Targeting Focal Adhesion Kinase Using Inhibitors of Protein-Protein Interactions


Abstract
1. Search for Inhibitors of Protein-Protein Interaction
1.1. Introduction
1.2. Successful Examples
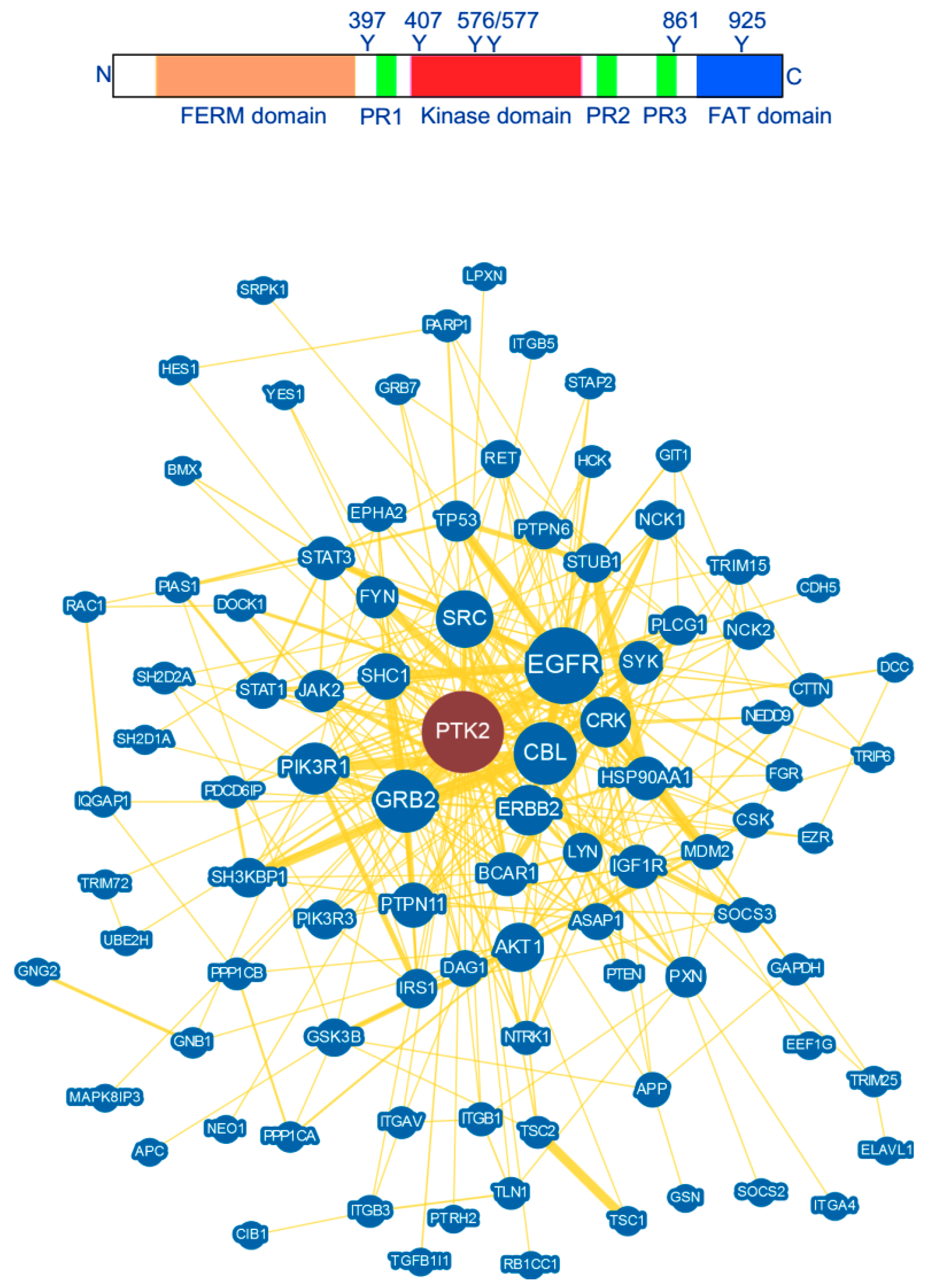
2. FAK Structure and Interaction
3. FAK Structural Determinant for the Search of Potent FAK Inhibitors
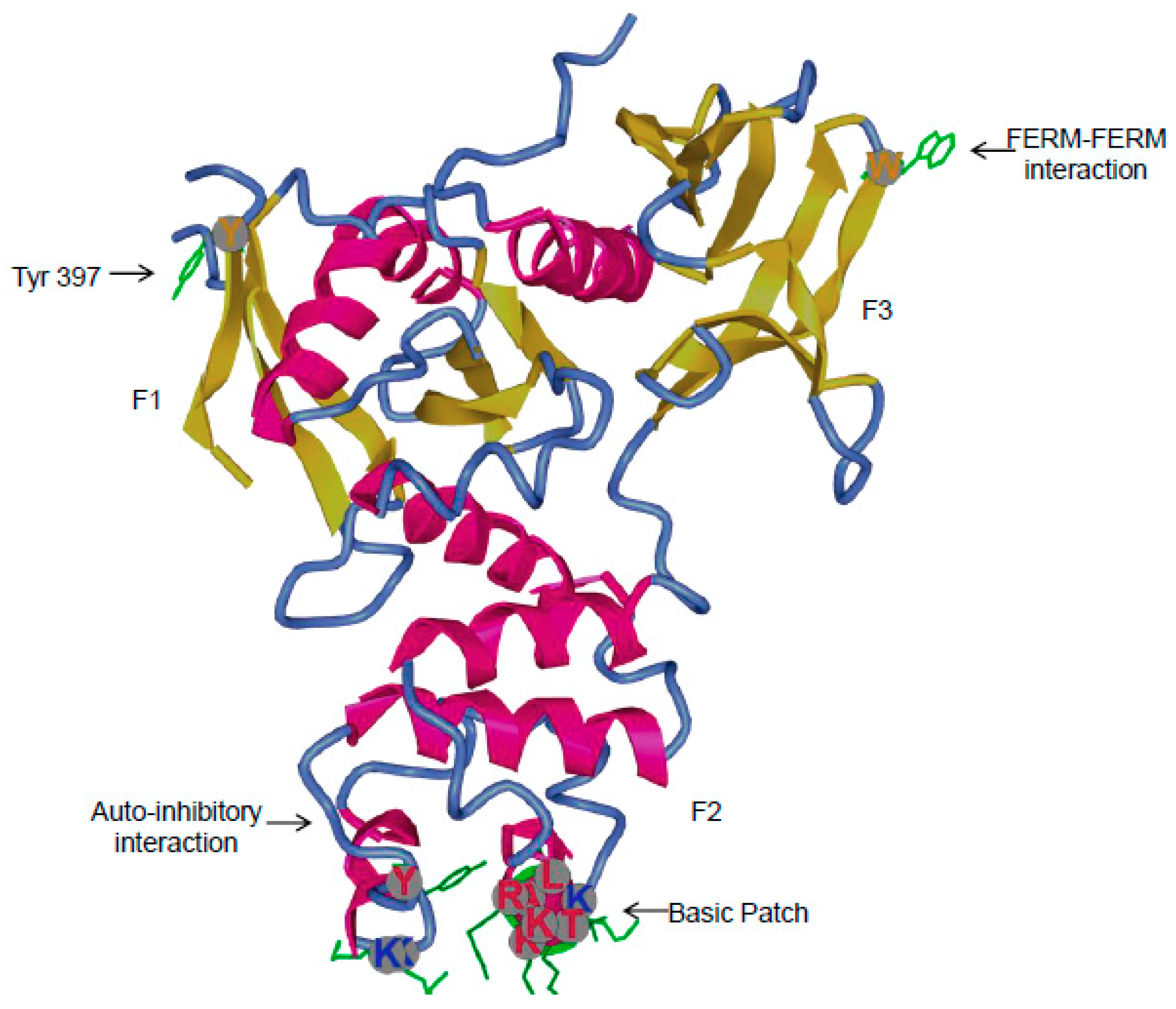
3.1. Major Interactions at the FERM Domain
3.1.1. FAK Interaction with Growth Factor Receptors and Mechanism of FAK Activation
3.1.2. FAK Control of Cell Polarity and Migration
3.1.3. FAK Functions in the Nucleus
3.1.4. Inhibitor of FERM Interactions
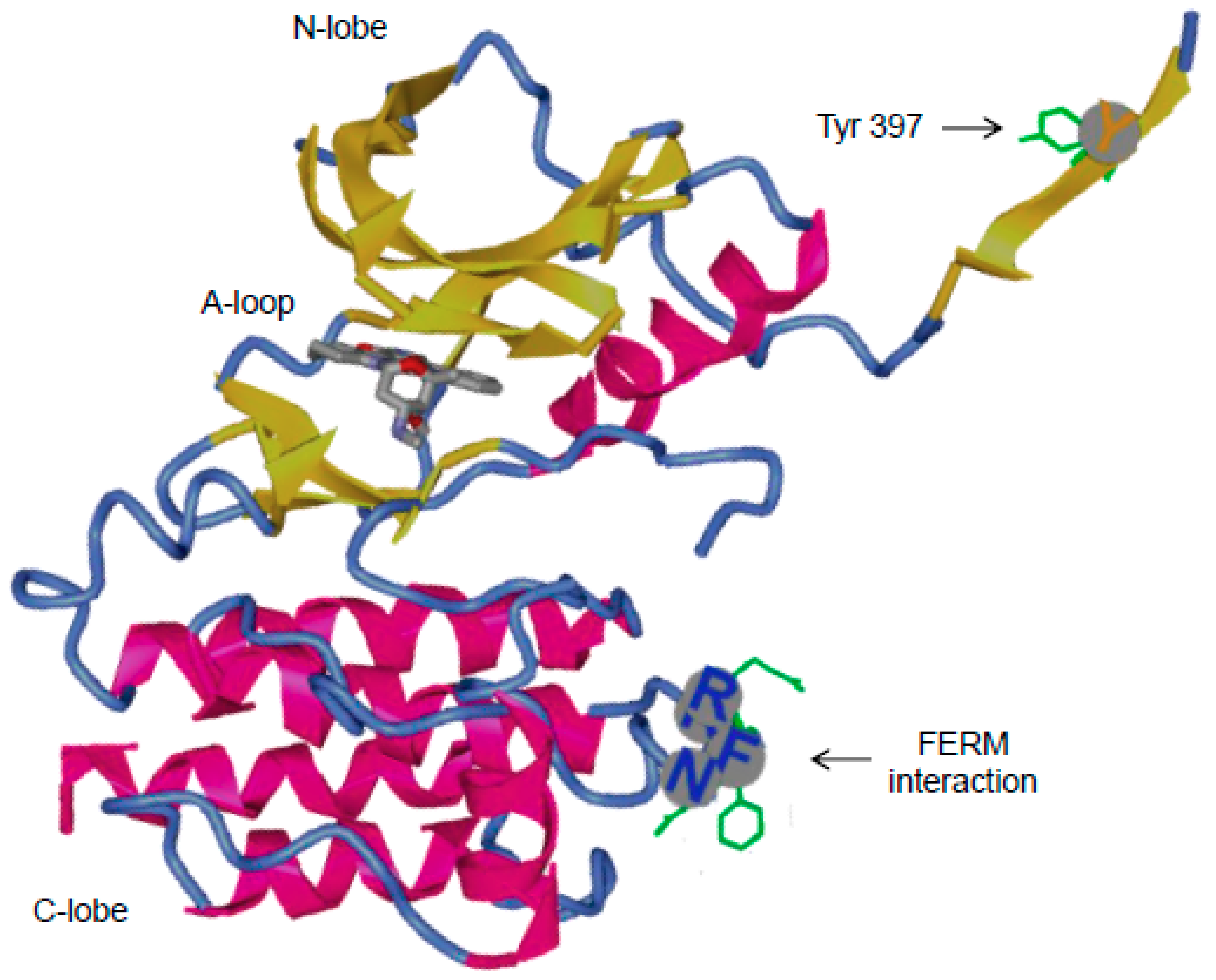
3.2. Major Interactions at the Kinase Domain
3.2.1. Role of Tyr397 in FAK Catalytic Activity and Interactions with Binding Partners
3.2.2. FAK Inhibitors: Targeting the ATP Binding Site
3.2.3. Inhibiting FAK P-Tyr Binding to SH2-Containing Protein
3.3. Major Interactions at the Prolin-Rich Domains
3.3.1. Role of the FAK PR Domain in the Invasion Pathway
3.3.2. FAK as a Regulator of the Rho-GTPase Family
3.3.3. Role of FAK Ser732
3.3.4. Inhibiting the FAK PR Domain Binding to SH3-Containing Protein
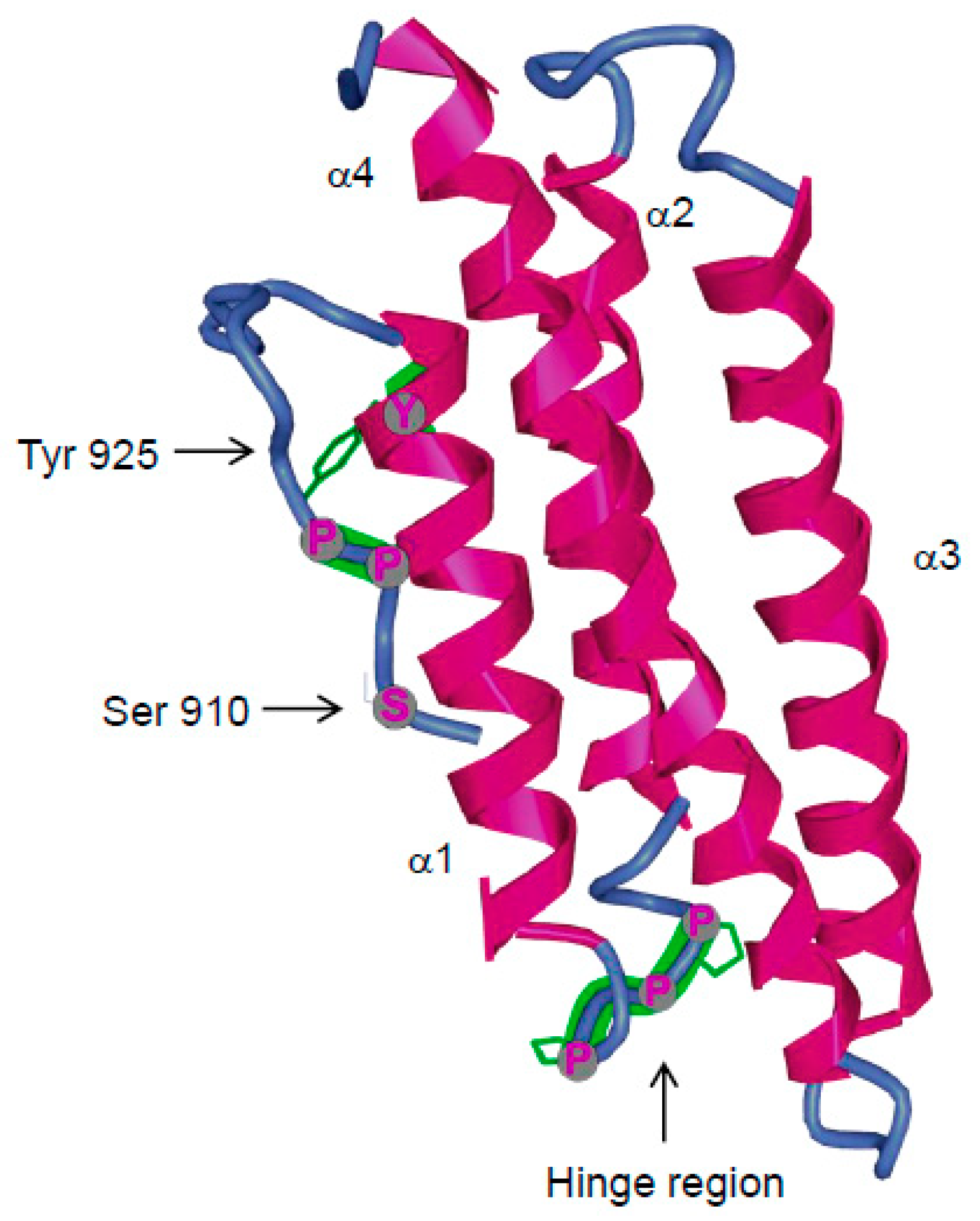
3.4. Major Interactions at the FAT Domain
3.4.1. FAK Binding to Paxillin and the Targeting to Focal Adhesions
3.4.2. FAK Phosphorylation at Tyr925
3.4.3. FAK Phosphorylation at Ser910
3.4.4. FAK as a Regulator of the Rho-GTPase Family
3.4.5. Inhibitors of FAT Interactions
4. Conclusions
Funding
Conflicts of Interest
References
- Jin, L.; Wang, W.; Fang, G. Targeting protein-protein interaction by small molecules. Ann. Rev. Pharmacol. Toxicol. 2014, 54, 435–456. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, J.R. Drugs, leads and drug-likeness: An analysis of some recently launched drugs. Bioorg. Med. Chem. Lett. 2002, 12, 1647–1650. [Google Scholar] [CrossRef]
- Conte, M.P.; Longhi, C.; Petrone, G.; Buonfiglio, V.; Di Santo, S.; Seganti, L.; Velenti, P. The anti-invasive effect of bovine lactoferrin requires an interaction with surface proteins of Listeria Monocytogenes. Int. J. Immunopathol. Pharmacol. 1999, 12, 149–155. [Google Scholar] [PubMed]
- Smith, J.; Diez, G.; Klemm, A.H.; Schewkunow, V.; Goldmann, W.H. CapZ-lipid membrane interactions: A computer analysis. Theor. Biol. Med. Model. 2006, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Fuller, J.C.; Burgoyne, N.J.; Jackson, R.M. Predicting druggable binding sites at the protein-protein interface. Drug Discov. Today 2009, 14, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Clackson, T.; Wells, J.A. A hot spot of binding energy in a hormone-receptor interface. Science 1995, 267, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, P.J.; Huth, J.R.; Tse, C. Predicting protein druggability. Drug Discov. Today 2005, 10, 1675–1682. [Google Scholar] [CrossRef]
- Bogan, A.A.; Thorn, K.S. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998, 280, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.C.; Coleman, R.G.; Smyth, K.T.; Cao, Q.; Soulard, P.; Caffrey, D.R.; Salzberg, A.C.; Huang, E.S. Structure-based maximal affinity model predicts small-molecule druggability. Nat. Biotechnol. 2007, 25, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Hall, D.R.; Chuang, G.Y.; Cencic, R.; Brenke, R.; Grove, L.E.; Beglov, D.; Pelletier, J.; Whitty, A.; Vajda, S. Structural conservation of druggable hot spots in protein-protein interfaces. Proc. Natl. Acad. Sci. USA 2011, 108, 13528–13533. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Narayanapillai, S.; Zhang, W.; Sham, Y.Y.; Xing, C. Rapid identification of Keap1-Nrf2 small-molecule inhibitors through structure-based virtual screening and hit-based substructure search. J. Med. Chem. 2014, 57, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.H.; Christoffer, C.W.; Kihara, D. In silico structure-based approaches to discover protein-protein interaction-targeting drugs. Methods 2017, 131, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumour suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Fry, D.C.; Emerson, S.D.; Palme, S.; Vu, B.T.; Liu, C.M.; Podlaski, F. NMR structure of a complex between MDM2 and a small molecule inhibitor. J. Biomol. NMR 2004, 30, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Zhang, Z.; Liu, J.J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- Tisato, V.; Voltan, R.; Gonelli, A.; Secchiero, P.; Zauli, G. MDM2/X inhibitors under clinical evaluation: Perspectives for the management of hematological malignancies and pediatric cancer. J. Hematol. Oncol. 2017, 10, 133. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.A.; Huang, D.; Roberts, A. Targeting BCL2 for the treatment of lymphoid malignancies. Semin. Hematol. 2014, 51, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Levitzki, A. Tyrosine kinase inhibitors: Views of selectivity, sensitivity and clinical performance. Ann. Rev. Pharmacol. Toxicol. 2013, 53, 161–185. [Google Scholar] [CrossRef] [PubMed]
- Kanner, S.B.; Reynolds, A.B.; Vines, R.R.; Parsons, J.T. Monoclonal antibodies to individual tyrosine-phosphorylated protein substrates of oncogene-encoded tyrosine kinases. Proc. Natl. Acad. Sci. USA 1990, 87, 3328–3332. [Google Scholar] [CrossRef] [PubMed]
- Lipfert, L.; Haimovich, B.; Schaller, M.D.; Cobb, B.S.; Parsons, J.T.; Brugge, J.S. Integrin-dependent phosphorylation and activation of the protein tyrosine kinase pp125FAK in platelets. J. Cell Biol. 1992, 119, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.D.; Borgman, C.A.; Cobb, B.S.; Vines, R.R.; Reynolds, A.B.; Parsons, J.T. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc. Natl. Acad. Sci. USA 1992, 89, 5192–5196. [Google Scholar] [CrossRef] [PubMed]
- Weiner, T.M.; Liu, E.T.; Craven, R.J.; Cance, W.G. Expression of focal adhesion kinase gene and invasive cancer. Lancet 1993, 342, 1024–1025. [Google Scholar] [CrossRef]
- Owens, L.V.; Xu, L.; Craven, R.J.; Dent, G.A.; Weiner, T.M.; Kornberg, L.; Liu, E.T.; Cance, W.G. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res. 1995, 55, 2752–2755. [Google Scholar] [PubMed]
