,
Abstract
1. Introduction
2. Results
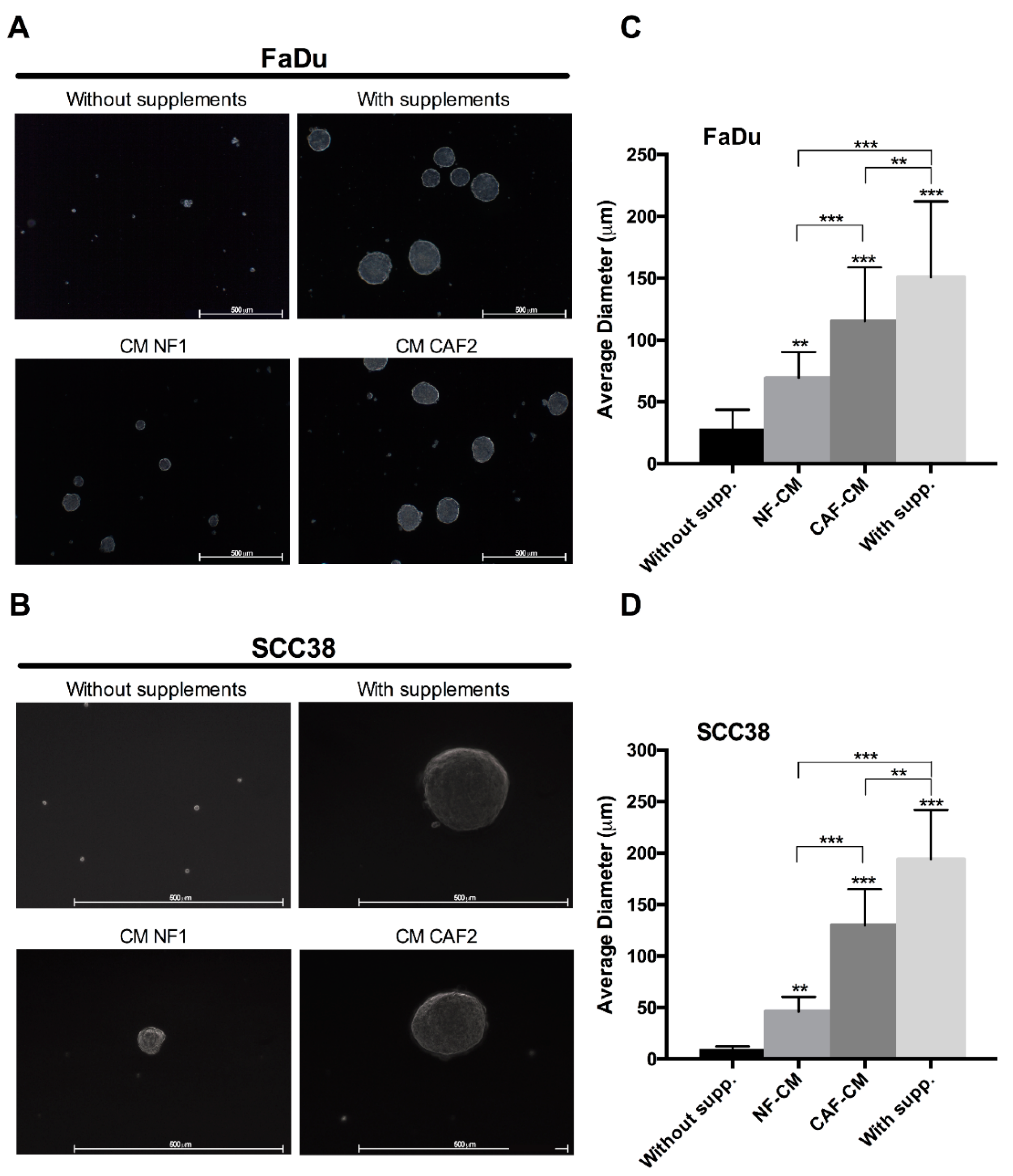
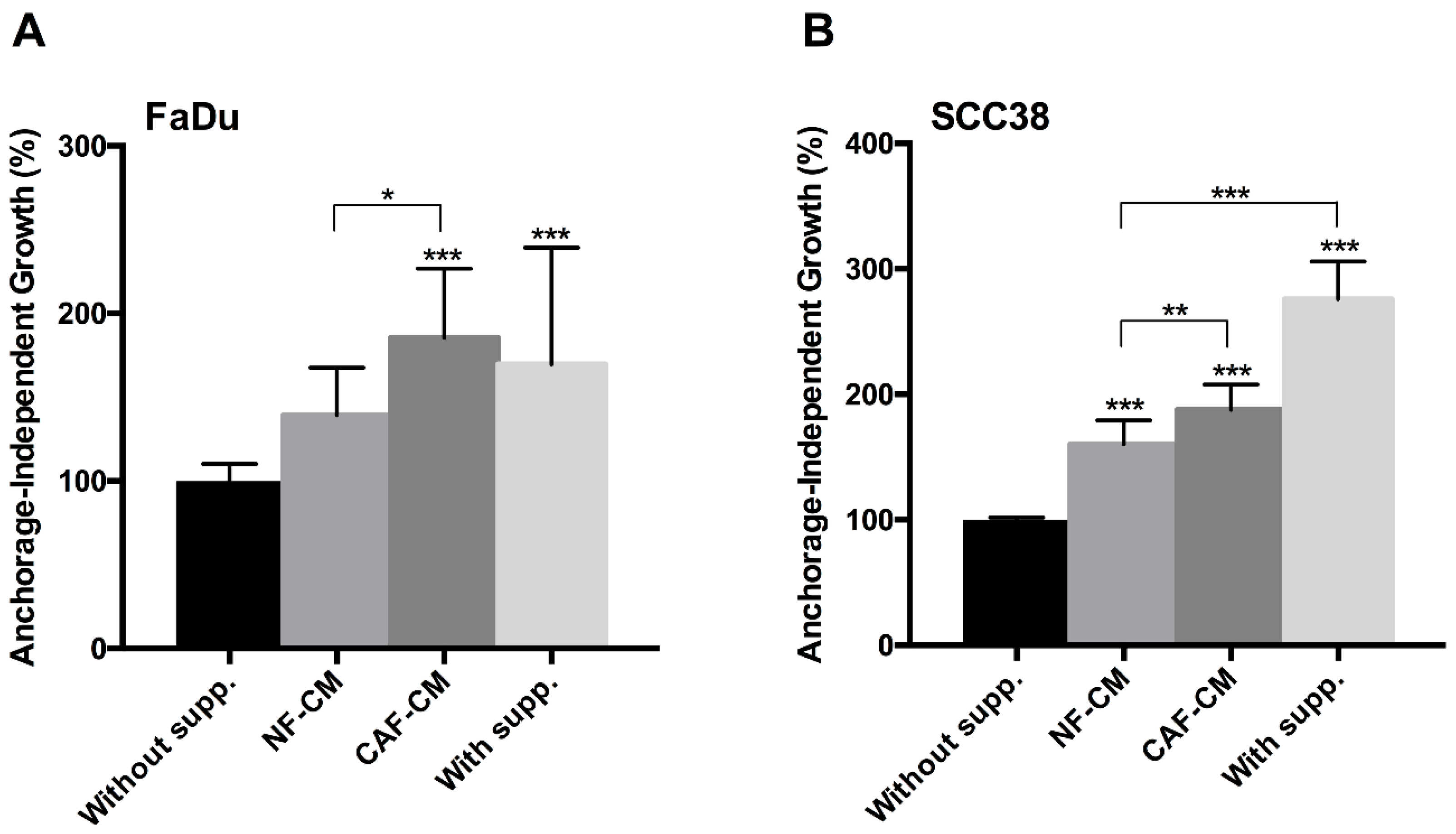
2.1. Fibroblast-Secreted Factors Sustain Cancer Stem Properties of HNSCC Cells
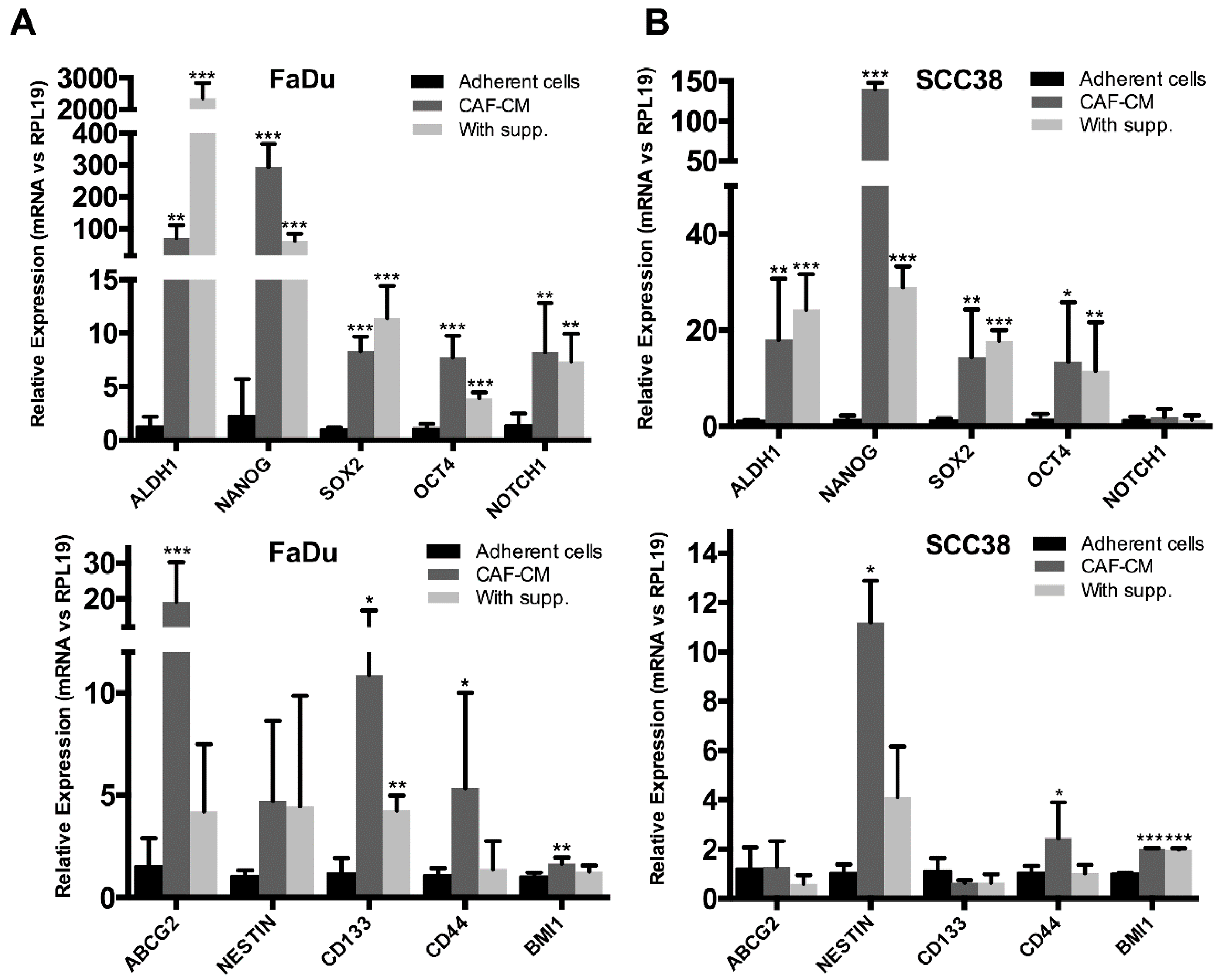
2.2. Fibroblast-Secreted Factors Induced the Expression of Stemness-Related Genes in HNSCC Cells
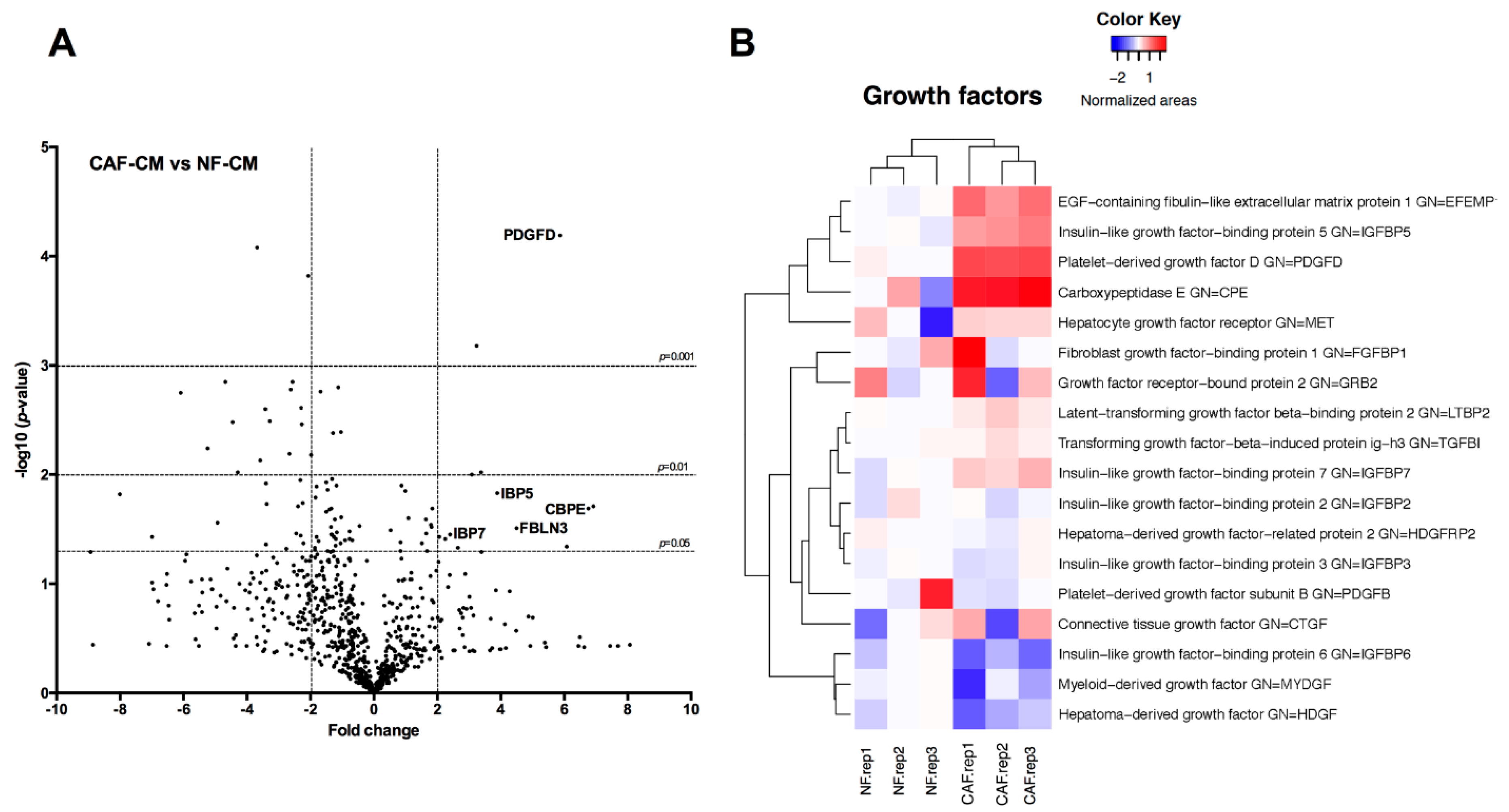
2.3. Identification of Fibroblast-Secreted Proteins by Mass Spectrometry
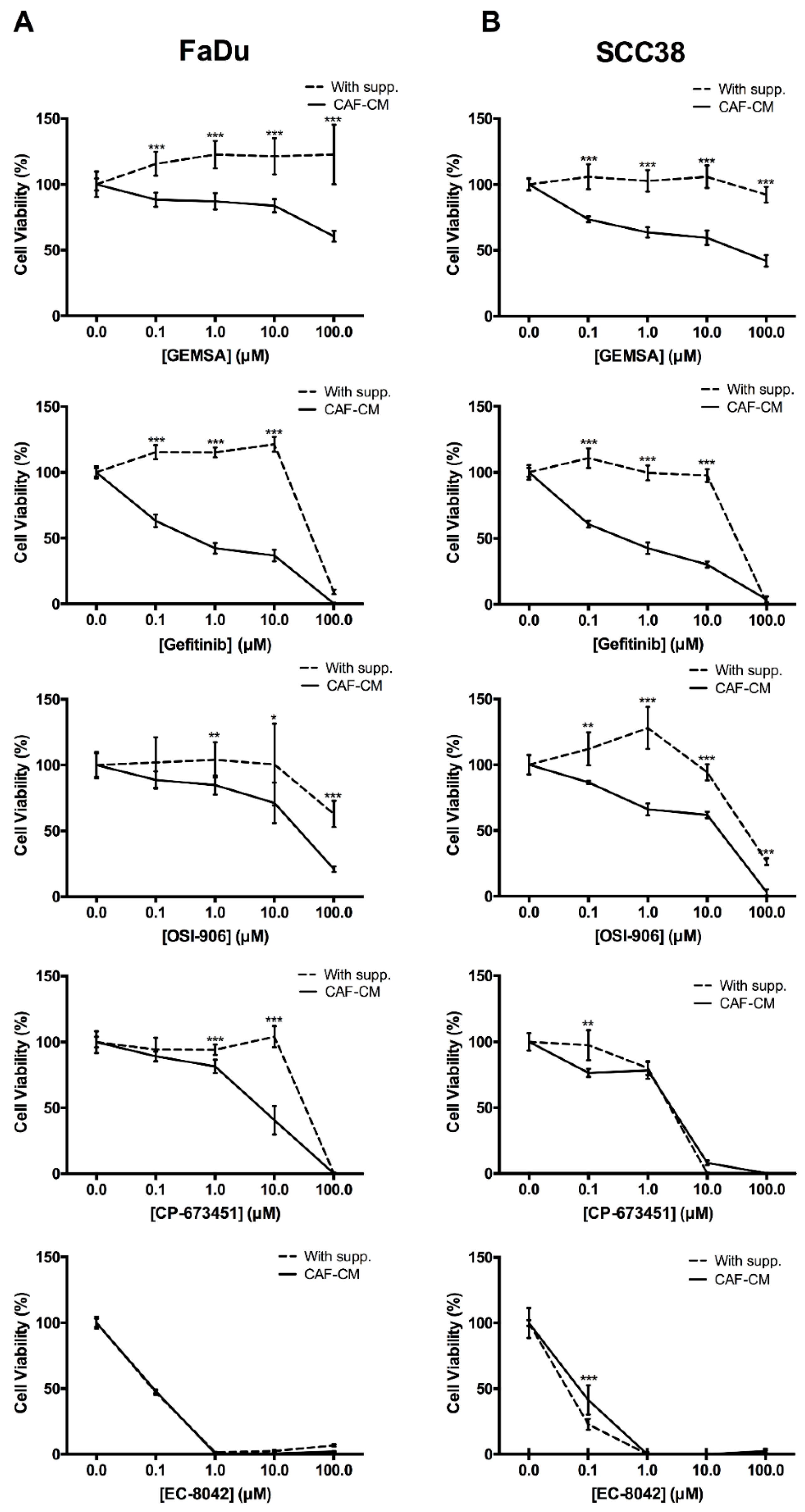
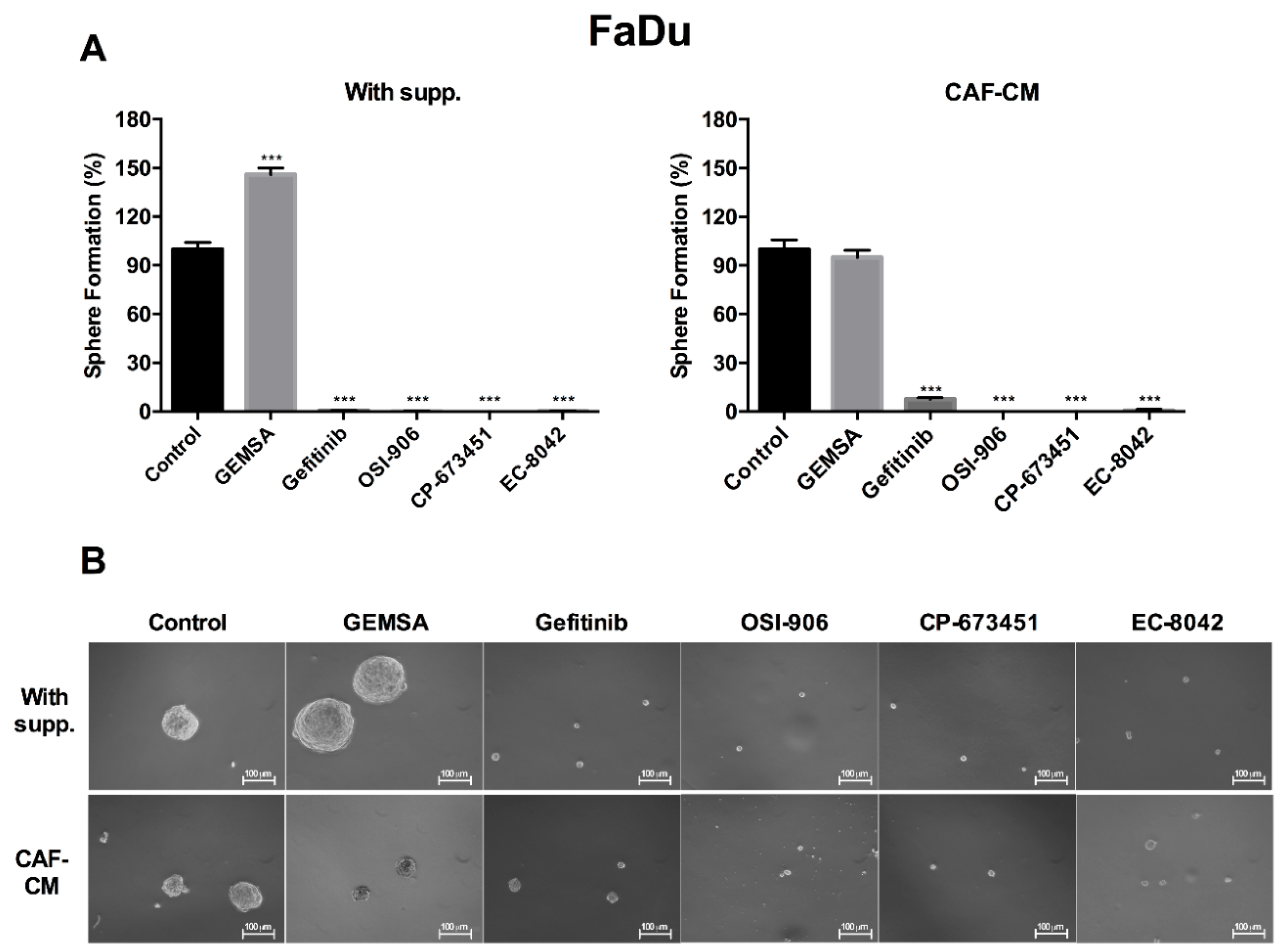
2.4. Targeting EGFR, IGFR, and PDGFR Signaling Effectively Inhibited CAF-Promoted Stemness in HNSCC Cells
3. Discussion
4. Materials and Methods
4.1. Drugs
4.2. Cell Culture
4.3. Conditioned Media Production
4.4. Anchorage-Independent Cell Growth
4.5. Orosphere Formation Assay
4.6. RNA Extraction and Real-Time RT-PCR
4.7. Secretome Analysis by Mass Spectrometry
4.8. Mass Spectrometry Data Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CAFs | Cancer-associated fibroblasts |
| CSC | Cancer stem cells |
| HNSCC | Head and neck squamous cell carcinomas |
| EMT | Epithelial-mesenchymal transition |
| NFs | Normal fibroblasts |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| IGF | Insulin-like growth factor |
| IGFR | Insulin-like growth factor receptor |
| PDGF | Platelet-derived growth factor |
| PDGFR | Platelet-derived growth factor receptor |
| ECM | Extracellular matrix |
| TME | Tumor microenvironment |
| MMPs | Matrix metalloproteinases |
| MS | Mass spectrometry |
| GEMSA | 2-Guanidinoethylmercaptosuccinic acid |
| FBS | Fetal bovine serum |
| HEPES | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid |
| CM | Conditioned media |
| polyHEMA | poly-2-hydroxyethyl methacrylate |
| FGF | Fibroblast growth factor |
| SPE | Solid phase extraction |
| CAN | Acetonitrile |
| TFA | Trifluoroacetic acid |
| FA | Formic acid |
| CBPE | Carboxypeptidase E |
| PDGFD | Platelet-derived growth factor D |
| FBLN3 | EGF-containing fibulin-like extracellular matrix protein-1 |
| IBPs | Insulin-like growth factor binding protein |
| IBP5 | Insulin-like growth factor binding protein-5 |
| IBP7 | Insulin-like growth factor binding protein-7 |
| HGF | Hepatocyte growth factor |
| IL-6 | Interleukin-6 |
References
