G9a Correlates with VLA-4 Integrin and Influences the Migration of Childhood Acute Lymphoblastic Leukemia Cells

 , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Patient Characteristics
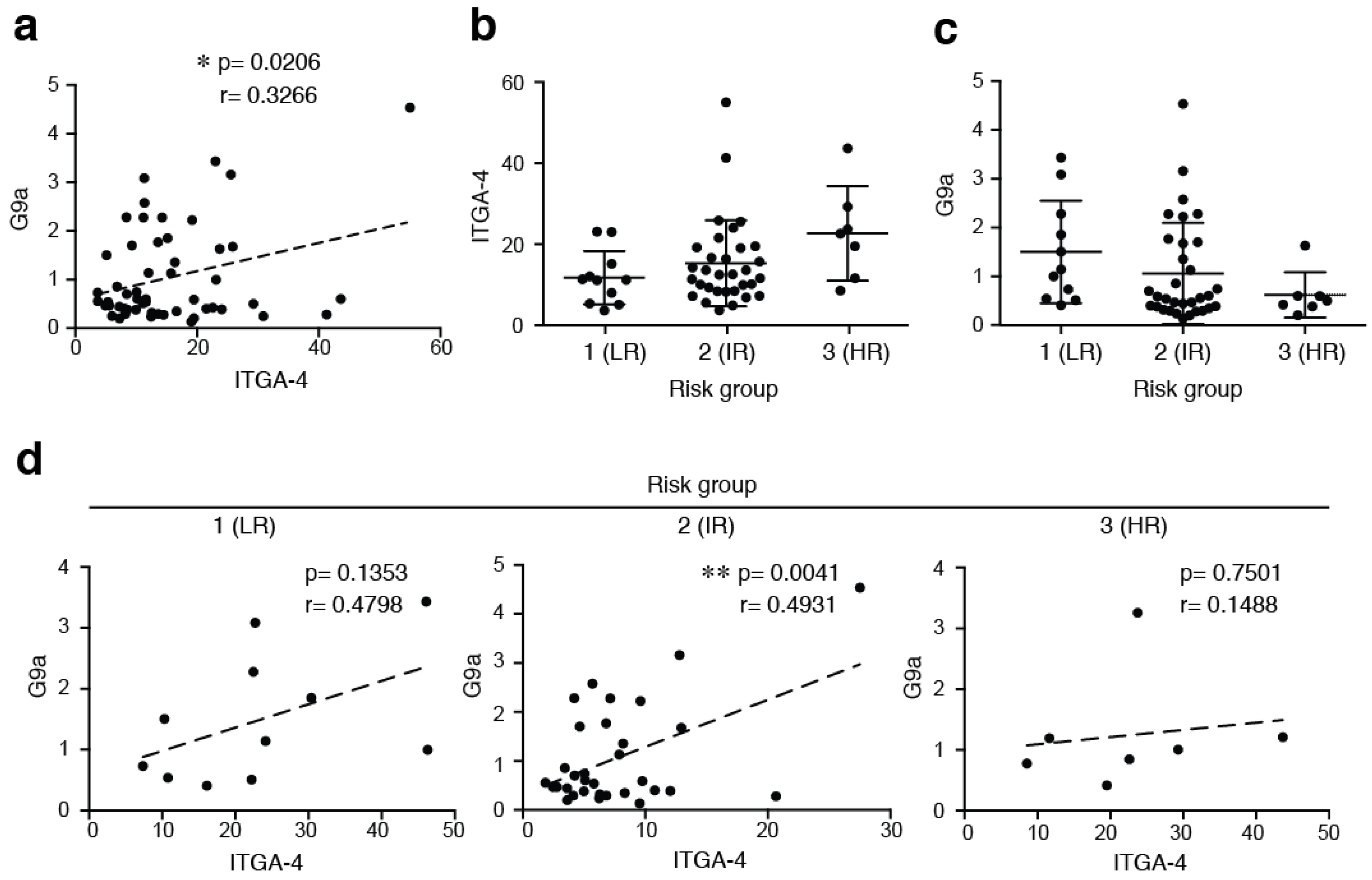
2.2. The Expression of G9a Correlates with α4 Integrin Expression in ALL Cells
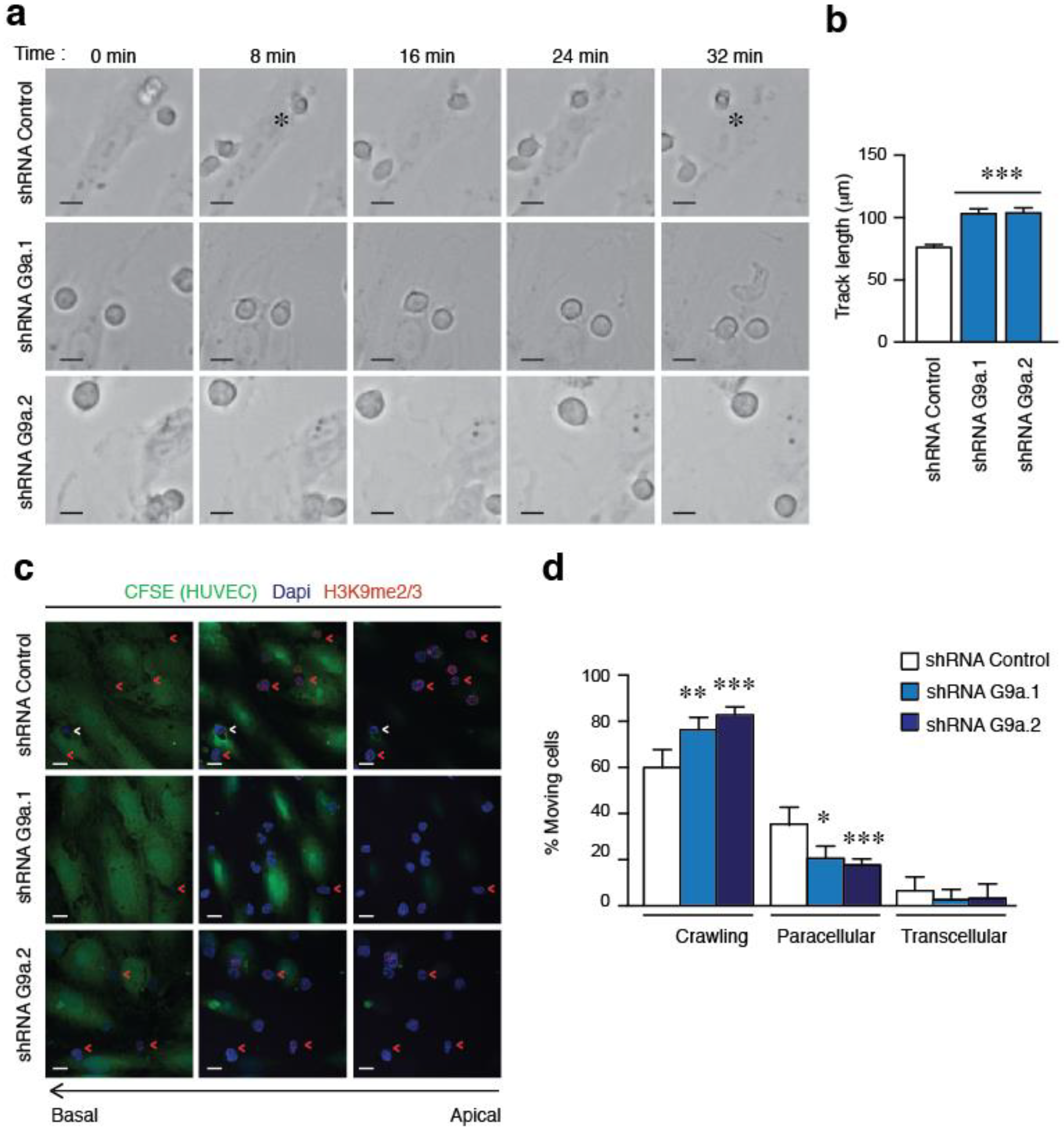
2.3. G9a Depletion Abrogates ALL Transendothelial Migration
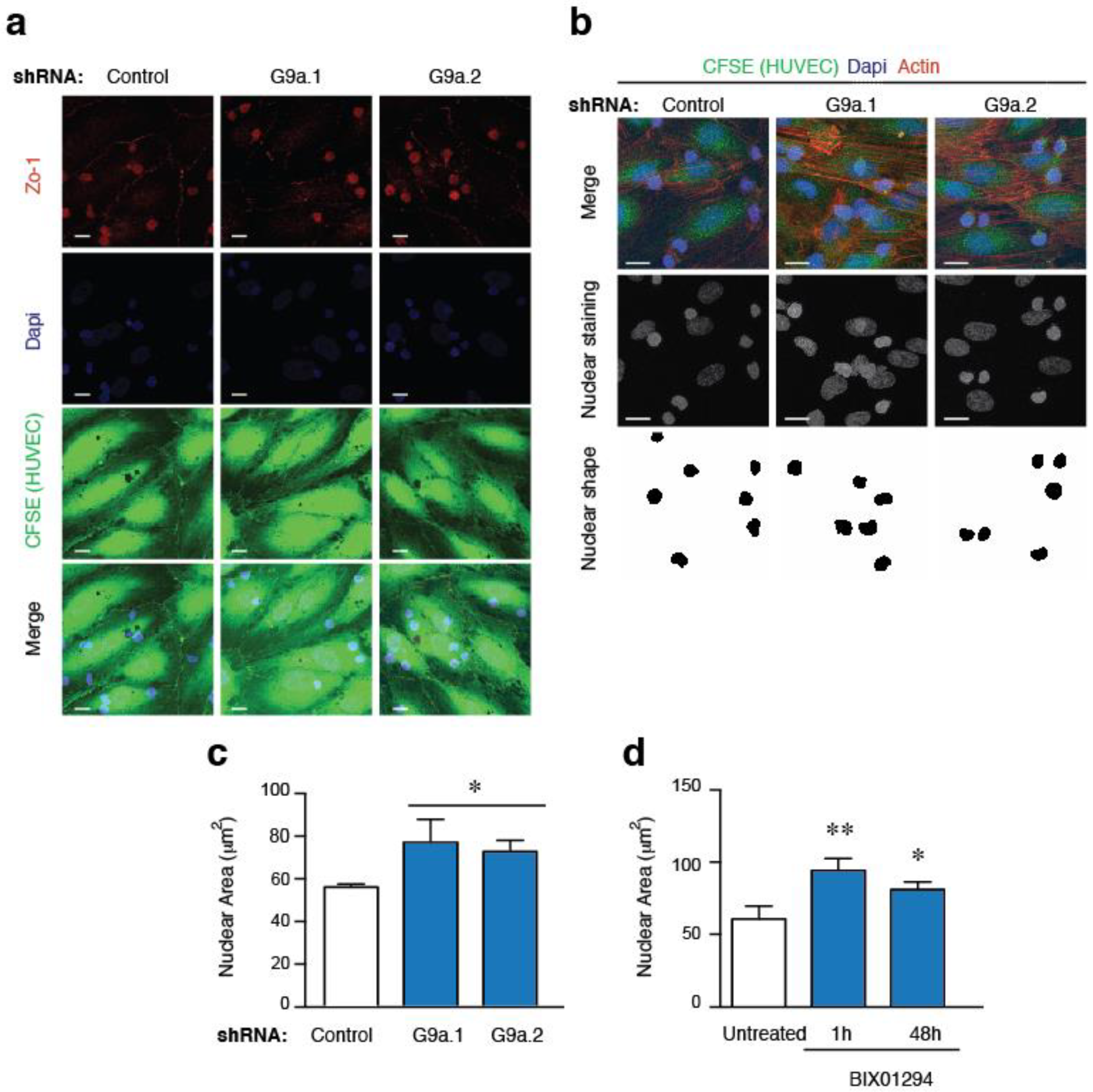
2.4. G9a Activity Does Not Affect Integrin Expression and Only Partially the Actin Polymerization in ALL Cells
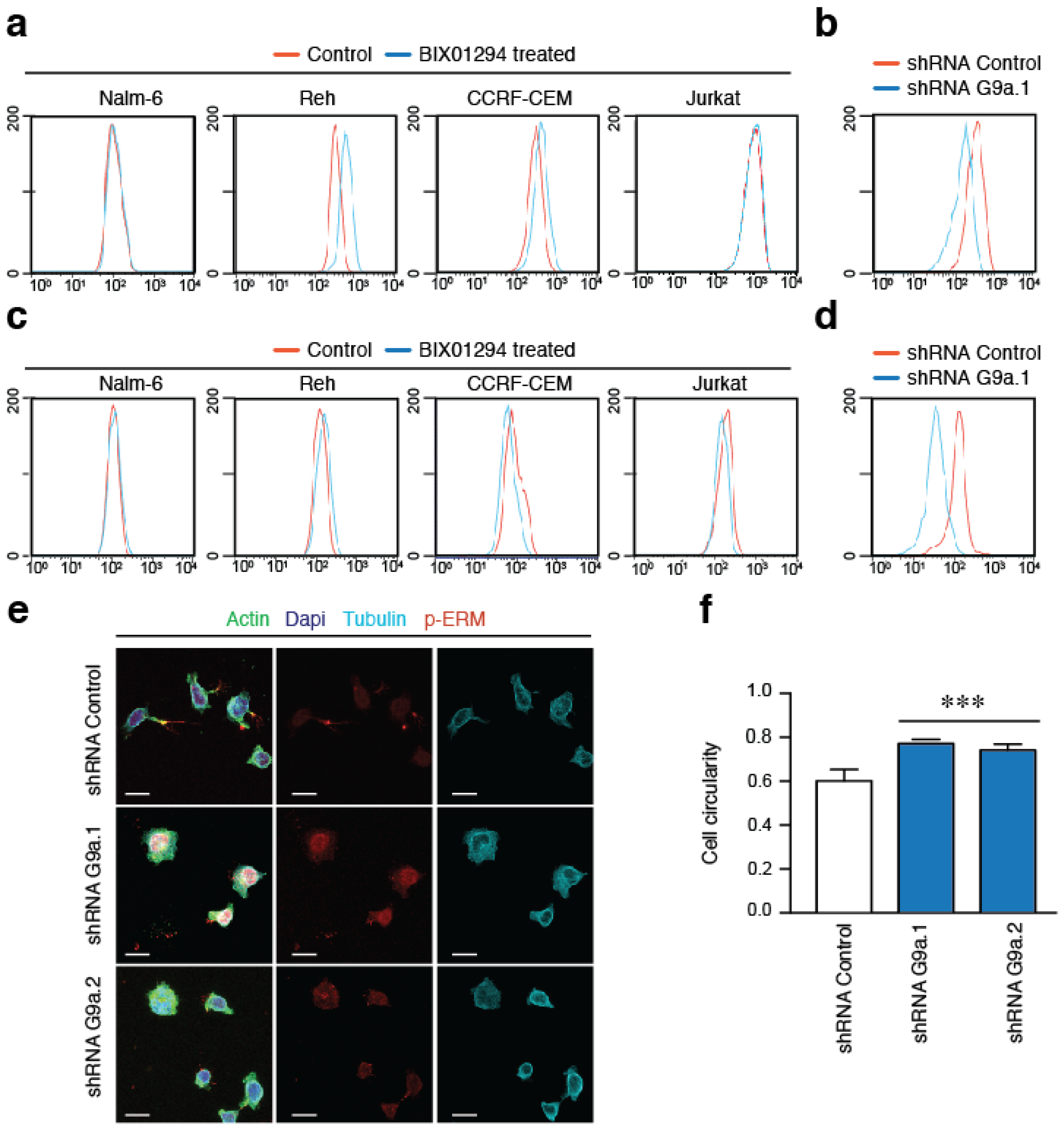
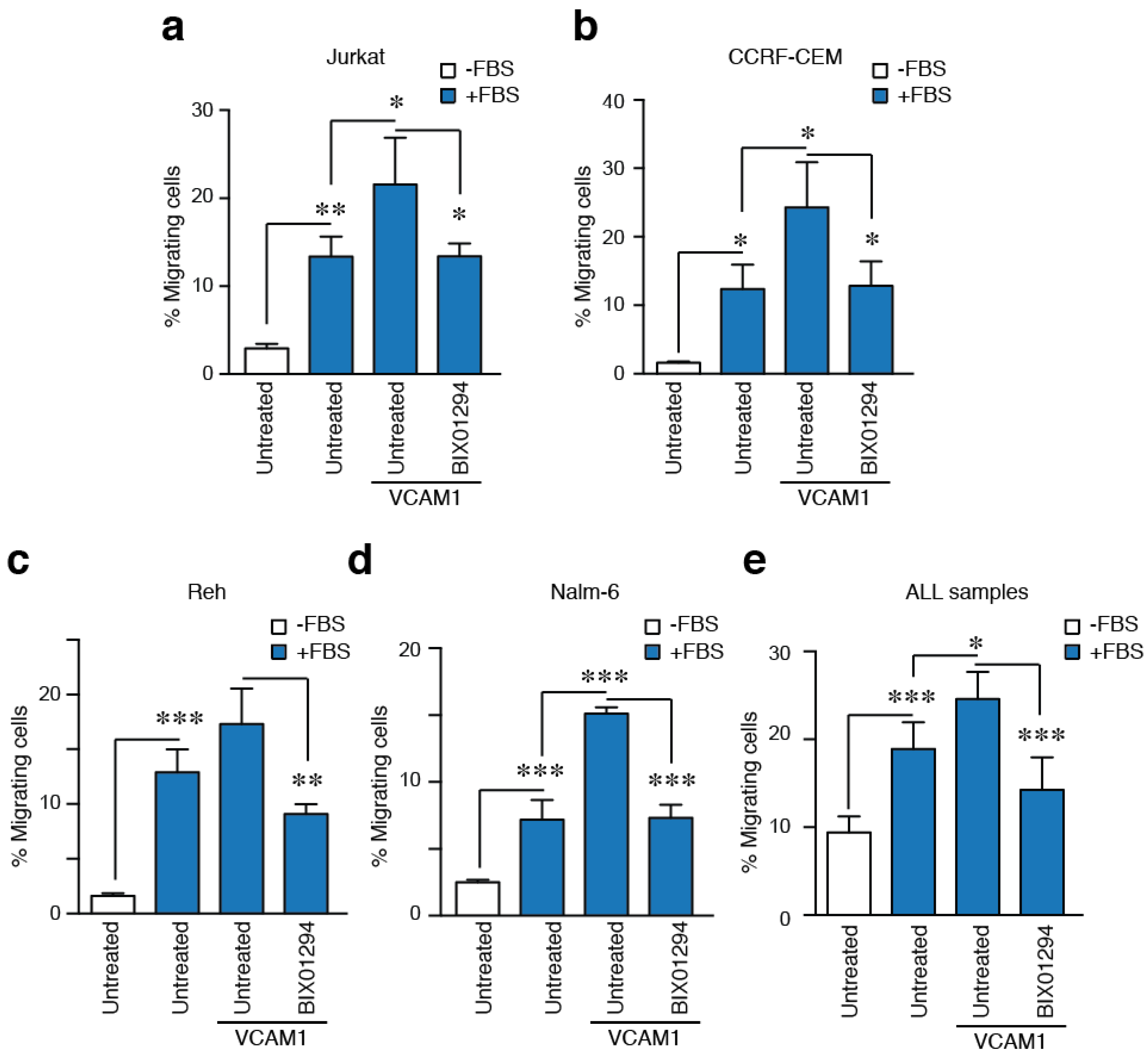
2.5. G9a Activity Modulates VLA-4-Mediated ALL Cell Migration
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Ethics Approval and Consent to Participate
4.3. Cell Lines
4.4. Reagents and Antibodies
4.5. RT- Real-Time PCR (qPCR)
4.6. Production of Lentivirus
4.7. Immunofluorescence and Transendothelial Cell Migration
4.8. Flow Cytometry Analysis
4.9. Transwell Invasion
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Inaba, H.; Greaves, M.; Mullighan, C.G. Acute lymphoblastic leukaemia. Lancet 2013, 381, 1943–1955. [Google Scholar] [CrossRef]
- Hunger, S.P.; Mullighan, C.G. Acute lymphoblastic leukemia in children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.V. New and emerging prognostic and predictive genetic biomarkers in B-cell precursor acute lymphoblastic leukemia. Haematologica 2016, 101, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Bhojwani, D.; Pui, C.H. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013, 14, e205–e217. [Google Scholar] [CrossRef]
- Conter, V.; Bartram, C.R.; Valsecchi, M.G.; Schrauder, A.; Panzer-Grümayer, R.; Möricke, A.; Aricò, M.; Zimmermann, M.; Mann, G.; De Rossi, G.; et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: Results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood 2010, 115, 3206–3214. [Google Scholar] [CrossRef] [PubMed]