- Sieg, D.J.; Hauck, C.R.; Ilic, D.; Klingbeil, C.K.; Schaefer, E.; Damsky, C.H.; Schlaepfer, D.D. FAK integrates growth-factor and integrin signals to promote cell migration. Nat. Cell Biol. 2000, 2, 249–256. [Google Scholar] [CrossRef] [PubMed]
- McLean, G.W.; Komiyama, N.H.; Serrels, B.; Asano, H.; Reynolds, L.; Conti, F.; Hodivala-Dilke, K.; Metzger, D.; Chambon, P.; Grant, S.G.; et al. Specific deletion of focal adhesion kinase suppresses tumor formation and blocks malignant progression. Genes Dev. 2004, 18, 2998–3003. [Google Scholar] [CrossRef] [PubMed]
- Cance, W.G.; Kurenova, E.; Marlowe, T.; Golubovskaya, V. Disrupting the scaffold to improve focal adhesion kinase-targeted cancer therapeutics. Sci. Signal. 2013, 6, pe10. [Google Scholar] [CrossRef] [PubMed]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID. A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [PubMed]
- Chatr-Aryamontri, A.; Breitkreutz, B.J.; Oughtred, R.; Boucher, L.; Heinicke, S.; Chen, D.; Stark, C.; Breitkreutz, A.; Kolas, N.; O’Donnell, L.; et al. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2014, 43, D470–D478. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.D.; Parsons, J.T. Focal adhesion kinase: An integrin-linked protein tyrosine kinase. Trends Cell Biol. 1993, 3, 258–262. [Google Scholar] [CrossRef]
- Schaller, M.D.; Otey, C.A.; Hildebrand, J.D.; Parsons, J.T. Focal adhesion kinase and paxillin bind to peptides mimicking beta integrin cytoplasmic domains. J. Cell Biol. 1995, 130, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.A.; Shen, T.L.; Guan, J.L. Regulation of focal adhesion kinase by its amino-terminal domain through an autoinhibitory interaction. Mol. Cell. Biol. 2003, 23, 8030–8041. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.L.; Chu, P.Y.; Lai, I.R.; Wang, M.Y.; Tseng, H.Y.; Guan, J.L.; Liou, J.Y.; Shen, T.L. An EGFR/Src-dependent beta4 integrin/FAK complex contributes to malignancy of breast cancer. Sci. Rep. 2015, 5, 16408. [Google Scholar] [CrossRef] [PubMed]
- Lietha, D.; Cai, X.; Ceccarelli, D.F.; Li, Y.; Schaller, M.D.; Eck, M.J. Structural basis for the autoinhibition of focal adhesion kinase. Cell 2007, 129, 1177–1187. [Google Scholar] [CrossRef] [PubMed]
- Frame, M.C.; Patel, H.; Serrels, B.; Lietha, D.; Eck, M.J. The FERM domain: Organizing the structure and function of FAK. Nat. Rev. Mol. Cell Biol. 2010, 11, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Jung, O.; Choi, S.; Jang, S.B.; Lee, S.A.; Lim, S.T.; Choi, Y.J.; Kim, H.J.; Kim, D.H.; Kwak, T.K.; Kim, H.; et al. Tetraspan TM4SF5-dependent direct activation of FAK and metastatic potential of hepatocarcinoma cells. J. Cell Sci. 2012, 125, 5960–5973. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Chen, H.C. Direct interaction of focal adhesion kinase (FAK) with Met is required for FAK to promote hepatocyte growth factor-induced cell invasion. Mol. Cell. Biol. 2006, 26, 5155–5167. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Menacho, I.; Morandi, A.; Mologni, L.; Boender, P.; Gambacorti-Passerini, C.; Magee, A.I.; Hofstra, R.M.; Knowles, P.; McDonald, N.Q.; Isacke, C.M. Focal adhesion kinase (FAK) binds RET kinase via its FERM domain, priming a direct and reciprocal RET-FAK transactivation mechanism. J. Biol. Chem. 2011, 286, 17292–17302. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Bloom, D.A.; Cance, W.G.; Kurenova, E.V.; Golubovskaya, V.M.; Hochwald, S.N. FAK and IGF-IR interact to provide survival signals in human pancreatic adenocarcinoma cells. Carcinogenesis 2008, 29, 1096–1107. [Google Scholar] [CrossRef] [PubMed]
- Marlowe, T.A.; Lenzo, F.L.; Figel, S.A.; Grapes, A.T.; Cance, W.G. Oncogenic Receptor Tyrosine Kinases Directly Phosphorylate Focal Adhesion Kinase (FAK) as a Resistance Mechanism to FAK-Kinase Inhibitors. Mol. Cancer Ther. 2016, 15, 3028–3039. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.H.; Chan, P.C.; Chen, C.L.; Chen, H.C. Phosphorylation of focal adhesion kinase on tyrosine 194 by Met leads to its activation through relief of autoinhibition. Oncogene 2011, 30, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Nam, J.O.; Jean, C.; Lawson, C.; Walsh, C.T.; Goka, E.; Lim, S.T.; Tomar, A.; Tancioni, I.; Uryu, S.; et al. VEGF-induced vascular permeability is mediated by FAK. Dev. Cell 2012, 22, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Poullet, P.; Gautreau, A.; Kadare, G.; Girault, J.A.; Louvard, D.; Arpin, M. Ezrin interacts with focal adhesion kinase and induces its activation independently of cell-matrix adhesion. J. Biol. Chem. 2001, 276, 37686–37691. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Lietha, D.; Ceccarelli, D.F.; Karginov, A.V.; Rajfur, Z.; Jacobson, K.; Hahn, K.M.; Eck, M.J.; Schaller, M.D. Spatial and temporal regulation of focal adhesion kinase activity in living cells. Mol. Cell. Biol. 2008, 28, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Goni, G.M.; Epifano, C.; Boskovic, J.; Camacho-Artacho, M.; Zhou, J.; Bronowska, A.; Martin, M.T.; Eck, M.J.; Kremer, L.; Grater, F.; et al. Phosphatidylinositol 4,5-bisphosphate triggers activation of focal adhesion kinase by inducing clustering and conformational changes. Proc. Natl. Acad. Sci. USA 2014, 111, E3177–E3186. [Google Scholar] [CrossRef] [PubMed]
- Ezratty, E.J.; Partridge, M.A.; Gundersen, G.G. Microtubule-induced focal adhesion disassembly is mediated by dynamin and focal adhesion kinase. Nat. Cell Biol. 2005, 7, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.A.; Guan, J.L. Mechanisms of focal adhesion kinase regulation. Curr. Cancer Drug Targets 2005, 5, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, V.; Fischer, R.S.; Waterman, C.M. The FAK-Arp2/3 interaction promotes leading edge advance and haptosensing by coupling nascent adhesions to lamellipodia actin. Mol. Biol. Cell 2016, 27, 1085–1100. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Suetsugu, S.; Cooper, L.A.; Takenawa, T.; Guan, J.L. Focal adhesion kinase regulation of N-WASP subcellular localization and function. J. Biol. Chem. 2004, 279, 9565–9576. [Google Scholar] [CrossRef] [PubMed]
