CX-4945 Induces Methuosis in Cholangiocarcinoma Cell Lines by a CK2-Independent Mechanism

 , ,
, ,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
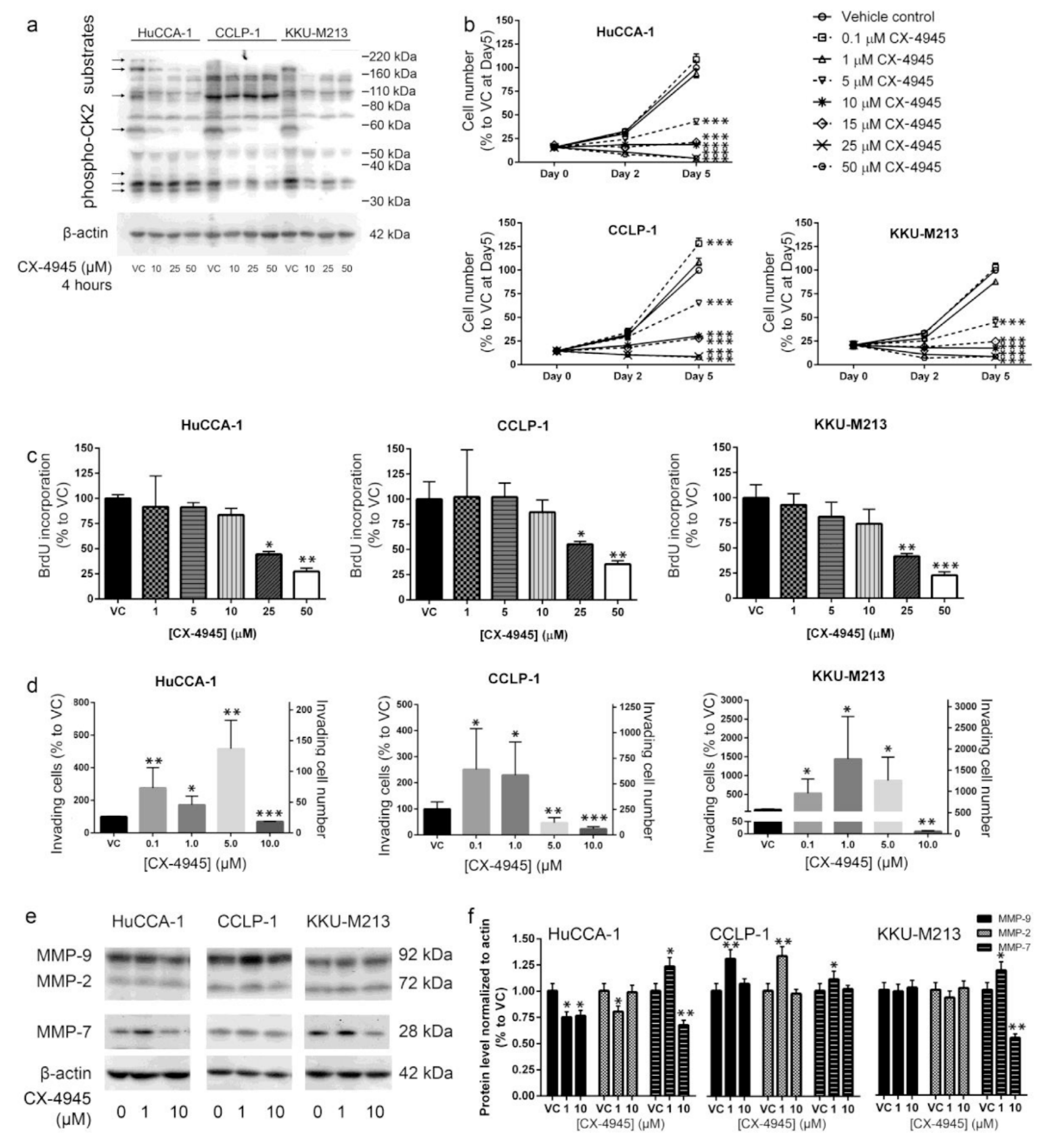
2.1. CX-4945 Inhibits CK2 Activity at High Dose
2.2. CX-4945 Treatment Inhibits CCA Cell Proliferation
2.3. CX-4945 Treatment Alters Cell Invasion
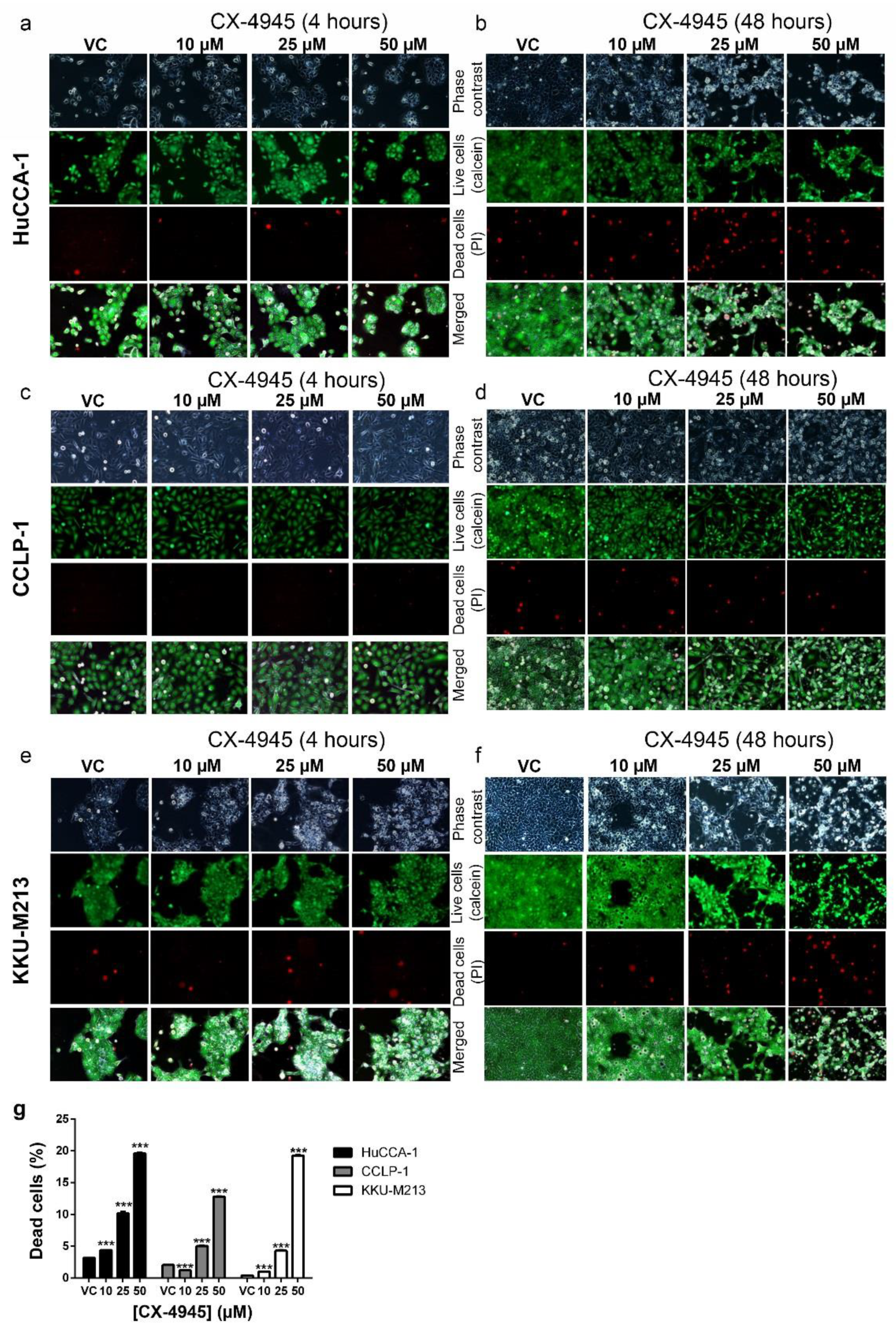
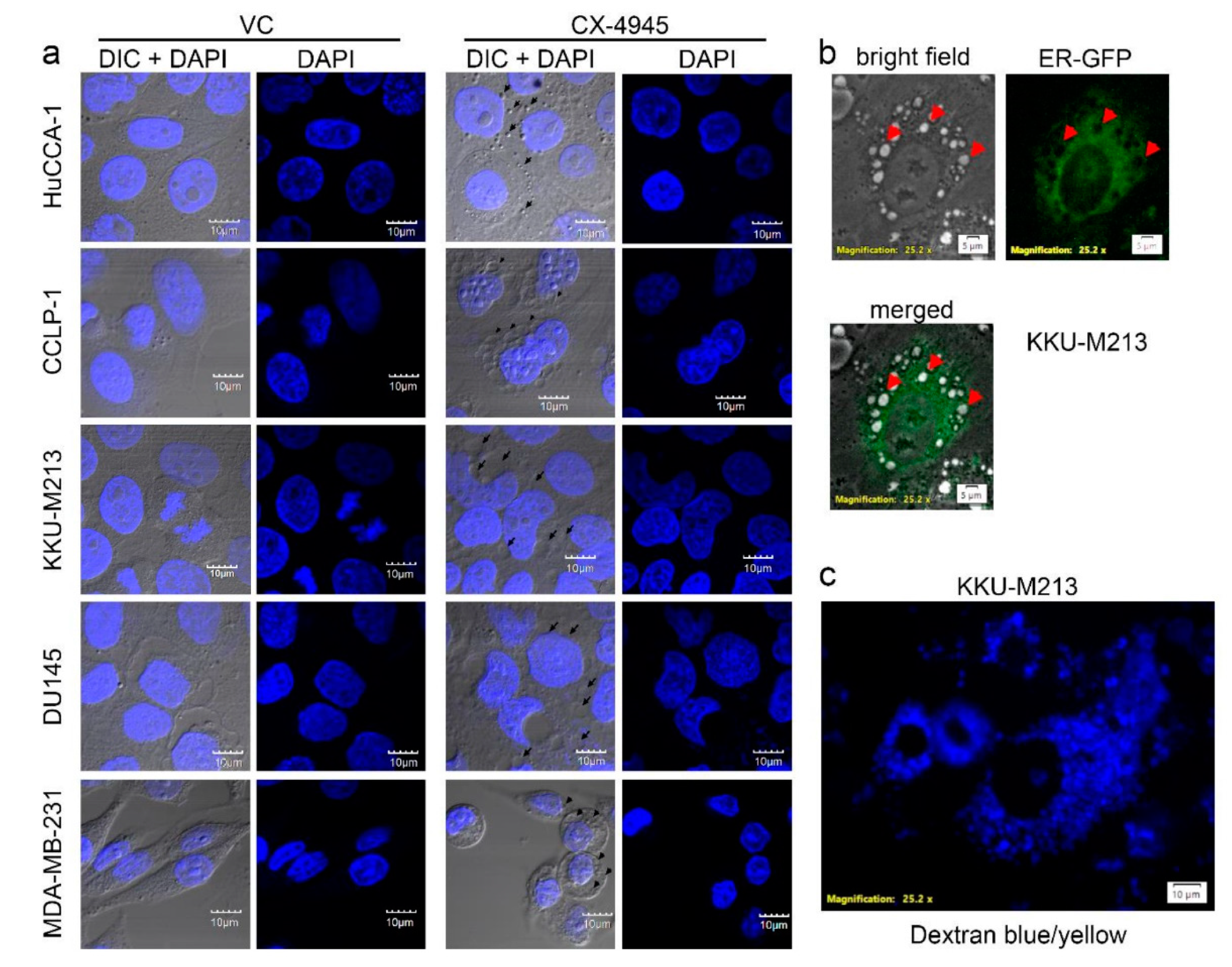
2.4. CX-4945 Treatment Induces Intensive Vacuolization
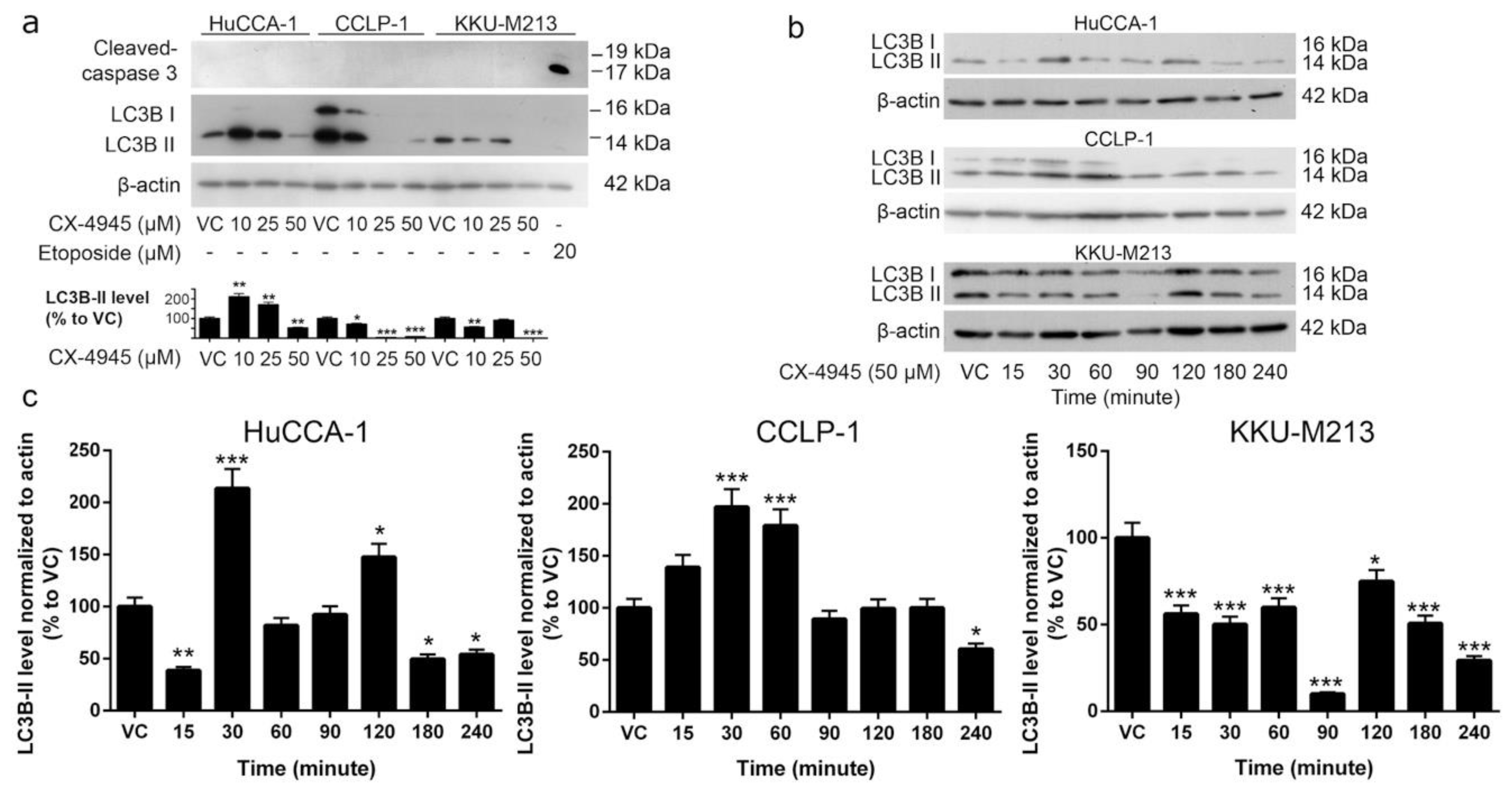
2.5. CX-4945 Induces Caspase-3 Independent Non-Autophagic Cell Death in CCA Cells
2.6. CX-4945 Induced Methuosis in KKU-M213
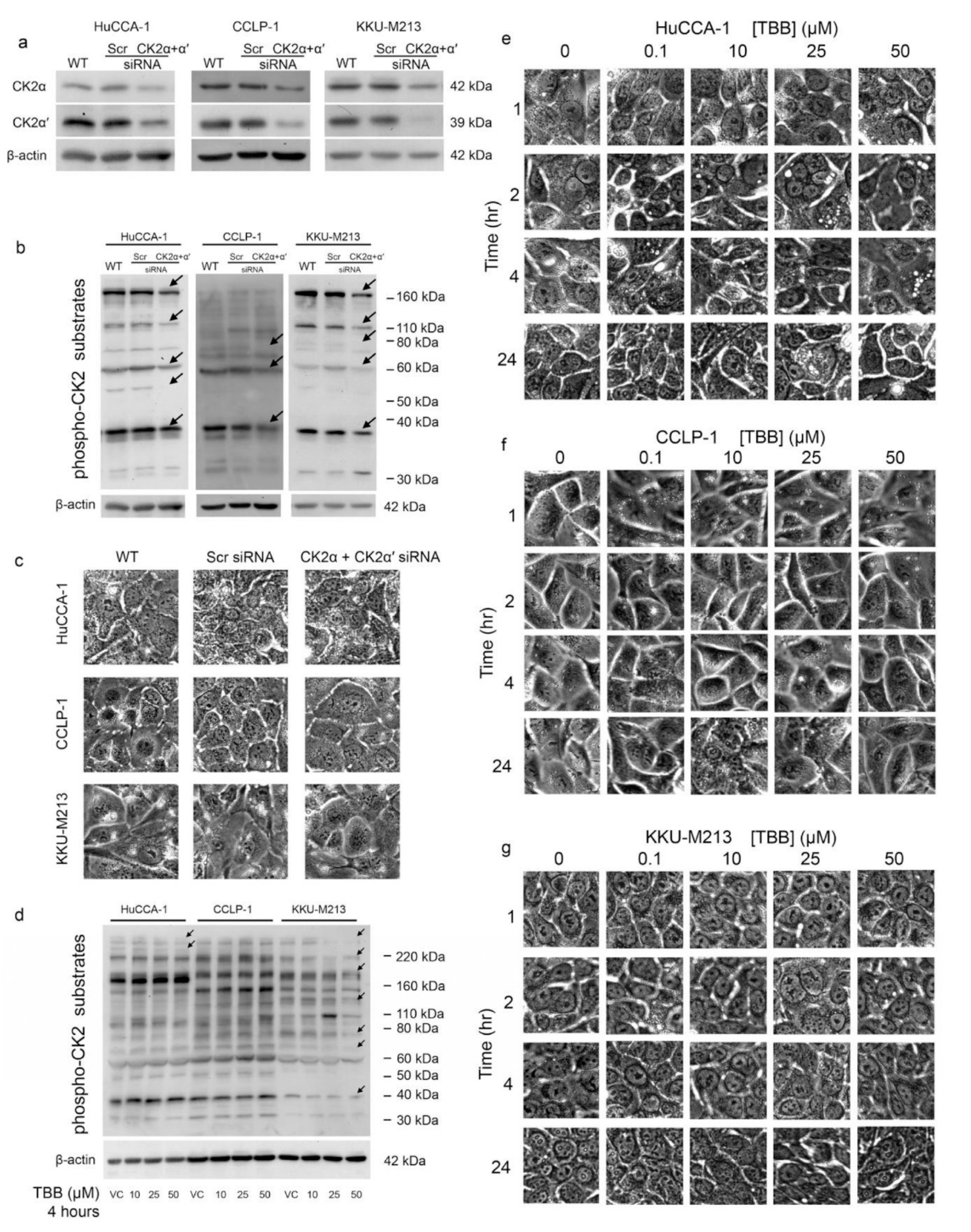
2.7. CX-4945 Induced Vacuolization Is CK2-Independent
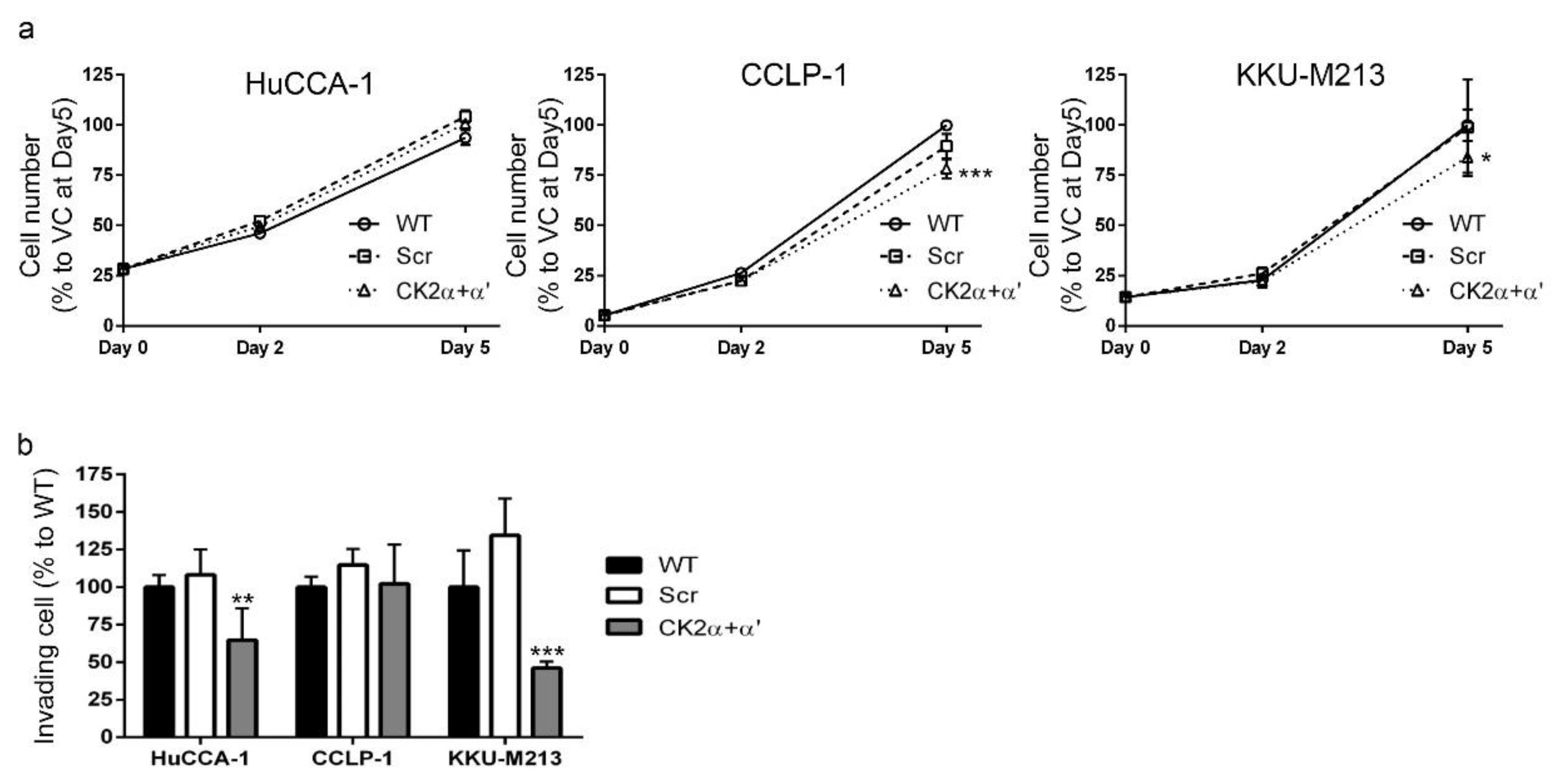
2.8. CK2 Knockdown Inhibited CCA Cell Proliferation and Cell Invasion
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. MTT Assay
4.3. BrdU Incorporation Assay
4.4. Cell Invasion Assay
4.5. Cell Viability Assay
4.6. Western Blot Analysis
4.7. siRNA Transfection
4.8. Organelle Labeling and Imaging
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Njei, B. Changing pattern of epidemiology in intrahepatic cholangiocarcinoma. Hepatology 2014, 60, 1107–1108. [Google Scholar] [CrossRef] [PubMed]
- Pinter, M.; Hucke, F.; Zielonke, N.; Waldhor, T.; Trauner, M.; Peck-Radosavljevic, M.; Sieghart, W. Incidence and mortality trends for biliary tract cancers in austria. Liver Int. 2014, 34, 1102–1108. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Takikawa, H. Geoepidemiology of primary sclerosing cholangitis: A critical review. J. Autoimmun. 2013, 46, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Dixon, E. Epidemiology and risk factors: Intrahepatic cholangiocarcinoma. Hepatobiliary Surg. Nutr. 2017, 6, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Furst, T.; Keiser, J.; Utzinger, J. Global burden of human food-borne trematodiasis: A systematic review and meta-analysis. Lancet Infect. Dis. 2012, 12, 210–221. [Google Scholar] [CrossRef]
- Kaewpitoon, S.J.; Rujirakul, R.; Ueng-Arporn, N.; Matrakool, L.; Namwichaisiriku, N.; Churproong, S.; Wongkaewpothong, P.; Nimkuntod, P.; Sripa, B.; Kaewpitoon, N. Community-based cross-sectional study of carcinogenic human liver fluke in elderly from Surin province, Thailand. Asian Pac. J. Cancer Prev. 2012, 13, 4285–4288. [Google Scholar] [CrossRef] [PubMed]
- Sithithaworn, P.; Yongvanit, P.; Duenngai, K.; Kiatsopit, N.; Pairojkul, C. Roles of liver fluke infection as risk factor for cholangiocarcinoma. J. Hepatobiliary Pancreat. Sci. 2014, 21, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Bridgewater, J.; Galle, P.R.; Khan, S.A.; Llovet, J.M.; Park, J.W.; Patel, T.; Pawlik, T.M.; Gores, G.J. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J. Hepatol. 2014, 60, 1268–1289. [Google Scholar] [CrossRef] [PubMed]
- Yoh, T.; Hatano, E.; Yamanaka, K.; Nishio, T.; Seo, S.; Taura, K.; Yasuchika, K.; Okajima, H.; Kaido, T.; Uemoto, S. Is surgical resection justified for advanced intrahepatic cholangiocarcinoma? Liver Cancer 2016, 5, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Beal, E.W.; Bagante, F.; Chakedis, J.; Weiss, M.; Popescu, I.; Marques, H.P.; Aldrighetti, L.; Maithel, S.K.; Pulitano, C.; et al. Early versus late recurrence of intrahepatic cholangiocarcinoma after resection with curative intent. Br. J. Surg. 2017. [Google Scholar] [CrossRef]
