Dynamics of p14ARF and Focal Adhesion Kinase-Mediated Autophagy in Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
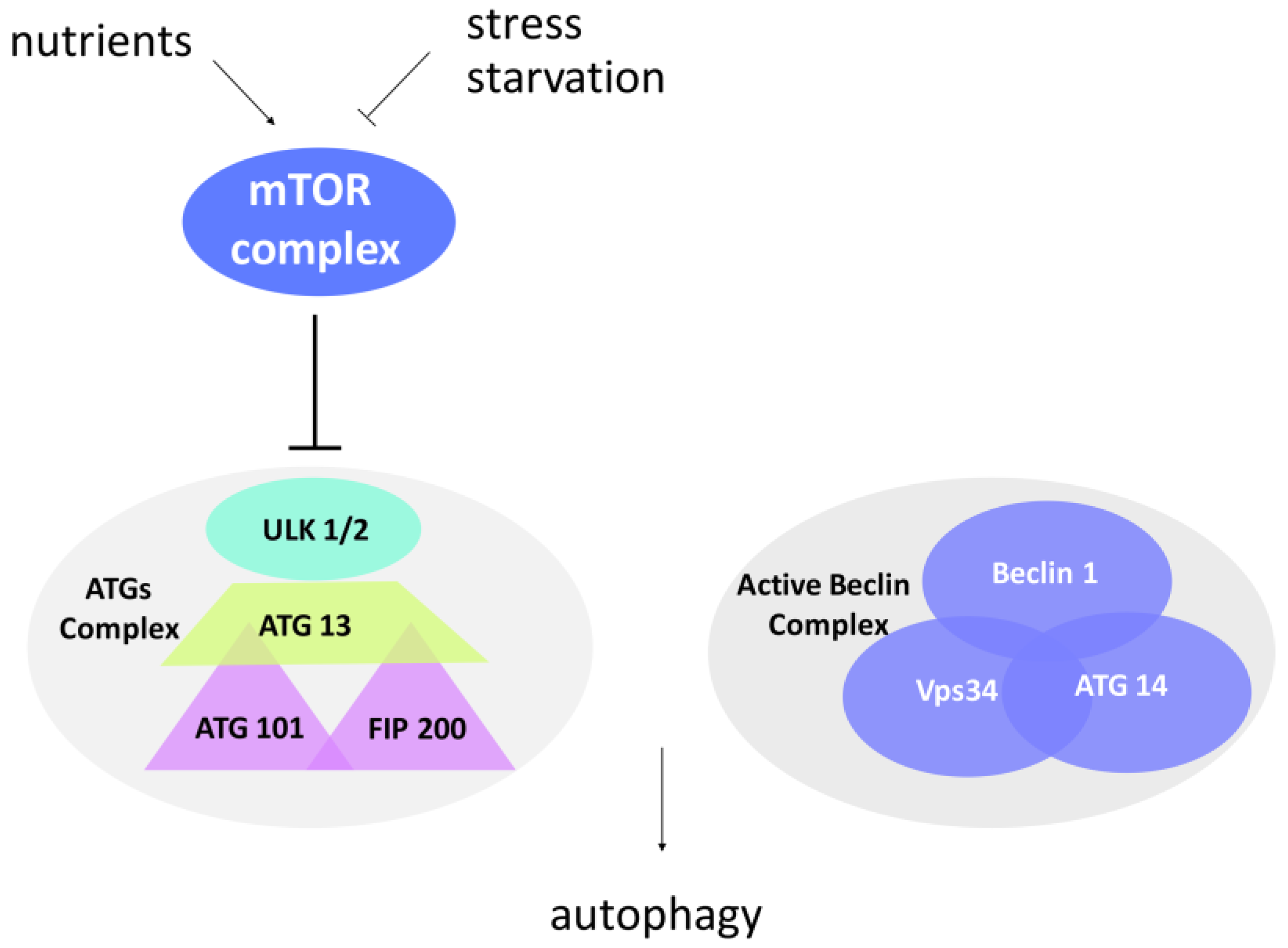
2. Autophagy