- Serrels, B.; Sandilands, E.; Serrels, A.; Baillie, G.; Houslay, M.D.; Brunton, V.G.; Canel, M.; Machesky, L.M.; Anderson, K.I.; Frame, M.C. A complex between FAK, RACK1 and PDE4D5 controls spreading initiation and cancer cell polarity. Curr. Biol. CB 2010, 20, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Kim, O.; Li, M.; Xiong, X.; Guan, J.L.; Kung, H.J.; Chen, H.; Shimizu, Y.; Qiu, Y. Regulation of the PH-domain-containing tyrosine kinase Etk by focal adhesion kinase through the FERM domain. Nat. Cell Biol. 2001, 3, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.; Olson, G.; Figel, S.; Gelman, I.; Cance, W.G.; Golubovskaya, V.M. Nanog increases focal adhesion kinase (FAK) promoter activity and expression and directly binds to FAK protein to be phosphorylated. J. Biol. Chem. 2012, 287, 18656–18673. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.T.; Chen, X.L.; Lim, Y.; Hanson, D.A.; Vo, T.T.; Howerton, K.; Larocque, N.; Fisher, S.J.; Schlaepfer, D.D.; Ilic, D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol. Cell 2008, 29, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Kadare, G.; Toutant, M.; Formstecher, E.; Corvol, J.C.; Carnaud, M.; Boutterin, M.C.; Girault, J.A. PIAS1-mediated sumoylation of focal adhesion kinase activates its autophosphorylation. J. Biol. Chem. 2003, 278, 47434–47440. [Google Scholar] [CrossRef] [PubMed]
- Kurenova, E.; Xu, L.H.; Yang, X.; Baldwin, A.S., Jr.; Craven, R.J.; Hanks, S.K.; Liu, Z.G.; Cance, W.G. Focal adhesion kinase suppresses apoptosis by binding to the death domain of receptor-interacting protein. Mol. Cell. Biol. 2004, 24, 4361–4371. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Finch, R.; Cance, W.G. Direct interaction of the N-terminal domain of focal adhesion kinase with the N-terminal transactivation domain of p53. J. Biol. Chem. 2005, 280, 25008–25021. [Google Scholar] [CrossRef] [PubMed]
- Seong, J.; Tajik, A.; Sun, J.; Guan, J.L.; Humphries, M.J.; Craig, S.E.; Shekaran, A.; Garcia, A.J.; Lu, S.; Lin, M.Z.; et al. Distinct biophysical mechanisms of focal adhesion kinase mechanoactivation by different extracellular matrix proteins. Proc. Natl. Acad. Sci. USA 2013, 110, 19372–19377. [Google Scholar] [CrossRef] [PubMed]
- Toutant, M.; Costa, A.; Studler, J.M.; Kadare, G.; Carnaud, M.; Girault, J.A. Alternative splicing controls the mechanisms of FAK autophosphorylation. Mol. Cell. Biol. 2002, 22, 7731–7743. [Google Scholar] [CrossRef] [PubMed]
- Brami-Cherrier, K.; Gervasi, N.; Arsenieva, D.; Walkiewicz, K.; Boutterin, M.C.; Ortega, A.; Leonard, P.G.; Seantier, B.; Gasmi, L.; Bouceba, T.; et al. FAK dimerization controls its kinase-dependent functions at focal adhesions. EMBO J. 2014, 33, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Alanko, J.; Mai, A.; Jacquemet, G.; Schauer, K.; Kaukonen, R.; Saari, M.; Goud, B.; Ivaska, J. Integrin endosomal signalling suppresses anoikis. Nat. Cell Biol. 2015, 17, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cao, H.; Chen, J.; McNiven, M.A. A direct interaction between the large GTPase dynamin-2 and FAK regulates focal adhesion dynamics in response to active Src. Mol. Biol. Cell 2011, 22, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, D.F.; Song, H.K.; Poy, F.; Schaller, M.D.; Eck, M.J. Crystal structure of the FERM domain of focal adhesion kinase. J. Biol. Chem. 2006, 281, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.; Webb, B.A.; Chimenti, M.S.; Jacobson, M.P.; Barber, D.L. PH sensing by FAK-His58 regulates focal adhesion remodeling. J. Cell Biol. 2013, 202, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Serrels, B.; Serrels, A.; Brunton, V.G.; Holt, M.; McLean, G.W.; Gray, C.H.; Jones, G.E.; Frame, M.C. Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat. Cell Biol. 2007, 9, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Tomar, A.; Lim, S.T.; Lim, Y.; Schlaepfer, D.D. A FAK-p120RasGAP-p190RhoGAP complex regulates polarity in migrating cells. J. Cell Sci. 2009, 122, 1852–1862. [Google Scholar] [CrossRef] [PubMed]
- Delyon, J.; Servy, A.; Laugier, F.; Andre, J.; Ortonne, N.; Battistella, M.; Mourah, S.; Bensussan, A.; Lebbe, C.; Dumaz, N. PDE4D promotes FAK-mediated cell invasion in BRAF-mutated melanoma. Oncogene 2017, 36, 3252–3262. [Google Scholar] [CrossRef] [PubMed]
- Hungerford, J.E.; Compton, M.T.; Matter, M.L.; Hoffstrom, B.G.; Otey, C.A. Inhibition of pp125FAK in cultured fibroblasts results in apoptosis. J. Cell Biol. 1996, 135, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.H.; Owens, L.V.; Sturge, G.C.; Yang, X.; Liu, E.T.; Craven, R.J.; Cance, W.G. Attenuation of the expression of the focal adhesion kinase induces apoptosis in tumor cells. Cell Growth Differ. 1996, 7, 413–418. [Google Scholar] [PubMed]
- McLean, G.W.; Carragher, N.O.; Avizienyte, E.; Evans, J.; Brunton, V.G.; Frame, M.C. The role of focal-adhesion kinase in cancer—A new therapeutic opportunity. Nat. Rev. Cancer 2005, 5, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Meurice, N.; Wang, L.; Lipinski, C.A.; Yang, Z.; Hulme, C.; Loftus, J.C. Structural conservation in band 4.1, ezrin, radixin, moesin (FERM) domains as a guide to identify inhibitors of the proline-rich tyrosine kinase 2. J. Med. Chem. 2010, 53, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Finch, R.; Zheng, M.; Kurenova, E.V.; Cance, W.G. The 7-amino-acid site in the proline-rich region of the N-terminal domain of p53 is involved in the interaction with FAK and is critical for p53 functioning. Biochem. J. 2008, 411, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Ho, B.; Zheng, M.; Magis, A.; Ostrov, D.; Morrison, C.; Cance, W.G. Disruption of focal adhesion kinase and p53 interaction with small molecule compound R2 reactivated p53 and blocked tumor growth. BMC Cancer 2013, 13, 342. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Palma, N.L.; Zheng, M.; Ho, B.; Magis, A.; Ostrov, D.; Cance, W.G. A small-molecule inhibitor, 5’-O-tritylthymidine, targets FAK and Mdm-2 interaction and blocks breast and colon tumorigenesis in vivo. Anti-Cancer Agents Med. Chem. 2013, 13, 532–545. [Google Scholar] [CrossRef]