- Koontongkaew, S. The tumor microenvironment contribution to development, growth, invasion and metastasis of head and neck squamous cell carcinomas. J. Cancer 2013, 4, 66–83. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Koczorowska, M.M.; Tholen, S.; Bucher, F.; Lutz, L.; Kizhakkedathu, J.N.; De Wever, O.; Wellner, U.F.; Biniossek, M.L.; Stahl, A.; Lassmann, S.; et al. Fibroblast activation protein-alpha, a stromal cell surface protease, shapes key features of cancer associated fibroblasts through proteome and degradome alterations. Mol. Oncol. 2016, 10, 40–58. [Google Scholar] [CrossRef] [PubMed]
- De Wever, O.; Mareel, M. Role of tissue stroma in cancer cell invasion. J. Pathol. 2003, 200, 429–447. [Google Scholar] [CrossRef] [PubMed]
- Bello, I.O.; Vered, M.; Dayan, D.; Dobriyan, A.; Yahalom, R.; Alanen, K.; Nieminen, P.; Kantola, S.; Laara, E.; Salo, T. Cancer-associated fibroblasts, a parameter of the tumor microenvironment, overcomes carcinoma-associated parameters in the prognosis of patients with mobile tongue cancer. Oral Oncol. 2011, 47, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Vered, M.; Dobriyan, A.; Dayan, D.; Yahalom, R.; Talmi, Y.P.; Bedrin, L.; Barshack, I.; Taicher, S. Tumor-host histopathologic variables, stromal myofibroblasts and risk score, are significantly associated with recurrent disease in tongue cancer. Cancer Sci. 2010, 101, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.X.; Li, X.L.; Dong, C.F. Epigenetic and metabolic regulation of breast cancer stem cells. J. Zhejiang Univ. Sci. B 2015, 16, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Patel, M.R.; Prescher, J.A.; Patsialou, A.; Qian, D.; Lin, J.; Wen, S.; Chang, Y.F.; Bachmann, M.H.; Shimono, Y.; et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc. Natl. Acad. Sci. USA 2010, 107, 18115–18120. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Xu, T.; Goldkorn, A. Cancer cells cyclically lose and regain drug-resistant highly tumorigenic features characteristic of a cancer stem-like phenotype. Mol. Cancer Ther. 2011, 10, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Weiland, A.; Roswall, P.; Hatzihristidis, T.C.; Pietras, K.; Ostman, A.; Strell, C. Fibroblast-dependent regulation of the stem cell properties of cancer cells. Neoplasma 2012, 59, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Marimuthu, A.; Chavan, S.; Sathe, G.; Sahasrabuddhe, N.A.; Srikanth, S.M.; Renuse, S.; Ahmad, S.; Radhakrishnan, A.; Barbhuiya, M.A.; Kumar, R.V.; et al. Identification of head and neck squamous cell carcinoma biomarker candidates through proteomic analysis of cancer cell secretome. Biochim. Biophys. Acta 2013, 1834, 2308–2316. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Hsu, C.W.; Chen, C.D.; Yu, C.J.; Chang, K.P.; Tai, D.I.; Liu, H.P.; Su, W.H.; Chang, Y.S.; Yu, J.S. Candidate serological biomarkers for cancer identified from the secretomes of 23 cancer cell lines and the human protein atlas. Mol. Cell. Proteom. 2010, 9, 1100–1117. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Li, X.; Li, L.; Wang, L.; Du, Z.; Yang, Y.; Zhao, J.; Li, Y. Silencing of carboxypeptidase E inhibits cell proliferation, tumorigenicity, and metastasis of osteosarcoma cells. Onco. Targets Ther. 2016, 9, 2795–2803. [Google Scholar] [PubMed]
- Liu, A.; Shao, C.; Jin, G.; Liu, R.; Hao, J.; Shao, Z.; Liu, Q.; Hu, X. Downregulation of CPE regulates cell proliferation and chemosensitivity in pancreatic cancer. Tumour Biol. 2014, 35, 12459–12465. [Google Scholar] [CrossRef] [PubMed]
- Beattie, J.; Allan, G.J.; Lochrie, J.D.; Flint, D.J. Insulin-like growth factor-binding protein-5 (IGFBP-5): A critical member of the IGF axis. Biochem. J. 2006, 395, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kong, D.; Li, Y.; Sarkar, F.H. PDGF-D signaling: A novel target in cancer therapy. Curr. Drug Targets 2009, 10, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhang, G.; Miao, X.B.; Deng, X.B.; Wu, Y.; Liu, Y.; Jin, Z.R.; Li, X.Q.; Liu, Q.Z.; Sun, D.X.; et al. Cancer stem-like cell properties are regulated by EGFR/AKT/beta-catenin signaling and preferentially inhibited by gefitinib in nasopharyngeal carcinoma. FEBS J. 2013, 280, 2027–2041. [Google Scholar] [CrossRef] [PubMed]
- Tornin, J.; Martinez-Cruzado, L.; Santos, L.; Rodriguez, A.; Nunez, L.E.; Oro, P.; Hermosilla, M.A.; Allonca, E.; Fernandez-Garcia, M.T.; Astudillo, A.; et al. Inhibition of SP1 by the mithramycin analog EC-8042 efficiently targets tumor initiating cells in sarcoma. Oncotarget 2016, 7, 30935–30950. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Sporn, M.B. The tumour microenvironment as a target for chemoprevention. Nat. Rev. Cancer 2007, 7, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Curry, J.M.; Sprandio, J.; Cognetti, D.; Luginbuhl, A.; Bar-ad, V.; Pribitkin, E.; Tuluc, M. Tumor microenvironment in head and neck squamous cell carcinoma. Semin. Oncol. 