- Primrose, J.N.; Fox, R.; Palmer, D.H.; Prasad, R.; Mirza, D.; Anthoney, D.A.; Corrie, P.; Falk, S.; Wasan, H.S.; Ross, P.J.; et al. Adjuvant capecitabine for biliary tract cancer: The bilcap randomized study. J. Clin. Oncol. 2017, 35, 4006. [Google Scholar] [CrossRef]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, D.W. Protein kinase ck2: Structure, regulation and role in cellular decisions of life and death. Biochem. J 2003, 369, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ortega, C.E.; Seidner, Y.; Dominguez, I. Mining CK2 in cancer. PLoS ONE 2014, 9, e115609. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Feng, C.; He, Y.; Ding, W.; Sheng, J.; Arshad, M.; Zhang, X.; Li, P. Phosphorylation of apoptosis repressor with caspase recruitment domain by protein kinase CK2 contributes to chemotherapy resistance by inhibiting doxorubicin induced apoptosis. Oncotarget 2015, 6, 27700–27713. [Google Scholar] [CrossRef] [PubMed]
- Kendall, J.J.; Chaney, K.E.; Patel, A.V.; Rizvi, T.A.; Largaespada, D.A.; Ratner, N. CK2 blockade causes mpnst cell apoptosis and promotes degradation of beta-catenin. Oncotarget 2016, 7, 53191–53203. [Google Scholar] [CrossRef] [PubMed]
- So, K.S.; Rho, J.K.; Choi, Y.J.; Kim, S.Y.; Choi, C.M.; Chun, Y.J.; Lee, J.C. AKT/mTOR down-regulation by CX-4945, a CK2 inhibitor, promotes apoptosis in chemorefractory non-small cell lung cancer cells. Anticancer Res. 2015, 35, 1537–1542. [Google Scholar] [PubMed]
- Quotti Tubi, L.; Gurrieri, C.; Brancalion, A.; Bonaldi, L.; Bertorelle, R.; Manni, S.; Pavan, L.; Lessi, F.; Zambello, R.; Trentin, L.; et al. Inhibition of protein kinase CK2 with the clinical-grade small ATP-competitive compound CX-4945 or by rna interference unveils its role in acute myeloid leukemia cell survival, p53-dependent apoptosis and daunorubicin-induced cytotoxicity. J. Hematol. Oncol. 2013, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Yang, B.; Shi, S.; Jiang, X. RNA interference (RNAi) mediated stable knockdown of protein casein kinase 2-alpha (CK2alpha) inhibits migration and invasion and enhances cisplatin-induced apoptosis in HEp-2 laryngeal carcinoma cells. Acta Histochem. 2014, 116, 1000–1006. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.-W.; Woo, J.H.; Kim, Y.-H.; Lee, Y.-S.; Park, J.W.; Bae, Y.-S. Downregulation of protein kinase CKII is associated with cellular senescence. FEBS Lett. 2006, 580, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Youn, H.; Gao, X.; Huang, B.; Zhou, F.; Li, B.; Han, H. Casein kinase 2 inhibition attenuates androgen receptor function and cell proliferation in prostate cancer cells. Prostate 2012, 72, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, X.; Yang, C.; Li, P.; Yuan, W.; Deng, X.; Cheng, Y.; Li, P.; Yang, H.; Tao, J.; et al. Targeting protein kinase CK2 suppresses bladder cancer cell survival via the glucose metabolic pathway. Oncotarget 2016, 7, 87361–87372. [Google Scholar] [CrossRef] [PubMed]