- Almamun, M.; Levinson, B.T.; van Swaay, A.C.; Johnson, N.T.; McKay, S.D.; Arthur, G.L.; Davis, J.W.; Taylor, K.H. Integrated methylome and transcriptome analysis reveals novel regulatory elements in pediatric acute lymphoblastic leukemia. Epigenetics 2015, 10, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Van Vlierberghe, P.; Pieters, R.; Beverloo, H.B.; Meijerink, J.P. Molecular-genetic insights in paediatric T-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2008, 143, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Gerlitz, G.; Bustin, M. The role of chromatin structure in cell migration. Trends Cell Biol. 2011, 21, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Gerlitz, G.; Bustin, M. Efficient cell migration requires global chromatin condensation. J. Cell Sci. 2010, 123, 2207–2217. [Google Scholar] [CrossRef] [PubMed]
- Shinkai, Y.; Tachibana, M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011, 25, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Scheer, S.; Zaph, C. The Lysine Methyltransferase G9a in Immune Cell Differentiation and Function. Front. Immunol. 2017, 8, 429. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zou, Y.; Lin, L.; Ma, X.; Huang, X. Effect of BIX-01294 on proliferation, apoptosis and histone methylation of acute T lymphoblastic leukemia cells. Leuk. Res. 2017, 62, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Hemler, M.E.; Elices, M.J.; Parker, C.; Takada, Y. Structure of the integrin VLA-4 and its cell-cell and cell-matrix adhesion functions. Immunol. Rev. 1990, 114, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Redondo-Muñoz, J.; Ugarte-Berzal, E.; García-Marco, J.A.; del Cerro, M.H.; van den Steen, P.E.; Opdenakker, G.; Terol, M.J.; García-Pardo, A. α4β1 integrin and 190-kDa CD44v constitute a cell surface docking complex for gelatinase B/MMP-9 in chronic leukemic but not in normal B cells. Blood 2008, 112, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Bayless, K.J.; Davis, G.E. Identification of dual α4β1 integrin binding sites within a 38 amino acid domain in the N-terminal thrombin fragment of human osteopontin. J. Biol. Chem. 2001, 276, 13483–13489. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Hof, J.; Kirschner-Schwabe, R.; Bastian, L.; Eckert, C.; Prada, J.; Henze, G.; von Stackelberg, A.; Seeger, K. High VLA-4 expression is associated with adverse outcome and distinct gene expression changes in childhood B-cell precursor acute lymphoblastic leukemia at first relapse. Haematologica 2011, 96, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cook, P.C.; Zindy, E.; Williams, C.J.; Jowitt, T.A.; Streuli, C.H.; MacDonald, A.S.; Redondo-Muñoz, J. Integrin α4β1 controls G9a activity that regulates epigenetic changes and nuclear properties required for lymphocyte migration. Nucleic Acids Res. 2016, 44, 3031–3044. [Google Scholar] [CrossRef] [PubMed]
- Mozzetta, C.; Boyarchuk, E.; Pontis, J.; Ait-Si-Ali, S. Sound of silence: The properties and functions of repressive Lys methyltransferases. Nat. Rev. Mol. Cell Biol. 2015, 16, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Nourshargh, S.; Hordijk, P.L.; Sixt, M. Breaching multiple barriers: Leukocyte motility through venular walls and the interstitium. Nat. Rev. Mol. Cell Biol. 2010, 11, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Katakai, T.; Hara, T.; Gonda, H.; Sugai, M.; Shimizy, A. Roles of p-ERM and Rho-ROCK signaling in lymphocyte polarity and uropod formation. J. Cell Biol. 2004, 167, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Gaynes, J.S.; Jonart, L.M.; Zamora, E.A.; Naumann, J.A.; Gossai, N.P.; Gordon, P.M. The central nervous system microenvironment influences the leukemia transcriptome and enhances leukemia chemo-resistance. Haematologica 2017, 102, e136–e139. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, F.; Lonetti, A.; Evangelisti, C.; Buontempo, F.; Orsini, E.; Evangelisti, C.; Cappellini, A.; Neri, L.M.; McCubrey, J.A.; Martelli, A.M. Advances in understanding the acute lymphoblastic leukemia bone marrow microenvironment: From biology to therapeutic targeting. Biochim. Biophys. Acta 2016, 1863, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Benyoucef, A.; Palii, C.G.; Wang, C.; Porter, C.J.; Chu, A.; Dai, F.; Tremblay, V.; Rakopoulos, P.; Singh, K.; Huang, S.; et al. UTX inhibition as selective epigenetic therapy against TAL1-driven T-cell acute lymphoblastic leukemia. Genes Dev. 2016, 30, 508–521. [Google Scholar] [CrossRef] [PubMed]
- Montaño, A.; Forero-Castro, M.; Marchena-Mendoza, D.; Benito, R.; Hernández-Rivas, J.M. New challenges in targeting signaling pathways in acute lymphoblastic leukemia by NGS approaches: An update. Cancers 2018, 10, 110. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Chagraoui, J.; Krosl, J.; Gendron, P.; Wilhelm, B.; Lemieux, S.; Boucher, G.; Chagnon, P.; Drouin, S.; Lambert, R.; et al. A key role for EZH2 and associated genes in mouse and human adult T-cell acute leukemia. Genes Dev. 2012, 26, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Hieda, M.; Nishioka, Y.; Matsumoto, A.; Higashi, S.; Kimura, H.; Yamamoto, H.; Mori, M.; Matsuura, S.; Matsuura, N. Cancer-associated upregulation of histone H3 lysine 9 trimethylation promotes cell motility in vitro and drives tumor formation in vivo. Cancer Sci. 2013, 104, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Mutonga, M.; Tamura, K.; Malnassy, G.; Fulton, N.; de Albuquerque, A.; Hamamoto, R.; Stock, W.; Nakamura, Y.; Alachkar, H. Targeting Suppressor of Variegation 3-9 Homologue 2 (SUV39H2) in Acute Lymphoblastic Leukemia (ALL). Transl. Oncol. 2015, 8, 368–375. [Google Scholar] [CrossRef] [PubMed]