2014, 41, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.M.; Fusenig, N.E. Friends or foes—Bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Donnarumma, E.; Fiore, D.; Nappa, M.; Roscigno, G.; Adamo, A.; Iaboni, M.; Russo, V.; Affinito, A.; Puoti, I.; Quintavalle, C.; et al. Cancer-associated fibroblasts release exosomal microRNAs that dictate an aggressive phenotype in breast cancer. Oncotarget 2017, 8, 19592–19608. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, M.; Principe, S.; Jackson, H.W.; Luga, V.; Fang, H.; Molyneux, S.D.; Shao, Y.W.; Aiken, A.; Waterhouse, P.D.; Karamboulas, C.; et al. Loss of the Timp gene family is sufficient for the acquisition of the CAF-like cell state. Nat. Cell Biol. 2014, 16, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; De Sousa, E.M.F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, E.; Bianchini, F.; Masieri, L.; Serni, S.; Torre, E.; Calorini, L.; Chiarugi, P. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010, 70, 6945–6956. [Google Scholar] [CrossRef] [PubMed]
- Ishitoya, J.; Toriyama, M.; Oguchi, N.; Kitamura, K.; Ohshima, M.; Asano, K.; Yamamoto, T. Gene amplification and overexpression of EGF receptor in squamous cell carcinomas of the head and neck. Br. J. Cancer 1989, 59, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Grandis, J.R.; Tweardy, D.J. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993, 53, 3579–3584. [Google Scholar] [PubMed]
- Ang, K.K.; Berkey, B.A.; Tu, X.; Zhang, H.Z.; Katz, R.; Hammond, E.H.; Fu, K.K.; Milas, L. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002, 62, 7350–7356. [Google Scholar] [PubMed]
- Firth, S.M.; Baxter, R.C. Cellular actions of the insulin-like growth factor binding proteins. Endocr. Rev. 2002, 23, 824–854. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.I.; Clemmons, D.R. Insulin-like growth factors and their binding proteins: Biological actions. Endocr. Rev. 1995, 16, 3–34. [Google Scholar] [PubMed]
- Unger, C.; Kramer, N.; Unterleuthner, D.; Scherzer, M.; Burian, A.; Rudisch, A.; Stadler, M.; Schlederer, M.; Lenhardt, D.; Riedl, A.; et al. Stromal-derived IGF2 promotes colon cancer progression via paracrine and autocrine mechanisms. Oncogene 2017, 36, 5341–5355. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.; Baylink, D.J. IGF-binding proteins are multifunctional and act via IGF-dependent and -independent mechanisms. J. Endocrinol. 2002, 175, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.K.; Haun, R.S. Insulin-like growth factor binding protein-5 influences pancreatic cancer cell growth. World J. Gastroenterol. 2009, 15, 3355–3366. [Google Scholar] [CrossRef] [PubMed]