- Gowda, C.; Sachdev, M.; Muthusami, S.; Kapadia, M.; Petrovic-Dovat, L.; Hartman, M.; Ding, Y.; Song, C.; Payne, J.L.; Tan, B.H.; et al. Casein kinase II (CK2) as a therapeutic target for hematological malignancies. Curr. Pharm. Des. 2017, 23, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Li, D.; Zhou, Y.; Landesman-Bollag, E.; Zhang, G.; Anderson, N.M.; Tang, K.C.; Roderick, J.E.; Kelliher, M.A.; Seldin, D.C.; et al. CK2 inhibitor CX-4945 destabilizes NOTCH1 and synergizes with JQ1 against human T-acute lymphoblastic leukemic cells. Haematologica 2017, 102, e17–e21. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Chen, J.; Guo, S.; Wang, Y.; Zhou, Q.; Li, Z.; Yang, X.; Yu, X.; Zhang, Z.; Zhou, F.; et al. CX4945 suppresses the growth of castration-resistant prostate cancer cells by reducing AR-V7 expression. World J. Urol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Gray, G.K.; McFarland, B.C.; Rowse, A.L.; Gibson, S.A.; Benveniste, E.N. Therapeutic CK2 inhibition attenuates diverse prosurvival signaling cascades and decreases cell viability in human breast cancer cells. Oncotarget 2014, 5, 6484–6496. [Google Scholar] [CrossRef] [PubMed]
- Pierre, F.; Chua, P.C.; O’Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K.; Stefan, E.; et al. Pre-clinical characterization of CX-4945, a potent and selective small molecule inhibitor of CK2 for the treatment of cancer. Mol. Cell. Biochem. 2011, 356, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Lustri, A.M.; Di Matteo, S.; Fraveto, A.; Costantini, D.; Cantafora, A.; Napoletano, C.; Bragazzi, M.C.; Giuliante, F.; De Rose, A.M.; Berloco, P.B.; et al. TGF-beta signaling is an effective target to impair survival and induce apoptosis of human cholangiocarcinoma cells: A study on human primary cell cultures. PLoS ONE 2017, 12, e0183932. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B.B.; Issinger, O.G.; Guerra, B. Regulation of DNA-dependent protein kinase by protein kinase CK2 in human glioblastoma cells. Oncogene 2010, 29, 6016–6026. [Google Scholar] [CrossRef] [PubMed]
- Willmore, E.; de Caux, S.; Sunter, N.J.; Tilby, M.J.; Jackson, G.H.; Austin, C.A.; Durkacz, B.W. A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood 2004, 103, 4659–4665. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Choi, K.; Kang, H.; Lee, S.Y.; Chi, S.W.; Lee, M.S.; Song, J.; Im, D.; Choi, Y.; Cho, S. Identification of a novel function of CX-4945 as a splicing regulator. PLoS ONE 2014, 9, e94978. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the nomenclature committee on cell death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Lockshin, R.A.; Zakeri, Z. Apoptosis, autophagy, and more. Int. J. Biochem. Cell Biol. 2004, 36, 2405–2419. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Maltese, W.A.; Overmeyer, J.H. Methuosis: Nonapoptotic cell death associated with vacuolization of macropinosome and endosome compartments. Am. J. Pathol. 2014, 184, 1630–1642. [Google Scholar] [CrossRef] [PubMed]