- San José-Enériz, E.; Agirre, X.; Rabal, O.; Vilas-Zornoza, A.; Sanchez-Arias, J.A.; Miranda, E.; Ugarte, A.; Roa, S.; Paiva, B.; Estella-Hermoso de Mendoza, A.; et al. Discovery of first-in-class reversible dual small molecule inhibitors against G9a and DNMTs in hematological malignancies. Nat. Commun. 2017, 8, 15424. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.H.; Oh, S.H.; Park, C.J.; Lee, B.R.; Kim, Y.J.; Cho, Y.U.; Jang, S.; Lee, J.H.; Kim, N.; Park, S.H.; et al. VLA-4 and CXCR4 expression levels show contrasting prognostic impact (favorable and unfavorable; respectively) in acute myeloid leukemia. Ann. Hematol. 2015, 94, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.Y.; Park, C.J.; Park, S.H.; Cho, Y.U.; Jang, S.; Seo, E.J.; Kim, N.; Kim, D.Y.; Koh, K.N.; Im, H.J.; et al. High CXCR4 and low VLA-4 expression predicts poor survival in adults with acute lymphoblastic leukemia. Leuk. Res. 2014, 38, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Su, G.; Ji, J.; Yang, X.; Miao, M.; Mo, P.; Li, M.; Xu, J.; Li, W.; Yu, C. Histone demethylase JMJD1A promotes colorectal cancer growth and metastasis by enhancing Wnt/β-catenin signaling. J. Biol. Chem. 2018, 293, 10606–10619. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.K.; Huang, Y.Q.; Zou, Y.; Zheng, X.K.; Ma, X.D. Silencing of LSD1 gene modulates histone methylation and acetylation and induces the apoptosis of JeKo-1 and MOLT-4 cells. Int. J. Mol. Med. 2017, 40, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.T.; Gang, E.J.; Geng, H.; Park, E.; Huantes, S.; Chudziak, D.; Dauber, K.; Schaefer, P.; Scharman, C.; Shimada, H.; et al. Integrin alpha4 blockade sensitizes drug resistant pre-B acute lymphoblastic leukemia to chemotherapy. Blood 2013, 121, 1814–1818. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.T.; Gang, E.J.; Shishido, S.N.; Kim, H.N.; Pham, J.; Khazal, S.; Osborne, A.; Esguerra, Z.A.; Kwok, E.; Jang, J.; et al. Effects of the small-molecule inhibitor of integrin α4, TBC3486, on pre-B-ALL cells. Leukemia 2014, 28, 2101–2104. [Google Scholar] [CrossRef] [PubMed]