- De Bont, J.M.; van Doorn, J.; Reddingius, R.E.; Graat, G.H.; Passier, M.M.; den Boer, M.L.; Pieters, R. Various components of the insulin-like growth factor system in tumor tissue, cerebrospinal fluid and peripheral blood of pediatric medulloblastoma and ependymoma patients. Int. J. Cancer 2008, 123, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, J.; Zhu, B.; Duan, Y.; Chen, F.; Nian, W.; Sun, J.; Zhang, B.; Tong, Z.; Chen, Z. IGFBP7 functions as a potential lymphangiogenesis inducer in non-small cell lung carcinoma. Oncol. Rep. 2016, 35, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Bartram, I.; Erben, U.; Ortiz-Tanchez, J.; Blunert, K.; Schlee, C.; Neumann, M.; Heesch, S.; Baldus, C.D. Inhibition of IGF1-R overcomes IGFBP7-induced chemotherapy resistance in T-ALL. BMC Cancer 2015, 15, e663. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Inokuchi, M.; Takagi, Y.; Otsuki, S.; Fujimori, Y.; Yanaka, Y.; Kobayashi, K.; Higuchi, K.; Kojima, K.; Kawano, T. Relationship between expression of IGFBP7 and clinicopathological variables in gastric cancer. J. Clin. Pathol. 2015, 68, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ahmad, A.; Li, Y.; Kong, D.; Azmi, A.S.; Banerjee, S.; Sarkar, F.H. Emerging roles of PDGF-D signaling pathway in tumor development and progression. Biochim. Biophys. Acta 2010, 1806, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Banerjee, S.; Ahmad, A.; Li, Y.; Wang, Z.; Sethi, S.; Sarkar, F.H. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PLoS ONE 2010, 5, e12445. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Li, Y.; Wang, Z.; Banerjee, S.; Ahmad, A.; Kim, H.R.; Sarkar, F.H. miR-200 regulates PDGF-D-mediated epithelial-mesenchymal transition, adhesion, and invasion of prostate cancer cells. Stem Cells 2009, 27, 1712–1721. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Wang, Z.; Sarkar, S.H.; Li, Y.; Banerjee, S.; Saliganan, A.; Kim, H.R.; Cher, M.L.; Sarkar, F.H. Platelet-derived growth factor-D overexpression contributes to epithelial-mesenchymal transition of PC3 prostate cancer cells. Stem Cells 2008, 26, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Wang, Z.; Kong, D.; Ali, R.; Ali, S.; Banerjee, S.; Sarkar, F.H. Platelet-derived growth factor-D contributes to aggressiveness of breast cancer cells by up-regulating Notch and NF-kappaB signaling pathways. Breast Cancer Res. Treat. 