- Overmeyer, J.H.; Kaul, A.; Johnson, E.E.; Maltese, W.A. Active ras triggers death in glioblastoma cells through hyperstimulation of macropinocytosis. Mol. Cancer Res. 2008, 6, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Overmeyer, J.H.; Young, A.M.; Bhanot, H.; Maltese, W.A. A chalcone-related small molecule that induces methuosis, a novel form of non-apoptotic cell death, in glioblastoma cells. Mol. Cancer 2011, 10, 69. [Google Scholar] [CrossRef] [PubMed]
- Weerasinghe, P.; Buja, L.M. Oncosis: An important non-apoptotic mode of cell death. Exp. Mol. Pathol. 2012, 93, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, J.; Christofferson, D.E.; Ng, A.; Yao, J.; Degterev, A.; Xavier, R.J.; Yuan, J. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 2008, 135, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, S.; de Belle, I.; Bredesen, D.E. An alternative, nonapoptotic form of programmed cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 14376–14381. [Google Scholar] [CrossRef] [PubMed]
- Majno, G.; Joris, I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am. J. Pathol. 1995, 146, 3–15. [Google Scholar] [PubMed]
- Li, Z.; Mbah, N.E.; Maltese, W.A. Vacuole-inducing compounds that disrupt endolysosomal trafficking stimulate production of exosomes by glioblastoma cells. Mol. Cell. Biochem. 2018, 439, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chon, H.J.; Bae, K.J.; Lee, Y.; Kim, J. The casein kinase 2 inhibitor, CX-4945, as an anti-cancer drug in treatment of human hematological malignancies. Front. Pharmacol. 2015, 6, 70. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.W.; So, K.S.; Kim, S.C.; Park, K.M.; Lee, Y.J.; Kim, S.W.; Choi, C.M.; Rho, J.K.; Choi, Y.J.; Lee, J.C. Autophagy induced by CX-4945, a casein kinase 2 inhibitor, enhances apoptosis in pancreatic cancer cell lines. Pancreas 2017, 46, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Quotti Tubi, L.; Canovas Nunes, S.; Brancalion, A.; Doriguzzi Breatta, E.; Manni, S.; Mandato, E.; Zaffino, F.; Macaccaro, P.; Carrino, M.; Gianesin, K.; et al. Protein kinase CK2 regulates AKT, NF-kappaB and STAT3 activation, stem cell viability and proliferation in acute myeloid leukemia. Leukemia 2017, 31, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Han, J.; Kannabiran, V.; Mohan, S.; Cheng, H.; Friedman, J.; Zhang, L.; VanWaes, C.; Chen, Z. MEK inhibitor PD-0325901 overcomes resistance to CK2 inhibitor CX-4945 and exhibits anti-tumor activity in head and neck cancer. Int. J. Biol. Sci. 2015, 11, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Long, H.; Yang, Y.L.; Wang, Y.; Hsieh, D.; Li, W.; Au, A.; Stoppler, H.J.; Xu, Z.; Jablons, D.M.; et al. Inhibition of CK2alpha down-regulates Notch1 signalling in lung cancer cells. J. Cell. Mol. Med. 2013, 17, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Wadey, K.S.; Brown, B.A.; Sala-Newby, G.B.; Jayaraman, P.S.; Gaston, K.; George, S.J. Protein kinase CK2 inhibition suppresses neointima formation via a proline-rich homeodomain-dependent mechanism. Vascul. Pharmacol. 2017, 99, 34–44. [Google Scholar] [CrossRef] [PubMed]