- Duchartre, Y.; Bachl, S.; Kim, H.N.; Gang, E.J.; Lee, S.; Liu, H.C.; Shung, K.; Xu, R.; Kruse, A.; Tachas, G.; et al. Effects of CD49d-targeted antisense-oligonucleotide on α4 integrin expression and function of acute lymphoblastic leukemia cells: Results of in vitro and in vivo studies. PLoS ONE 2017, 12, e0187684. [Google Scholar] [CrossRef] [PubMed]
- Wigton, E.J.; Thompson, S.B.; Long, R.A.; Jacobelli, J. Myosin-IIA regulates leukemia engraftment and brain infiltration in a mouse model of acute lymphoblastic leukemia. J. Leukoc. Biol. 2016, 100, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Infante, E.; Heasman, S.J.; Ridley, A.J. Statins inhibit T-acute lymphoblastic leukemia cell adhesion and migration through Rap1b. J. Leukoc. Biol. 2011, 89, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.R.; Ma, Y.H. MIP-1α enhances Jurkat cell transendothelial migration by up-regulating endothelial adhesion molecules VCAM-1 and ICAM-1. Leuk. Res. 2014, 38, 1327–1331. [Google Scholar] [CrossRef] [PubMed]
- McGregor, A.L.; Hsia, C.R.; Lammerding, J. Squish and squeeze—The nucleus as a physical barrier during migration in confined environments. Curr. Opin. Cell Biol. 2016, 40, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Luo, Q.; Sun, J.; Song, G. Nucleus and nucleus-cytoskeleton connections in 3D cell migration. Exp. Cell Res. 2016, 348, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Wolf, K.; Te Lindert, M.; Krause, M.; Alexander, S.; Te Riet, J.; Willis, A.L.; Hoffman, R.M.; Figdor, C.G.; Weiss, S.J.; Friedl, P. Physical limits of cell migration: Control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J. Cell Biol. 2013, 201, 1069–1084. [Google Scholar] [CrossRef] [PubMed]