2011, 126, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kong, D.; Banerjee, S.; Li, Y.; Adsay, N.V.; Abbruzzese, J.; Sarkar, F.H. Down-regulation of platelet-derived growth factor-D inhibits cell growth and angiogenesis through inactivation of Notch-1 and nuclear factor-kappaB signaling. Cancer Res. 2007, 67, 11377–11385. [Google Scholar] [CrossRef] [PubMed]
- Okada, A.; Yaguchi, T.; Kanno, T.; Gotoh, A.; Nakano, T.; Nishizaki, T. PDGF-D/PDGF-betabeta receptor-regulated chemotaxis of malignant mesothelioma cells. Cell Physiol. Biochem. 2012, 29, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, E.; Song, Y.H.; Krishnappa, S.; Alt, E. Epithelial-mesenchymal transition in breast cancer lines is mediated through PDGF-D released by tissue-resident stem cells. Int. J. Cancer 2012, 131, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Sarkar, F.H.; Ahmed, Q.; Bandyopadhyay, S.; Nahleh, Z.A.; Semaan, A.; Sakr, W.; Munkarah, A.; Ali-Fehmi, R. Molecular markers of epithelial-to-mesenchymal transition are associated with tumor aggressiveness in breast carcinoma. Transl. Oncol. 2011, 4, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Lansford, C.D.; Grenman, R., Bier; Somers, K.D.; Kim, S.-Y.; Whiteside, T.L.; Clayman, G.L.; Welkoborsky, H.-J.; Carey, T.E. Head and Neck Cancer. In Human Cell Culture, Vol. II; Kluwer Academic Publisher: Dordrecht, The Netherlands, 1999. [Google Scholar]
- Leigh, I.M.; Watt, F.M. Keratinocyte Methods; Cambridge University Press: Cambridge, UK, 1994. [Google Scholar]
- Martinez-Cruzado, L.; Tornin, J.; Santos, L.; Rodriguez, A.; García-Castro, J.; Morís, F.; Rodriguez, R. Aldh1 Expression and Activity Increase During Tumor Evolution in Sarcoma Cancer Stem Cell Populations. Sci. Rep. 2016, 6, e27878. [Google Scholar] [CrossRef] [PubMed]
- Weiswald, L.B.; Bellet, D.; Dangles-Marie, V. Spherical cancer models in tumor biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Cutillas, P.R.; Vanhaesebroeck, B. Quantitative profile of five murine core proteomes using label-free functional proteomics. Mol. Cell. Proteom. 2007, 6, 1560–1573. [Google Scholar] [CrossRef] [PubMed]
- Alcolea, M.P.; Casado, P.; Rodriguez-Prados, J.C.; Vanhaesebroeck, B.; Cutillas, P.R. Phosphoproteomic analysis of leukemia cells under basal and drug-treated conditions identifies markers of kinase pathway activation and mechanisms of resistance. Mol. Cell. Proteom. 2012, 11, 453–466. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UNIPROT_ID | Gene Name | Fold Change CAFs | p-Value |