- von Morgen, P.; Burdova, K.; Flower, T.G.; O’Reilly, N.J.; Boulton, S.J.; Smerdon, S.J.; Macurek, L.; Horejsi, Z. MRE11 stability is regulated by CK2-dependent interaction with R2TP complex. Oncogene 2017, 36, 4943–4950. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Lin, C.Y.; He, H.H.; Witwicki, R.M.; Tabassum, D.P.; Roberts, J.M.; Janiszewska, M.; Huh, S.J.; Liang, Y.; Ryan, J.; et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 2016, 529, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Greene, T.T.; Tokuyama, M.; Knudsen, G.M.; Kunz, M.; Lin, J.; Greninger, A.L.; DeFilippis, V.R.; DeRisi, J.L.; Raulet, D.H.; Coscoy, L. A herpesviral induction of RAE-1 NKG2D ligand expression occurs through release of HDAC mediated repression. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Li, F.L.; Liu, J.P.; Bao, R.X.; Yan, G.; Feng, X.; Xu, Y.P.; Sun, Y.P.; Yan, W.; Ling, Z.Q.; Xiong, Y.; et al. Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin-induced apoptosis. Nat. Commun. 2018, 9, 508. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Ghoshal, S.; Tyagi, R.; Chakraborty, A. Global IP6K1 deletion enhances temperature modulated energy expenditure which reduces carbohydrate and fat induced weight gain. Mol. Metab. 2017, 6, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Calamita, P.; Miluzio, A.; Russo, A.; Pesce, E.; Ricciardi, S.; Khanim, F.; Cheroni, C.; Alfieri, R.; Mancino, M.; Gorrini, C.; et al. SBDS-deficient cells have an altered homeostatic equilibrium due to translational inefficiency which explains their reduced fitness and provides a logical framework for intervention. PLoS Genet. 2017, 13, e1006552. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, W.; Sun, X.; Xu, D.; Wang, C.; Zhang, Q.; Wang, H.; Luo, W.; Chen, Y.; Chen, H.; et al. AMPK regulates autophagy by phosphorylating BECN1 at threonine 388. Autophagy 2016, 12, 1447–1459. [Google Scholar] [CrossRef] [PubMed]
- Johanns, M.; Lai, Y.C.; Hsu, M.F.; Jacobs, R.; Vertommen, D.; Van Sande, J.; Dumont, J.E.; Woods, A.; Carling, D.; Hue, L.; et al. AMPK antagonizes hepatic glucagon-stimulated cyclic AMP signalling via phosphorylation-induced activation of cyclic nucleotide phosphodiesterase 4B. Nat. Commun. 2016, 7, 10856. [Google Scholar] [CrossRef] [PubMed]
- Di Magno, L.; Basile, A.; Coni, S.; Manni, S.; Sdruscia, G.; D’Amico, D.; Antonucci, L.; Infante, P.; De Smaele, E.; Cucchi, D.; et al. The energy sensor AMPK regulates hedgehog signaling in human cells through a unique Gli1 metabolic checkpoint. Oncotarget 2016, 7, 9538–9549. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Zhu, M.J.; Dodson, M.V.; Du, M. AMP-activated protein kinase stimulates warburg-like glycolysis and activation of satellite cells during muscle regeneration. J. Biol. Chem. 2015, 290, 26445–26456. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Wang, L.; Wang, C.; Yang, Y.; Hu, D.; Ding, R. Effects of high-intensity interval versus continuous moderate-intensity aerobic exercise on apoptosis, oxidative stress and metabolism of the infarcted myocardium in a rat model. Mol. Med. Rep. 2015, 12, 2374–2382. [Google Scholar] [CrossRef] [PubMed]
- Obba, S.; Hizir, Z.; Boyer, L.; Selimoglu-Buet, D.; Pfeifer, A.; Michel, G.; Hamouda, M.A.; Goncalves, D.; Cerezo, M.; Marchetti, S.; et al. The PRKAA1/AMPkalpha1 pathway triggers autophagy during CSF1-induced human monocyte differentiation and is a potential target in CMML. Autophagy 2015, 11, 1114–1129. [Google Scholar] [CrossRef] [PubMed]