- Lim, S.T.; Miller, N.L.; Chen, X.L.; Tancioni, I.; Walsh, C.T.; Lawson, C.; Uryu, S.; Weis, S.M.; Cheresh, D.A.; Schlaepfer, D.D. Nuclear-localized focal adhesion kinase regulates inflammatory VCAM-1 expression. J. Cell Biol. 2012, 197, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Kurenova, E.; Ucar, D.; Golubovskaya, V.; Magis, A.; Ostrov, D.; Cance, W.G.; Hochwald, S.N. Targeting of the protein interaction site between FAK and IGF-1R. Biochem. Biophys. Res. Commun. 2009, 388, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Ucar, D.A.; Kurenova, E.; Garrett, T.J.; Cance, W.G.; Nyberg, C.; Cox, A.; Massoll, N.; Ostrov, D.A.; Lawrence, N.; Sebti, S.M.; et al. Disruption of the protein interaction between FAK and IGF-1R inhibits melanoma tumor growth. Cell Cycle 2012, 11, 3250–3259. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.D.; Hildebrand, J.D.; Shannon, J.D.; Fox, J.W.; Vines, R.R.; Parsons, J.T. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol. Cell. Biol. 1994, 14, 1680–1688. [Google Scholar] [CrossRef] [PubMed]
- Hamadi, A.; Bouali, M.; Dontenwill, M.; Stoeckel, H.; Takeda, K.; Ronde, P. Regulation of focal adhesion dynamics and disassembly by phosphorylation of FAK at tyrosine 397. J. Cell Sci. 2005, 118, 4415–4425. [Google Scholar] [CrossRef] [PubMed]
- Webb, D.J.; Donais, K.; Whitmore, L.A.; Thomas, S.M.; Turner, C.E.; Parsons, J.T.; Horwitz, A.F. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 2004, 6, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.T. Focal adhesion kinase: The first ten years. J. Cell Sci. 2003, 116, 1409–1416. [Google Scholar] [CrossRef] [PubMed]
- Calalb, M.B.; Polte, T.R.; Hanks, S.K. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: A role for Src family kinases. Mol. Cell. Biol. 1995, 15, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Calalb, M.B.; Zhang, X.; Polte, T.R.; Hanks, S.K. Focal adhesion kinase tyrosine-861 is a major site of phosphorylation by Src. Biochem. Biophys. Res. Commun. 1996, 228, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, D.D.; Hunter, T. Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol. Cell. Biol. 1996, 16, 5623–5633. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.; Park, H.; Jeon, J.; Han, I.; Kim, J.; Jho, E.H.; Oh, E.S. Focal adhesion kinase is negatively regulated by phosphorylation at tyrosine 407. J. Biol. Chem. 2007, 282, 10398–10404. [Google Scholar] [CrossRef] [PubMed]
- Bellis, S.L.; Miller, J.T.; Turner, C.E. Characterization of tyrosine phosphorylation of paxillin in vitro by focal adhesion kinase. J. Biol. Chem. 1995, 270, 17437–17441. [Google Scholar] [CrossRef] [PubMed]
- Cary, L.A.; Han, D.C.; Polte, T.R.; Hanks, S.K.; Guan, J.L. Identification of p130Cas as a mediator of focal adhesion kinase-promoted cell migration. J. Cell Biol. 1998, 140, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Deramaudt, T.B.; Dujardin, D.; Hamadi, A.; Noulet, F.; Kolli, K.; De Mey, J.; Takeda, K.; Ronde, P. FAK phosphorylation at Tyr-925 regulates cross-talk between focal adhesion turnover and cell protrusion. Mol. Biol. Cell 2011, 22, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Hsia, D.A.; Mitra, S.K.; Hauck, C.R.; Streblow, D.N.; Nelson, J.A.; Ilic, D.; Huang, S.; Li, E.; Nemerow, G.R.; Leng, J.; et al. Differential regulation of cell motility and invasion by FAK. J. Cell Biol. 2003, 160, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Xing, Z.; Chen, H.C.; Nowlen, J.K.; Taylor, S.J.; Shalloway, D.; Guan, J.L. Direct interaction of v-Src with the focal adhesion kinase mediated by the Src SH2 domain. Mol. Biol. Cell 1994, 5, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Guan, J.L. Association of focal adhesion kinase with its potential substrate phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 1994, 91, 10148–10152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chattopadhyay, A.; Ji, Q.S.; Owen, J.D.; Ruest, P.J.; Carpenter, G.; Hanks, S.K. Focal adhesion kinase promotes phospholipase C-gamma1 activity. Proc. Natl. Acad. Sci. USA 1999, 96, 9021–9026. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.L.; Guan, J.L. Differential regulation of cell migration and cell cycle progression by FAK complexes with Src, PI3K, Grb7 and Grb2 in focal contacts. FEBS Lett. 2001, 499, 176–181. [Google Scholar] [CrossRef]
- Tamura, M.; Gu, J.; Matsumoto, K.; Aota, S.; Parsons, R.; Yamada, K.M. Inhibition of cell migration, spreading and focal adhesions by tumor suppressor PTEN. Science 1998, 280, 1614–1617. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Gu, J.; Danen, E.H.; Takino, T.; Miyamoto, S.; Yamada, K.M. PTEN interactions with focal adhesion kinase and suppression of the extracellular matrix-dependent phosphatidylinositol 3-kinase/Akt cell survival pathway. J. Biol. Chem. 1999, 274, 20693–20703. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Cote, J.F.; Vuori, K. Negative regulation of FAK signaling by SOCS proteins. EMBO J. 2003, 22, 5036–5046. [Google Scholar] [CrossRef] [PubMed]
- Han, D.C.; Guan, J.L. Association of focal adhesion kinase with Grb7 and its role in cell migration. J. Biol. Chem. 1999, 274, 24425–24430. [Google Scholar] [CrossRef] [PubMed]
- Hecker, T.P.; Ding, Q.; Rege, T.A.; Hanks, S.K.; Gladson, C.L. Overexpression of FAK promotes Ras activity through the formation of a FAK/p120RasGAP complex in malignant astrocytoma cells. Oncogene 2004, 23, 3962–3971. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, J.D.; Taylor, J.M.; Parsons, J.T. An SH3 domain-containing GTPase-activating protein for Rho and Cdc42 associates with focal adhesion kinase. Mol. Cell. Biol. 1996, 16, 3169–3178. [Google Scholar] [CrossRef] [PubMed]
- Abbi, S.; Ueda, H.; Zheng, C.; Cooper, L.A.; Zhao, J.; Christopher, R.; Guan, J.L. Regulation of focal adhesion kinase by a novel protein inhibitor FIP200. Mol. Biol. Cell 2002, 13, 3178–3191. [Google Scholar] [CrossRef] [PubMed]