- Madrazo, E.; Conde, A.C.; Redondo-Muñoz, J. Inside the cell: Integrins as new governors of nuclear alterations? Cancers 2017, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Tajik, A.; Chen, J.; Jia, Q.; Chowdhury, F.; Wang, L.; Chen, J.; Zhang, S.; Hong, Y.; Yi, H.; et al. Matrix softness regulates plasticity of tumour-repopulating cells via H3K9 demethylation and Sox2 expression. Nat. Commun. 2014, 5, 4619. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Zang, M.D.; Wang, H.X.; Zhang, B.G.; Wang, Z.Q.; Fan, Z.Y.; Wu, H.; Li, J.F.; Su, L.P.; Yan, M.; et al. G9A promotes gastric cancer metastasis by upregulating ITGB3 in a SET domain-independent manner. Cell Death Dis. 2018, 9, 278. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Condeelis, J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim. Biophys. Acta 2007, 1773, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Moeendarbary, E.; Isermann, P.; Davidson, P.M.; Wang, X.; Chen, M.B.; Burkart, A.K.; Lammerding, J.; Kamm, R.D.; Shenoy, V.B. A chemomechanical model for nuclear morphology and stresses during cell transendothelial migration. Biophys. J. 2016, 111, 1541–1552. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, S.; Yadav, S.K.; Morrell, S.; Roncato, F.; Klein, E.; Stoler-Barak, L.; Golani, O.; Feigelson, S.W.; Zemel, A.; Nourshargh, S.; et al. Leukocytes breach endothelial barriers by insertion of nuclear lobes and disassembly of endothelial actin filaments. Cell Rep. 2017, 18, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Alon, R.; van Buul, J.D. Leukocyte breaching of endothelial barriers: The actin link. Trends Immunol. 2017, 38, 606–615. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Characteristics | Number of Cases | Percentage (%) |
|---|---|---|---|
| Gender | Male | 27 | 54 |
| Female | 23 | 46 | |
| Risk group | 1 (LR 1) | 11 | 22 |
| 2 (IR 1) | 32 | 64 | |
| 3 (HR 1) | 7 | 14 | |
| WBC 1 (>50,000 cells/mm3) | Positive | 6 | 12 |
| Negative | 44 | 88 | |
| MRD 1 after induction | Positive | 22 | 44 |
| Negative | 28 | 56 | |
| Extramedullary disease | Positive | 22 | 44 |
| Negative | 28 | 56 | |
| Blast in BM 1 | Positive | 25 | 50 |
| Negative | 25 | 50 | |