- Ducommun, S.; Deak, M.; Sumpton, D.; Ford, R.J.; Nunez Galindo, A.; Kussmann, M.; Viollet, B.; Steinberg, G.R.; Foretz, M.; Dayon, L.; et al. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: Identification of mitochondrial fission factor as a new AMPK substrate. Cell. Signal. 2015, 27, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W.; Na, W.; Kabir, M.H.; Yi, E.; Kwon, S.; Yeom, J.; Ahn, J.W.; Choi, H.H.; Lee, Y.; Seo, K.W.; et al. WIP1, a homeostatic regulator of the DNA damage response, is targeted by HIPK2 for phosphorylation and degradation. Mol. Cell 2013, 51, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Huang, Y.; Gunst, S.J. P21-activated kinase (Pak) regulates airway smooth muscle contraction by regulating paxillin complexes that mediate actin polymerization. J. Physiol. 2016, 594, 4879–4900. [Google Scholar] [CrossRef] [PubMed]
- Ahlskog, J.K.; Larsen, B.D.; Achanta, K.; Sorensen, C.S. ATM/ATR-mediated phosphorylation of PALB2 promotes RAD51 function. EMBO Rep. 2016, 17, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; de Renty, C.; Li, Y.; Xiao, H.; Kemp, M.G.; Han, Z.; DePamphilis, M.L.; Zhu, W. And-1 coordinates with claspin for efficient Chk1 activation in response to replication stress. EMBO J. 2015, 34, 2096–2110. [Google Scholar] [CrossRef] [PubMed]
- Rath, A.; Hromas, R.; De Benedetti, A. Fidelity of end joining in mammalian episomes and the impact of Metnase on joint processing. BMC Mol. Biol. 2014, 15, 6. [Google Scholar] [CrossRef] [PubMed]
- Hori, T.; Uemoto, S.; Chen, F.; Gardner, L.B.; Baine, A.M.; Hata, T.; Kogure, T.; Nguyen, J.H. Oxidative stress and extracellular matrices after hepatectomy and liver transplantation in rats. World J. Hepatol. 2014, 6, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Hori, T.; Gardner, L.B.; Hata, T.; Chen, F.; Baine, A.M.; Uemoto, S.; Nguyen, J.H. Pretreatment of liver grafts in vivo by gamma-aminobutyric acid receptor regulation reduces cold ischemia/warm reperfusion injury in rat. Ann. Transpl. 2013, 18, 299–313. [Google Scholar] [CrossRef]
- Gardner, L.B.; Hori, T.; Chen, F.; Baine, A.M.; Hata, T.; Uemoto, S.; Nguyen, J.H. Effect of specific activation of gamma-aminobutyric acid receptor in vivo on oxidative stress-induced damage after extended hepatectomy. Hepatol. Res. 2012, 42, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Ando, F.; Mori, S.; Yui, N.; Morimoto, T.; Nomura, N.; Sohara, E.; Rai, T.; Sasaki, S.; Kondo, Y.; Kagechika, H.; et al. AKAPs-PKA disruptors increase AQP2 activity independently of vasopressin in a model of nephrogenic diabetes insipidus. Nat. Commun. 2018, 9, 1411. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Ceddia, R.P.; Collins, S. Cardiac natriuretic peptides promote adipose ‘browning’ through mTOR complex-1. Mol. Metab. 