|---|---|---|---|
| HNRPL_HUMAN | heterogeneous nuclear ribonucleoprotein L(HNRNPL) | 6.92 | 0.020 |
| CBPE_HUMAN | carboxypeptidase E(CPE) | 6.76 | 0.020 |
| CO7_HUMAN | complement C7(C7) | 6.08 | 0.046 |
| PDGFD_HUMAN | platelet derived growth factor D(PDGFD) | 5.87 | >0.001 |
| FBLN3_HUMAN | EGF containing fibulin like extracellular matrix protein 1(EFEMP1) | 4.50 | 0.031 |
| IBP5_HUMAN | insulin like growth factor binding protein 5(IGFBP5) | 3.89 | 0.015 |
| DDAH1_HUMAN | dimethylarginine dimethylaminohydrolase 1(DDAH1) | 3.38 | 0.010 |
| PGM1_HUMAN | phosphoglucomutase 1(PGM1) | 3.24 | 0.001 |
| GREM1_HUMAN | gremlin 1, DAN family BMP antagonist(GREM1) | 3.09 | 0.010 |
| IF4A1_HUMAN | eukaryotic translation initiation factor 4A1(EIF4A1) | 2.65 | 0.047 |
| RS18_HUMAN | ribosomal protein S18(RPS18) | 2.40 | 0.036 |
| TCPQ_HUMAN | chaperonin containing TCP1 subunit 8(CCT8) | 2.25 | 0.039 |
| IBP7_HUMAN | insulin like growth factor binding protein 7(IGFBP7) | 2.06 | 0.037 |
| TCPD_HUMAN | chaperonin containing TCP1 subunit 4(CCT4) | −2.07 | >0.001 |
| MARCS_HUMAN | myristoylated alanine rich protein kinase C substrate(MARCKS) | −2.22 | 0.042 |
| ANXA2_HUMAN | annexin A2(ANXA2) | −2.24 | 0.018 |
| PEDF_HUMAN | serpin family F member 1(SERPINF1) | −2.27 | 0.003 |
| MMP3_HUMAN | matrix metallopeptidase 3(MMP3) | −2.29 | 0.002 |
| PSA5_HUMAN | proteasome subunit alpha 5(PSMA5) | −2.31 | 0.037 |
| CALR_HUMAN | calreticulin(CALR) | −2.32 | 0.011 |
| BASP1_HUMAN | brain abundant membrane attached signal protein 1(BASP1) | −2.39 | 0.020 |
| VASN_HUMAN | vasorin(VASN) | −2.45 | 0.034 |
| LUM_HUMAN | lumican(LUM) | −2.56 | 0.001 |
| CFAD_HUMAN | complement factor D(CFD) | −2.62 | 0.002 |
| LA_HUMAN | Sjogren syndrome antigen B(SSB) | −2.66 | 0.007 |
| UB2V1_HUMAN | TMEM189-UBE2V1 readthrough(TMEM189-UBE2V1) | −2.76 | 0.048 |
| PSG7_HUMAN | pregnancy specific beta-1-glycoprotein 7 (gene/pseudogene)(PSG7) | −3.28 | 0.003 |
| PTGDS_HUMAN | prostaglandin D2 synthase(PTGDS) | −3.38 | 0.019 |
| FBLN2_HUMAN | fibulin 2(FBLN2) | −3.40 | 0.012 |
| AN32B_HUMAN | acidic nuclear phosphoprotein 32 family member B(ANP32B) | −3.40 | 0.044 |
| ENPP2_HUMAN | ectonucleotide pyrophosphatase/phosphodiesterase 2(ENPP2) | −3.42 | 0.003 |
| MASP1_HUMAN | mannan binding lectin serine peptidase 1(MASP1) | −3.58 | 0.007 |