- Tavora, B.; Batista, S.; Reynolds, L.E.; Jadeja, S.; Robinson, S.; Kostourou, V.; Hart, I.; Fruttiger, M.; Parsons, M.; Hodivala-Dilke, K.M. Endothelial FAK is required for tumour angiogenesis. EMBO Mol. Med. 2010, 2, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, A.N.; Lees, D.M.; Bodrug, N.; Lechertier, T.; Fernandez, I.; D’Amico, G.; Dukinfield, M.; Batista, S.; Tavora, B.; Serrels, B.; et al. Focal Adhesion Kinase (FAK) tyrosine 397E mutation restores the vascular leakage defect in endothelium-specific FAK-kinase dead mice. J. Pathol. 2017, 242, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.M.; Lim, S.T.; Lutu-Fuga, K.M.; Barnes, L.A.; Chen, X.L.; Gothert, J.R.; Shen, T.L.; Guan, J.L.; Schlaepfer, D.D.; Cheresh, D.A. Compensatory role for Pyk2 during angiogenesis in adult mice lacking endothelial cell FAK. J. Cell Biol. 2008, 181, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Jean, C.; Chen, X.L.; Nam, J.O.; Tancioni, I.; Uryu, S.; Lawson, C.; Ward, K.K.; Walsh, C.T.; Miller, N.L.; Ghassemian, M.; et al. Inhibition of endothelial FAK activity prevents tumor metastasis by enhancing barrier function. J. Cell Biol. 2014, 204, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, S.; Goel, S.; Kamoun, W.S.; Maru, Y.; Fukumura, D.; Duda, D.G.; Jain, R.K. Endothelial focal adhesion kinase mediates cancer cell homing to discrete regions of the lungs via E-selectin up-regulation. Proc. Natl. Acad. Sci. USA 2011, 108, 3725–3730. [Google Scholar] [CrossRef] [PubMed]
- Giannone, G.; Ronde, P.; Gaire, M.; Haiech, J.; Takeda, K. Calcium oscillations trigger focal adhesion disassembly in human U87 astrocytoma cells. J. Biol. Chem. 2002, 277, 26364–26371. [Google Scholar] [CrossRef] [PubMed]
- Giannone, G.; Ronde, P.; Gaire, M.; Beaudouin, J.; Haiech, J.; Ellenberg, J.; Takeda, K. Calcium rises locally trigger focal adhesion disassembly and enhance residency of focal adhesion kinase at focal adhesions. J. Biol. Chem. 2004, 279, 28715–28723. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Hjelmeland, A.B.; Keir, S.T.; Song, L.; Wickman, S.; Jackson, D.; Ohmori, O.; Bigner, D.D.; Friedman, H.S.; Rich, J.N. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Mol. Carcinog. 2007, 46, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.J.; LaFortune, T.; Honda, T.; Ohmori, O.; Hatakeyama, S.; Meyer, T.; Jackson, D.; de Groot, J.; Yung, W.K. Inhibition of both focal adhesion kinase and insulin-like growth factor-I receptor kinase suppresses glioma proliferation in vitro and in vivo. Mol. Cancer Ther. 2007, 6, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Halder, J.; Lin, Y.G.; Merritt, W.M.; Spannuth, W.A.; Nick, A.M.; Honda, T.; Kamat, A.A.; Han, L.Y.; Kim, T.J.; Lu, C.; et al. Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res. 2007, 67, 10976–10983. [Google Scholar] [CrossRef] [PubMed]
- Lietha, D.; Eck, M.J. Crystal structures of the FAK kinase in complex with TAE226 and related bis-anilino pyrimidine inhibitors reveal a helical DFG conformation. PLoS ONE 2008, 3, e3800. [Google Scholar] [CrossRef] [PubMed]
- Tanjoni, I.; Walsh, C.; Uryu, S.; Tomar, A.; Nam, J.O.; Mielgo, A.; Lim, S.T.; Liang, C.; Koenig, M.; Sun, C.; et al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol. Therapy 2010, 9, 764–777. [Google Scholar] [CrossRef]
- Roberts, W.G.; Ung, E.; Whalen, P.; Cooper, B.; Hulford, C.; Autry, C.; Richter, D.; Emerson, E.; Lin, J.; Kath, J.; et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008, 68, 1935–1944. [Google Scholar] [CrossRef] [PubMed]
- Stokes, J.B.; Adair, S.J.; Slack-Davis, J.K.; Walters, D.M.; Tilghman, R.W.; Hershey, E.D.; Lowrey, B.; Thomas, K.S.; Bouton, A.H.; Hwang, R.F.; et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol. Cancer Ther. 2011, 10, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Camidge, D.R.; Mileshkin, L.R.; Chen, E.X.; Hicks, R.J.; Rischin, D.; Fingert, H.; Pierce, K.J.; Xu, H.; Roberts, W.G.; et al. Safety, pharmacokinetic and pharmacodynamic phase I dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J. Clin. Oncol. 2012, 30, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Slack-Davis, J.K.; Martin, K.H.; Tilghman, R.W.; Iwanicki, M.; Ung, E.J.; Autry, C.; Luzzio, M.J.; Cooper, B.; Kath, J.C.; Roberts, W.G.; et al. Cellular characterization of a novel focal adhesion kinase inhibitor. J. Biol. Chem. 2007, 282, 14845–14852. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Gao, C.; Gao, X.; Zhang, D.H.; Kuan, S.F.; Burns, T.F.; Hu, J. Wnt/beta-Catenin Pathway Activation Mediates Adaptive Resistance to BRAF Inhibition in Colorectal Cancer. Mol. Cancer Ther. 2018, 17, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin beta1/FAK signaling. Cancer Cell 2015, 27, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Siddiquee, K.; Zhang, S.; Guida, W.C.; Blaskovich, M.A.; Greedy, B.; Lawrence, H.R.; Yip, M.L.; Jove, R.; McLaughlin, M.M.; Lawrence, N.J.; et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7391–7396. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Figel, S.; Ho, B.T.; Johnson, C.P.; Yemma, M.; Huang, G.; Zheng, M.; Nyberg, C.; Magis, A.; Ostrov, D.A.; et al. A small molecule focal adhesion kinase (FAK) inhibitor, targeting Y397 site: 1-(2-hydroxyethyl)-3,5,7-triaza-1-azoniatricyclo [3.3.1.1(3,7)] decane; bromide effectively inhibits FAK autophosphorylation activity and decreases cancer cell viability, clonogenicity and tumor growth in vivo. Carcinogenesis 2012, 33, 1004–1013. [Google Scholar] [PubMed]
- Heffler, M.; Golubovskaya, V.M.; Dunn, K.M.; Cance, W. Focal adhesion kinase autophosphorylation inhibition decreases colon cancer cell growth and enhances the efficacy of chemotherapy. Cancer Biol. Ther. 2013, 14, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Huang, G.; Ho, B.; Yemma, M.; Morrison, C.D.; Lee, J.; Eliceiri, B.P.; Cance, W.G. Pharmacologic blockade of FAK autophosphorylation decreases human glioblastoma tumor growth and synergizes with temozolomide. Mol. Cancer Ther. 2013, 12, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Medley, Q.G.; Buchbinder, E.G.; Tachibana, K.; Ngo, H.; Serra-Pages, C.; Streuli, M. Signaling between focal adhesion kinase and trio. J. Biol. Chem. 2003, 278, 13265–13270. [Google Scholar] [CrossRef] [PubMed]
- Yeo, M.G.; Partridge, M.A.; Ezratty, E.J.; Shen, Q.; Gundersen, G.G.; Marcantonio, E.E. Src SH2 arginine 175 is required for cell motility: Specific focal adhesion kinase targeting and focal adhesion assembly function. Mol. Cell. Biol. 2006, 26, 4399–4409. [Google Scholar] [CrossRef] [PubMed]