2018, 9, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Cho, T.Y. Biochemical and structural characterization of a novel cooperative binding mode by Pit-1 with CATT repeats in the macrophage migration inhibitory factor promoter. Nucleic Acids Res. 2018, 46, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.J.A.; Pantazaka, E.; Shelley, K.L.; Taylor, C.W. Prostaglandin E2 inhibits histamine-evoked Ca2+ release in human aortic smooth muscle cells through hyperactive cAMP signaling junctions and protein kinase A. Mol. Pharmacol. 2017, 92, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Mohl, B.P.; Emmott, E.; Roy, P. Phosphoproteomic analysis reveals the importance of kinase regulation during orbivirus infection. Mol. Cell. Proteom. 2017, 16, 1990–2005. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.; Myhre, C.L.; Lassen, P.S.; Metaxas, A.; Khan, A.M.; Lambertsen, K.L.; Babcock, A.A.; Finsen, B.; Larsen, M.R.; Kempf, S.J. TNFalpha affects CREB-mediated neuroprotective signaling pathways of synaptic plasticity in neurons as revealed by proteomics and phospho-proteomics. Oncotarget 2017, 8, 60223–60242. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Shikano, S. Differential phosphorylation signals control endocytosis of GPR15. Mol. Biol. Cell 2017, 28, 2267–2281. [Google Scholar] [CrossRef] [PubMed]
- Garcia, R.; Bravo, E.; Diez-Muniz, S.; Nombela, C.; Rodriguez-Pena, J.M.; Arroyo, J. A novel connection between the cell wall integrity and the PKA pathways regulates cell wall stress response in yeast. Sci. Rep. 2017, 7, 5703. [Google Scholar] [CrossRef] [PubMed]
- Hiday, A.C.; Edler, M.C.; Salek, A.B.; Morris, C.W.; Thang, M.; Rentz, T.J.; Rose, K.L.; Jones, L.M.; Baucum, A.J., 2nd. Mechanisms and consequences of dopamine depletion-induced attenuation of the spinophilin/neurofilament medium interaction. Neural Plast. 2017, 2017, 4153076. [Google Scholar] [CrossRef] [PubMed]
- Shyu, K.G.; Velusamy, M.; Hsia, C.W.; Yang, C.H.; Hsia, C.H.; Chou, D.S.; Jayakumar, T.; Sheu, J.R.; Li, J.Y. Novel iridium (III)derived organometallic compound for the inhibition of human platelet activation. Int. J. Mol. Med. 2018, 41, 2589–2600. [Google Scholar] [CrossRef] [PubMed]
- Monteverde, T.; Tait-Mulder, J.; Hedley, A.; Knight, J.R.; Sansom, O.J.; Murphy, D.J. Calcium signalling links MYC to NUAK1. Oncogene 2018, 37, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Zitouni, S.; Mechali, F.; Papin, C.; Choquet, A.; Roche, D.; Baldin, V.; Coux, O.; Bonne-Andrea, C. The stability of Fbw7alpha in M-phase requires its phosphorylation by PKC. PLoS ONE 2017, 12, e0183500. [Google Scholar] [CrossRef] [PubMed]
- Waschek, J.A.; Cohen, J.R.; Chi, G.C.; Proszynski, T.J.; Niewiadomski, P. PACAP promotes matrix-driven adhesion of cultured adult murine neural progenitors. ASN Neuro 2017, 9, 1759091417708720. [Google Scholar] [CrossRef] [PubMed]
- Tello-Lafoz, M.; Rodriguez-Rodriguez, C.; Kinna, G.; Loo, L.S.; Hong, W.; Collins, B.M.; Teasdale, R.D.; Merida, I. SNX27 links DGKzeta to the control of transcriptional and metabolic programs in T lymphocytes. Sci. Rep. 2017, 7, 16361. [Google Scholar] [CrossRef] [PubMed]