| EMIL2_HUMAN | elastin microfibril interfacer 2(EMILIN2) | −3.68 | >0.001 |
| CSPG4_HUMAN | chondroitin sulfate proteoglycan 4(CSPG4) | −4.29 | 0.010 |
| APOE_HUMAN | apolipoprotein E(APOE) | −4.45 | 0.003 |
| TENA_HUMAN | tenascin C(TNC) | −4.68 | 0.001 |
| PDIA6_HUMAN | protein disulfide isomerase family A member 6(PDIA6) | −4.93 | 0.028 |
| A2GL_HUMAN | leucine rich alpha-2-glycoprotein 1(LRG1) | −5.24 | 0.006 |
| RAB2A_HUMAN | RAB2A, member RAS oncogene family(RAB2A) | −6.09 | 0.002 |
| RL15_HUMAN | ribosomal protein L15(RPL15) | −6.99 | 0.037 |
| PSG4_HUMAN | pregnancy specific beta-1-glycoprotein 4(PSG4) | −8.01 | 0.015 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Álvarez-Teijeiro, S.; García-Inclán, C.; Villaronga, M.Á.; Casado, P.; Hermida-Prado, F.; Granda-Díaz, R.; Rodrigo, J.P.; Calvo, F.; Del-Río-Ibisate, N.; Gandarillas, A.; et al. Factors Secreted by Cancer-Associated Fibroblasts that Sustain Cancer Stem Properties in Head and Neck Squamous Carcinoma Cells as Potential Therapeutic Targets. Cancers 2018, 10, 334. https://doi.org/10.3390/cancers10090334
Álvarez-Teijeiro S, García-Inclán C, Villaronga MÁ, Casado P, Hermida-Prado F, Granda-Díaz R, Rodrigo JP, Calvo F, Del-Río-Ibisate N, Gandarillas A, et al. Factors Secreted by Cancer-Associated Fibroblasts that Sustain Cancer Stem Properties in Head and Neck Squamous Carcinoma Cells as Potential Therapeutic Targets. Cancers. 2018; 10(9):334. https://doi.org/10.3390/cancers10090334
Chicago/Turabian StyleÁlvarez-Teijeiro, Saúl, Cristina García-Inclán, M. Ángeles Villaronga, Pedro Casado, Francisco Hermida-Prado, Rocío Granda-Díaz, Juan P. Rodrigo, Fernando Calvo, Nagore Del-Río-Ibisate, Alberto Gandarillas, and et al. 2018. "Factors Secreted by Cancer-Associated Fibroblasts that Sustain Cancer Stem Properties in Head and Neck Squamous Carcinoma Cells as Potential Therapeutic Targets" Cancers 10, no. 9: 334. https://doi.org/10.3390/cancers10090334
APA StyleÁlvarez-Teijeiro, S., García-Inclán, C., Villaronga, M. Á., Casado, P., Hermida-Prado, F., Granda-Díaz, R., Rodrigo, J. P., Calvo, F., Del-Río-Ibisate, N., Gandarillas, A., Morís, F., Hermsen, M., Cutillas, P., & García-Pedrero, J. M. (2018). Factors Secreted by Cancer-Associated Fibroblasts that Sustain Cancer Stem Properties in Head and Neck Squamous Carcinoma Cells as Potential Therapeutic Targets. Cancers, 10(9), 334. https://doi.org/10.3390/cancers10090334