| ETV6/RUNX1 | Positive | 15 | 30 |
| Negative | 32 | 64 | |
| ND 1 | 3 | 6 | |
| BCR/ABL | Positive | 2 | 4 |
| Negative | 47 | 94 | |
| ND 1 | 1 | 2 | |
| MLL rearrangements | Positive | 2 | 4 |
| Negative | 47 | 94 | |
| ND 1 | 1 | 2 | |
| Hyperdiploidy | Positive | 13 | 26 |
| Negative | 36 | 72 | |
| ND 1 | 1 | 2 | |
| Relapse | Positive | 7 | 14 |
| Negative | 41 | 82 | |
| ND 1 | 2 | 4 | |
| Death | Positive | 6 | 12 |
| Negative | 43 | 86 | |
| ND 1 | 1 | 2 |
| Risk Group | G9a LE 1 | G9a HE 1 | Percentage (%) |
|---|---|---|---|
| 1 | 3 | 8 | 27.3/72.7 |
| 2 | 17 | 15 | 53.1/46.9 |
| 3 | 5 | 2 | 71.4/28.6 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madrazo, E.; Ruano, D.; Abad, L.; Alonso-Gómez, E.; Sánchez-Valdepeñas, C.; González-Murillo, Á.; Ramírez, M.; Redondo-Muñoz, J. G9a Correlates with VLA-4 Integrin and Influences the Migration of Childhood Acute Lymphoblastic Leukemia Cells. Cancers 2018, 10, 325. https://doi.org/10.3390/cancers10090325
Madrazo E, Ruano D, Abad L, Alonso-Gómez E, Sánchez-Valdepeñas C, González-Murillo Á, Ramírez M, Redondo-Muñoz J. G9a Correlates with VLA-4 Integrin and Influences the Migration of Childhood Acute Lymphoblastic Leukemia Cells. Cancers. 2018; 10(9):325. https://doi.org/10.3390/cancers10090325
Chicago/Turabian StyleMadrazo, Elena, David Ruano, Lorea Abad, Estefanía Alonso-Gómez, Carmen Sánchez-Valdepeñas, África González-Murillo, Manuel Ramírez, and Javier Redondo-Muñoz. 2018. "G9a Correlates with VLA-4 Integrin and Influences the Migration of Childhood Acute Lymphoblastic Leukemia Cells" Cancers 10, no. 9: 325. https://doi.org/10.3390/cancers10090325
APA StyleMadrazo, E., Ruano, D., Abad, L., Alonso-Gómez, E., Sánchez-Valdepeñas, C., González-Murillo, Á., Ramírez, M., & Redondo-Muñoz, J. (2018). G9a Correlates with VLA-4 Integrin and Influences the Migration of Childhood Acute Lymphoblastic Leukemia Cells. Cancers, 10(9), 325. https://doi.org/10.3390/cancers10090325