- Guinebault, C.; Payrastre, B.; Racaud-Sultan, C.; Mazarguil, H.; Breton, M.; Mauco, G.; Plantavid, M.; Chap, H. Integrin-dependent translocation of phosphoinositide 3-kinase to the cytoskeleton of thrombin-activated platelets involves specific interactions of p85 alpha with actin filaments and focal adhesion kinase. J. Cell Biol. 1995, 129, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, T.; Sakai, R.; Honda, H.; Ogawa, S.; Ueno, H.; Suzuki, T.; Aizawa, S.; Yazaki, Y.; Hirai, H. Requirements for localization of p130cas to focal adhesions. Mol. Cell. Biol. 1997, 17, 3884–3897. [Google Scholar] [CrossRef] [PubMed]
- Harte, M.T.; Hildebrand, J.D.; Burnham, M.R.; Bouton, A.H.; Parsons, J.T. P130Cas, a substrate associated with v-Src and v-Crk, localizes to focal adhesions and binds to focal adhesion kinase. J. Biol. Chem. 1996, 271, 13649–13655. [Google Scholar] [CrossRef] [PubMed]
- Donato, D.M.; Ryzhova, L.M.; Meenderink, L.M.; Kaverina, I.; Hanks, S.K. Dynamics and mechanism of p130Cas localization to focal adhesions. J. Biol. Chem. 2010, 285, 20769–20779. [Google Scholar] [CrossRef] [PubMed]
- Pratt, S.J.; Epple, H.; Ward, M.; Feng, Y.; Braga, V.M.; Longmore, G.D. The LIM protein Ajuba influences p130Cas localization and Rac1 activity during cell migration. J. Cell Biol. 2005, 168, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.H.; Mui, K.L.; Hsu, B.Y.; Liu, S.L.; Cretu, A.; Razinia, Z.; Xu, T.; Pure, E.; Assoian, R.K. A FAK-Cas-Rac-lamellipodin signaling module transduces extracellular matrix stiffness into mechanosensitive cell cycling. Sci. Signal. 2014, 7, ra57. [Google Scholar] [CrossRef] [PubMed]
- Carragher, N.O.; Westhoff, M.A.; Fincham, V.J.; Schaller, M.D.; Frame, M.C. A novel role for FAK as a protease-targeting adaptor protein: Regulation by p42 ERK and Src. Curr. Biol. CB 2003, 13, 1442–1450. [Google Scholar] [CrossRef]
- Kolli-Bouhafs, K.; Sick, E.; Noulet, F.; Gies, J.P.; De Mey, J.; Ronde, P. FAK competes for Src to promote migration against invasion in melanoma cells. Cell Death Dis. 2014, 5, e1379. [Google Scholar] [CrossRef] [PubMed]
- Alexander, N.R.; Branch, K.M.; Parekh, A.; Clark, E.S.; Iwueke, I.C.; Guelcher, S.A.; Weaver, A.M. Extracellular matrix rigidity promotes invadopodia activity. Curr. Biol. 2008, 18, 1295–1299. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; McNiven, M.A. Invasive matrix degradation at focal adhesions occurs via protease recruitment by a FAK-p130Cas complex. J. Cell Biol. 2012, 196, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Gan, B.; Yoo, Y.; Guan, J.L. FAK-mediated Src phosphorylation of endophilin A2 inhibits endocytosis of MT1-MMP and promotes ECM degradation. Dev. Cell 2005, 9, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Sandilands, E.; Serrels, B.; McEwan, D.G.; Morton, J.P.; Macagno, J.P.; McLeod, K.; Stevens, C.; Brunton, V.G.; Langdon, W.Y.; Vidal, M.; et al. Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat. Cell Biol. 2011, 14, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Schoenherr, C.; Byron, A.; Sandilands, E.; Paliashvili, K.; Baillie, G.S.; Garcia-Munoz, A.; Valacca, C.; Cecconi, F.; Serrels, B.; Frame, M.C. Ambra1 spatially regulates Src activity and Src/FAK-mediated cancer cell invasion via trafficking networks. eLife 2017, 6, e23172. [Google Scholar] [CrossRef] [PubMed]
- Randazzo, P.A.; Andrade, J.; Miura, K.; Brown, M.T.; Long, Y.Q.; Stauffer, S.; Roller, P.; Cooper, J.A. The Arf GTPase-activating protein ASAP1 regulates the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 2000, 97, 4011–4016. [Google Scholar] [CrossRef] [PubMed]
- Carmon, K.S.; Gong, X.; Yi, J.; Thomas, A.; Liu, Q. RSPO-LGR4 functions via IQGAP1 to potentiate Wnt signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E1221–E1229. [Google Scholar] [CrossRef] [PubMed]
- Iwanicki, M.P.; Vomastek, T.; Tilghman, R.W.; Martin, K.H.; Banerjee, J.; Wedegaertner, P.B.; Parsons, J.T. FAK, PDZ-RhoGEF and ROCKII cooperate to regulate adhesion movement and trailing-edge retraction in fibroblasts. J. Cell Sci. 2008, 121, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Tsai, L.H. Cdk5 phosphorylation of FAK regulates centrosome-associated miocrotubules and neuronal migration. Cell Cycle 2004, 3, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Tomar, A.; Lawson, C.; Ghassemian, M.; Schlaepfer, D.D. Cortactin as a target for FAK in the regulation of focal adhesion dynamics. PLoS ONE 2012, 7, e44041. [Google Scholar] [CrossRef] [PubMed]
- Genna, A.; Lapetina, S.; Lukic, N.; Twafra, S.; Meirson, T.; Sharma, V.P.; Condeelis, J.S.; Gil-Henn, H. Pyk2 and FAK differentially regulate invadopodia formation and function in breast cancer cells. J. Cell Biol. 2018, 217, 375–395. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.T.; Cortesio, C.L.; Huttenlocher, A. FAK alters invadopodia and focal adhesion composition and dynamics to regulate breast cancer invasion. J. Cell Biol. 2009, 185, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.R.; Du, Q.S.; Huang, Y.Z.; Ao, S.Z.; Mei, L.; Xiong, W.C. Regulation of CDC42 GTPase by proline-rich tyrosine kinase 2 interacting with PSGAP, a novel pleckstrin homology and Src homology 3 domain containing rhoGAP protein. J. Cell Biol. 2001, 152, 971–984. [Google Scholar] [CrossRef] [PubMed]
- Chikumi, H.; Fukuhara, S.; Gutkind, J.S. Regulation of G protein-linked guanine nucleotide exchange factors for Rho, PDZ-RhoGEF and LARG by tyrosine phosphorylation: Evidence of a role for focal adhesion kinase. J. Biol. Chem. 2002, 277, 12463–12473. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Sanada, K.; Samuels, B.A.; Shih, H.; Tsai, L.H. Serine 732 phosphorylation of FAK by Cdk5 is important for microtubule organization, nuclear movement and neuronal migration. Cell 2003, 114, 469–482. [Google Scholar] [CrossRef]
- Rea, K.; Sensi, M.; Anichini, A.; Canevari, S.; Tomassetti, A. EGFR/MEK/ERK/CDK5-dependent integrin-independent FAK phosphorylated on serine 732 contributes to microtubule depolymerization and mitosis in tumor cells. Cell Death Dis. 2013, 4, e815. [Google Scholar] [CrossRef] [PubMed]
- Le Boeuf, F.; Houle, F.; Sussman, M.; Huot, J. Phosphorylation of focal adhesion kinase (FAK) on Ser732 is induced by rho-dependent kinase and is essential for proline-rich tyrosine kinase-2-mediated phosphorylation of FAK on Tyr407 in response to vascular endothelial growth factor. Mol. Biol. Cell 2006, 17, 3508–3520. [Google Scholar] [CrossRef] [PubMed]