- Shirafuji, T.; Ueyama, T.; Tanaka, S.; Hide, I.; Saito, N.; Sakai, N. Validation of anti-cspalpha, snap25, tyrosine hydroxylase, ubiquitin, cleaved caspase 3, and pser pkc motif antibodies for utilization in western blotting. Acta Histochem. Cytochem. 2017, 50, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Lucien, F.; Pelletier, P.P.; Lavoie, R.R.; Lacroix, J.M.; Roy, S.; Parent, J.L.; Arsenault, D.; Harper, K.; Dubois, C.M. Hypoxia-induced mobilization of NHE6 to the plasma membrane triggers endosome hyperacidification and chemoresistance. Nat. Commun. 2017, 8, 15884. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Stumpf, M.; Muller, R.; Eichinger, L.; Glockner, G.; Noegel, A.A. The function of the inner nuclear envelope protein SUN1 in mRNA export is regulated by phosphorylation. Sci. Rep. 2017, 7, 9157. [Google Scholar] [CrossRef] [PubMed]
- Carmon, K.S.; Gong, X.; Yi, J.; Wu, L.; Thomas, A.; Moore, C.M.; Masuho, I.; Timson, D.J.; Martemyanov, K.A.; Liu, Q.J. LGR5 receptor promotes cell-cell adhesion in stem cells and colon cancer cells via the IQGAP1-Rac1 pathway. J. Biol. Chem. 2017, 292, 14989–15001. [Google Scholar] [CrossRef] [PubMed]
- So, K.S.; Kim, C.H.; Rho, J.K.; Kim, S.Y.; Choi, Y.J.; Song, J.S.; Kim, W.S.; Choi, C.M.; Chun, Y.J.; Lee, J.C. Autophagosome-mediated EGFR down-regulation induced by the CK2 inhibitor enhances the efficacy of EGFR-TKI on EGFR-mutant lung cancer cells with resistance by T790M. PLoS ONE 2014, 9, e114000. [Google Scholar] [CrossRef] [PubMed]
- Sripa, B.; Leungwattanawanit, S.; Nitta, T.; Wongkham, C.; Bhudhisawasdi, V.; Puapairoj, A.; Sripa, C.; Miwa, M. Establishment and characterization of an opisthorchiasis-associated cholangiocarcinoma cell line (KKU-100). World J. Gastroenterol. 2005, 11, 3392–3397. [Google Scholar] [CrossRef] [PubMed]
- Sirisinha, S.; Tengchaisri, T.; Boonpucknavig, S.; Prempracha, N.; Ratanarapee, S.; Pausawasdi, A. Establishment and characterization of a cholangiocarcinoma cell line from a thai patient with intrahepatic bile duct cancer. Asian Pac. J. Allergy Immunol. 1991, 9, 153–157. [Google Scholar] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lertsuwan, J.; Lertsuwan, K.; Sawasdichai, A.; Tasnawijitwong, N.; Lee, K.Y.; Kitchen, P.; Afford, S.; Gaston, K.; Jayaraman, P.-S.; Satayavivad, J. CX-4945 Induces Methuosis in Cholangiocarcinoma Cell Lines by a CK2-Independent Mechanism. Cancers 2018, 10, 283. https://doi.org/10.3390/cancers10090283
Lertsuwan J, Lertsuwan K, Sawasdichai A, Tasnawijitwong N, Lee KY, Kitchen P, Afford S, Gaston K, Jayaraman P-S, Satayavivad J. CX-4945 Induces Methuosis in Cholangiocarcinoma Cell Lines by a CK2-Independent Mechanism. Cancers. 2018; 10(9):283. https://doi.org/10.3390/cancers10090283
Chicago/Turabian StyleLertsuwan, Jomnarong, Kornkamon Lertsuwan, Anyaporn Sawasdichai, Nathapol Tasnawijitwong, Ka Ying Lee, Philip Kitchen, Simon Afford, Kevin Gaston, Padma-Sheela Jayaraman, and Jutamaad Satayavivad. 2018. "CX-4945 Induces Methuosis in Cholangiocarcinoma Cell Lines by a CK2-Independent Mechanism" Cancers 10, no. 9: 283. https://doi.org/10.3390/cancers10090283
APA StyleLertsuwan, J., Lertsuwan, K., Sawasdichai, A., Tasnawijitwong, N., Lee, K. Y., Kitchen, P., Afford, S., Gaston, K., Jayaraman, P.-S., & Satayavivad, J. (2018). CX-4945 Induces Methuosis in Cholangiocarcinoma Cell Lines by a CK2-Independent Mechanism. Cancers, 10(9), 283. https://doi.org/10.3390/cancers10090283