- Park, A.Y.; Shen, T.L.; Chien, S.; Guan, J.L. Role of focal adhesion kinase Ser-732 phosphorylation in centrosome function during mitosis. J. Biol. Chem. 2009, 284, 9418–9425. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Zhao, X.; Sun, S.; Luo, M.; Guan, J.L. Function of focal adhesion kinase scaffolding to mediate endophilin A2 phosphorylation promotes epithelial-mesenchymal transition and mammary cancer stem cell activities in vivo. J. Biol. Chem. 2013, 288, 3322–3333. [Google Scholar] [CrossRef] [PubMed]
- Ball, L.J.; Kuhne, R.; Schneider-Mergener, J.; Oschkinat, H. Recognition of proline-rich motifs by protein-protein-interaction domains. Angew. Chem. Int. Ed. Engl. 2005, 44, 2852–2869. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.D.; Parsons, J.T. Pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol. Cell. Biol. 1995, 15, 2635–2645. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, D.D.; Hanks, S.K.; Hunter, T.; van der Geer, P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature 1994, 372, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Appeddu, P.A.; Parsons, J.T.; Hildebrand, J.D.; Schaller, M.D.; Guan, J.L. Interaction of focal adhesion kinase with cytoskeletal protein talin. J. Biol. Chem. 1995, 270, 16995–16999. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, K.; Urano, T.; Fujita, H.; Ohashi, Y.; Kamiguchi, K.; Iwata, S.; Hirai, H.; Morimoto, C. Tyrosine phosphorylation of Crk-associated substrates by focal adhesion kinase. A putative mechanism for the integrin-mediated tyrosine phosphorylation of Crk-associated substrates. J. Biol. Chem. 1997, 272, 29083–29090. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, D.D.; Jones, K.C.; Hunter, T. Multiple Grb2-mediated integrin-stimulated signaling pathways to ERK2/mitogen-activated protein kinase: Summation of both c-Src- and focal adhesion kinase-initiated tyrosine phosphorylation events. Mol. Cell. Biol. 1998, 18, 2571–2585. [Google Scholar] [CrossRef] [PubMed]
- Garces, C.A.; Kurenova, E.V.; Golubovskaya, V.M.; Cance, W.G. Vascular endothelial growth factor receptor-3 and focal adhesion kinase bind and suppress apoptosis in breast cancer cells. Cancer Res. 2006, 66, 1446–1454. [Google Scholar] [CrossRef] [PubMed]
- Arold, S.T.; Hoellerer, M.K.; Noble, M.E. The structural basis of localization and signaling by the focal adhesion targeting domain. Structure 2002, 10, 319–327. [Google Scholar] [CrossRef]
- Gao, G.; Prutzman, K.C.; King, M.L.; Scheswohl, D.M.; DeRose, E.F.; London, R.E.; Schaller, M.D.; Campbell, S.L. NMR solution structure of the focal adhesion targeting domain of focal adhesion kinase in complex with a paxillin LD peptide: Evidence for a two-site binding model. J. Biol. Chem. 2004, 279, 8441–8451. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, I.; Vuori, K.; Liddington, R.C. The focal adhesion targeting (FAT) region of focal adhesion kinase is a four-helix bundle that binds paxillin. Nat. Struct. Biol. 2002, 9, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.; Lim, S.T.; Uryu, S.; Chen, X.L.; Calderwood, D.A.; Schlaepfer, D.D. FAK promotes recruitment of talin to nascent adhesions to control cell motility. J. Cell Biol. 2012, 196, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Hagel, M.; George, E.L.; Kim, A.; Tamimi, R.; Opitz, S.L.; Turner, C.E.; Imamoto, A.; Thomas, S.M. The adaptor protein paxillin is essential for normal development in the mouse and is a critical transducer of fibronectin signaling. Mol. Cell. Biol. 2002, 22, 901–915. [Google Scholar] [CrossRef] [PubMed]
- Scheswohl, D.M.; Harrell, J.R.; Rajfur, Z.; Gao, G.; Campbell, S.L.; Schaller, M.D. Multiple paxillin binding sites regulate FAK function. J. Mol. Signal. 2008, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Nishiya, N.; Tachibana, K.; Shibanuma, M.; Mashimo, J.I.; Nose, K. Hic-5-reduced cell spreading on fibronectin: Competitive effects between paxillin and Hic-5 through interaction with focal adhesion kinase. Mol. Cell. Biol. 2001, 21, 5332–5345. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Du, X.; Vass, W.C.; Papageorge, A.G.; Lowy, D.R.; Qian, X. Full activity of the deleted in liver cancer 1 (DLC1) tumor suppressor depends on an LD-like motif that binds talin and focal adhesion kinase (FAK). Proc. Natl. Acad. Sci. USA 2011, 108, 17129–17134. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.F.; Wei, L.; Zhou, X.; Lu, Z.Y.; Francis, K.; Hu, X.Y.; Liu, Y.; Xiong, W.C.; Zhang, X.; Banik, N.L.; et al. Formation of Kv2.1-FAK complex as a mechanism of FAK activation, cell polarization and enhanced motility. J. Cell. Physiol. 2008, 217, 544–557. [Google Scholar] [CrossRef] [PubMed]
- Petridou, N.I.; Skourides, P.A. FAK transduces extracellular forces that orient the mitotic spindle and control tissue morphogenesis. Nat. Commun. 2014, 5, 5240. [Google Scholar] [CrossRef] [PubMed]
- Katz, B.Z.; Romer, L.; Miyamoto, S.; Volberg, T.; Matsumoto, K.; Cukierman, E.; Geiger, B.; Yamada, K.M. Targeting membrane-localized focal adhesion kinase to focal adhesions: Roles of tyrosine phosphorylation and SRC family kinases. J. Biol. Chem. 2003, 278, 29115–29120. [Google Scholar] [CrossRef] [PubMed]
- Kadare, G.; Gervasi, N.; Brami-Cherrier, K.; Blockus, H.; El Messari, S.; Arold, S.T.; Girault, J.A. Conformational dynamics of the focal adhesion targeting domain control specific functions of focal adhesion kinase in cells. J. Biol. Chem. 2015, 290, 478–491. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Sinnett-Smith, J.; Rozengurt, E. Differential FAK phosphorylation at Ser-910, Ser-843 and Tyr-397 induced by angiotensin, I.I.; LPA and EGF in intestinal epithelial cells. Cell. Signal. 2007, 19, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Xia, Y.; Hawke, D.; Halle, M.; Tremblay, M.L.; Gao, X.; Zhou, X.Z.; Aldape, K.; Cobb, M.H.; Xie, K.; et al. FAK phosphorylation by ERK primes ras-induced tyrosine dephosphorylation of FAK mediated by PIN1 and PTP-PEST. Mol. Cell 2009, 35, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Lin, H.; Nie, Z.; Wu, J.; Canete-Soler, R.; Schlaepfer, W.W.; Schlaepfer, D.D. Direct interaction of focal adhesion kinase with p190RhoGEF. J. Biol. Chem. 2003, 278, 24865–24873. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jung, I.D.; Chang, W.K.; Park, C.G.; Cho, D.Y.; Shin, E.Y.; Seo, D.W.; Kim, Y.K.; Lee, H.W.; Han, J.W.; et al. P85 beta-PIX is required for cell motility through phosphorylations of focal adhesion kinase and p38 MAP kinase. Exp. Cell Res. 2005, 307, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Brunton, V.G.; Avizienyte, E.; Fincham, V.J.; Serrels, B.; Metcalf, C.A., 3rd; Sawyer, T.K.; Frame, M.C. Identification of Src-specific phosphorylation site on focal adhesion kinase: Dissection of the role of Src SH2 and catalytic functions and their consequences for tumor cell behavior. Cancer Res. 2005, 65, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.K.; Mikolon, D.; Molina, J.E.; Hsia, D.A.; Hanson, D.A.; Chi, A.; Lim, S.T.; Bernard-Trifilo, J.A.; Ilic, D.; Stupack, D.G.; et al. Intrinsic FAK activity and Y925 phosphorylation facilitate an angiogenic switch in tumors. Oncogene 2006, 25, 5969–5984. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.Y.; Sun, G.; Schlaepfer, D.D.; Pallen, C.J. Grb2 promotes integrin-induced focal adhesion kinase (FAK) autophosphorylation and directs the phosphorylation of protein tyrosine phosphatase alpha by the Src-FAK kinase complex. Mol. Cell. Biol. 2014, 34, 348–361. [Google Scholar] [CrossRef] [PubMed]
- Hunger-Glaser, I.; Salazar, E.P.; Sinnett-Smith, J.; Rozengurt, E. Bombesin, lysophosphatidic acid and epidermal growth factor rapidly stimulate focal adhesion kinase phosphorylation at Ser-910: Requirement for ERK activation. J. Biol. Chem. 2003, 278, 22631–22643. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.S.; Manser, E.; Loo, T.H.; Lim, L. Coupling of PAK-interacting exchange factor PIX to GIT1 promotes focal complex disassembly. Mol. Cell. Biol. 2000, 20, 6354–6363. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Yu, L.; Fan, J.; Rui, Z.; Hua, Z.; Zhang, Z.; Zhang, N.; Yin, G. Phosphorylation of GIT1 tyrosine 321 is required for association with FAK at focal adhesions and for PDGF-activated migration of osteoblasts. Mol. Cell. Biochem. 2012, 365, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.A.; Deakin, N.O.; Turner, C.E. Paxillin-kinase-linker tyrosine phosphorylation regulates directional cell migration. Mol. Biol. Cell 2009, 20, 4706–4719. [Google Scholar] [CrossRef] [PubMed]
- Schoenherr, C.; Serrels, B.; Proby, C.; Cunningham, D.L.; Findlay, J.E.; Baillie, G.S.; Heath, J.K.; Frame, M.C. Eps8 controls Src- and FAK-dependent phenotypes in squamous carcinoma cells. J. Cell Sci. 2014, 127, 5303–5316. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Schaller, M.D. Focal adhesion targeting: The critical determinant of FAK regulation and substrate phosphorylation. Mol. Biol. Cell 1999, 10, 2507–2518. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.D.; Borgman, C.A.; Parsons, J.T. Autonomous expression of a noncatalytic domain of the focal adhesion-associated protein tyrosine kinase pp125FAK. Mol. Cell. Biol. 1993, 13, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Tamura, M.; Pankov, R.; Danen, E.H.; Takino, T.; Matsumoto, K.; Yamada, K.M. Shc and FAK differentially regulate cell motility and directionality modulated by PTEN. J. Cell. Biol. 1999, 146, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Sieg, D.J.; Hauck, C.R.; Schlaepfer, D.D. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J. Cell Sci. 1999, 112, 2677–2691. [Google Scholar] [PubMed]
- Zak, T.J.; Koshman, Y.E.; Samarel, A.M.; Robia, S.L. Regulation of Focal Adhesion Kinase through a Direct Interaction with an Endogenous Inhibitor. Biochemistry 2017, 56, 4722–4731. [Google Scholar] [CrossRef] [PubMed]
- Deramaudt, T.B.; Dujardin, D.; Noulet, F.; Martin, S.; Vauchelles, R.; Takeda, K.; Ronde, P. Altering FAK-paxillin interactions reduces adhesion, migration and invasion processes. PLoS ONE 2014, 9, e92059. [Google Scholar] [CrossRef] [PubMed]
- Kurenova, E.V.; Hunt, D.L.; He, D.; Magis, A.T.; Ostrov, D.A.; Cance, W.G. Small molecule chloropyramine hydrochloride (C4) targets the binding site of focal adhesion kinase and vascular endothelial growth factor receptor 3 and suppresses breast cancer growth in vivo. J. Med. Chem. 2009, 52, 4716–4724. [Google Scholar] [CrossRef] [PubMed]
- Gogate, P.N.; Ethirajan, M.; Kurenova, E.V.; Magis, A.T.; Pandey, R.K.; Cance, W.G. Design, synthesis and biological evaluation of novel FAK scaffold inhibitors targeting the FAK-VEGFR3 protein-protein interaction. Eur. J. Med. Chem. 2014, 80, 154–166. [Google Scholar] [CrossRef] [PubMed]


| Interactions | References |
| Integrin, PIP2, Tetraspanin | [27,37,39,41,42,50,51] |
| PDGFR, EGFR, IGFR, RET, c-MET | [31,44,45,46,47] |
| Cadherin, Ezrin, Dynamin | [48,49,52] |
| Arp2/3, N-WASP, RACK1, ETK | [53,54,55,56,57] |
| PIAS1, RIP, p53, Mdm2, Nanog | [58,59,60,61,62] |

| Interactions | References |
| Src, PI3K, PLCγ, PTEN | [86,95,96,97,98,99,100] |
| Grb7, SOCS3, p120RasGAP | [71,101,102,103] |
| Catenin, p190RhoGAP | [48,71] |
| PDZ-RhoGEF, FIP200 | [104,105] |

| Interactions | References |
| Paxillin, Talin, p130Cas | [129,155,157,158,160,162] |
| Grb2, PI3K, VEGFR3, DLT1, Kv2.1 | [127,156,159,160,168,169] |
| PTPα, ERK, PIN1, p190RhoGEF | [93,173,174,175,176] |
| GIT1, Eps8 | [177,178,179] |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mousson, A.; Sick, E.; Carl, P.; Dujardin, D.; De Mey, J.; Rondé, P. Targeting Focal Adhesion Kinase Using Inhibitors of Protein-Protein Interactions. Cancers 2018, 10, 278. https://doi.org/10.3390/cancers10090278
Mousson A, Sick E, Carl P, Dujardin D, De Mey J, Rondé P. Targeting Focal Adhesion Kinase Using Inhibitors of Protein-Protein Interactions. Cancers. 2018; 10(9):278. https://doi.org/10.3390/cancers10090278
Chicago/Turabian StyleMousson, Antoine, Emilie Sick, Philippe Carl, Denis Dujardin, Jan De Mey, and Philippe Rondé. 2018. "Targeting Focal Adhesion Kinase Using Inhibitors of Protein-Protein Interactions" Cancers 10, no. 9: 278. https://doi.org/10.3390/cancers10090278
APA StyleMousson, A., Sick, E., Carl, P., Dujardin, D., De Mey, J., & Rondé, P. (2018). Targeting Focal Adhesion Kinase Using Inhibitors of Protein-Protein Interactions. Cancers, 10(9), 278. https://doi.org/10.3390/cancers10090278