Molecular Control of Autophagosome Formation
3. Regulation of Autophagy
3.1. ARF Role in Autophagy: A Tale of Two Isoforms
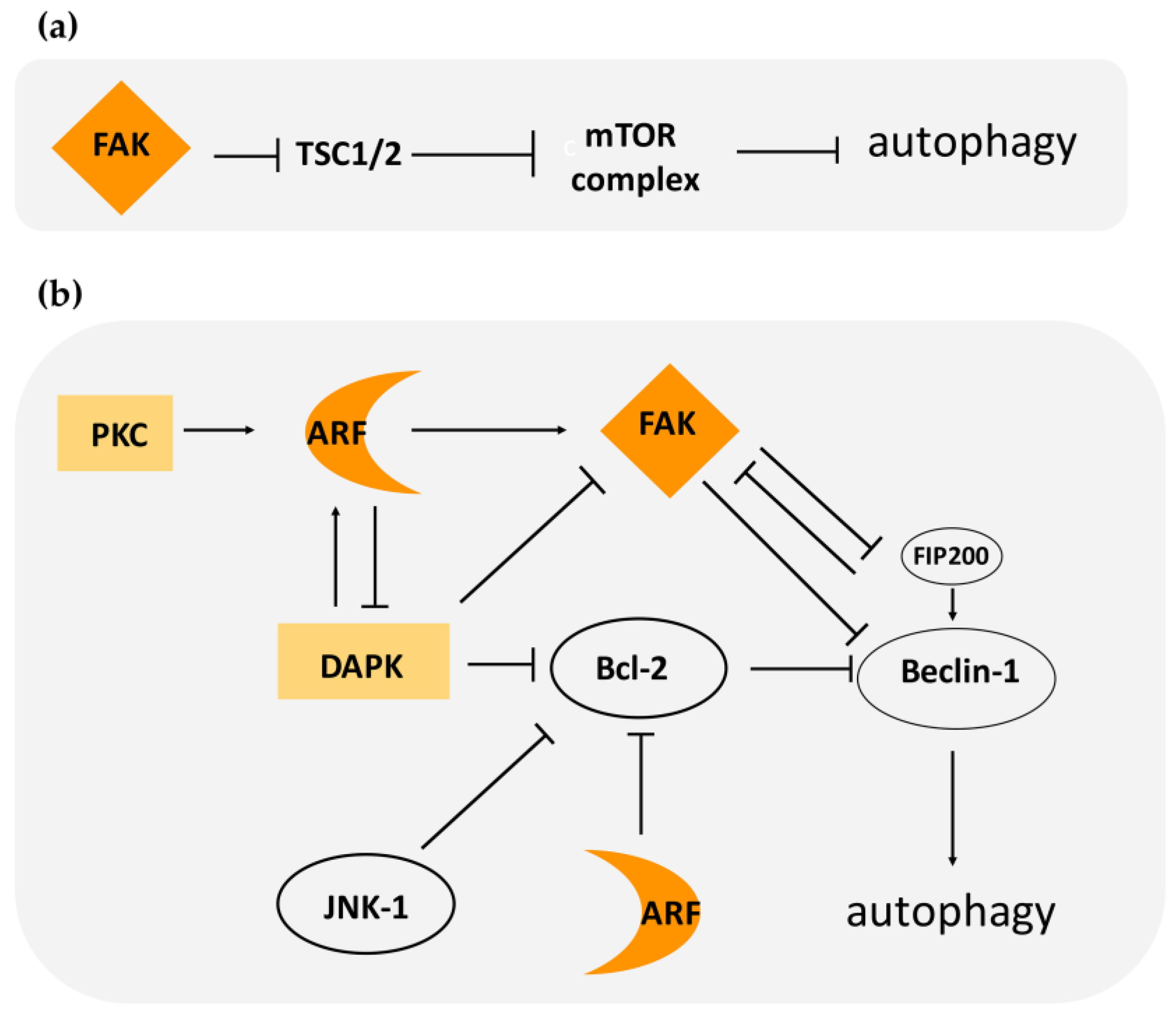
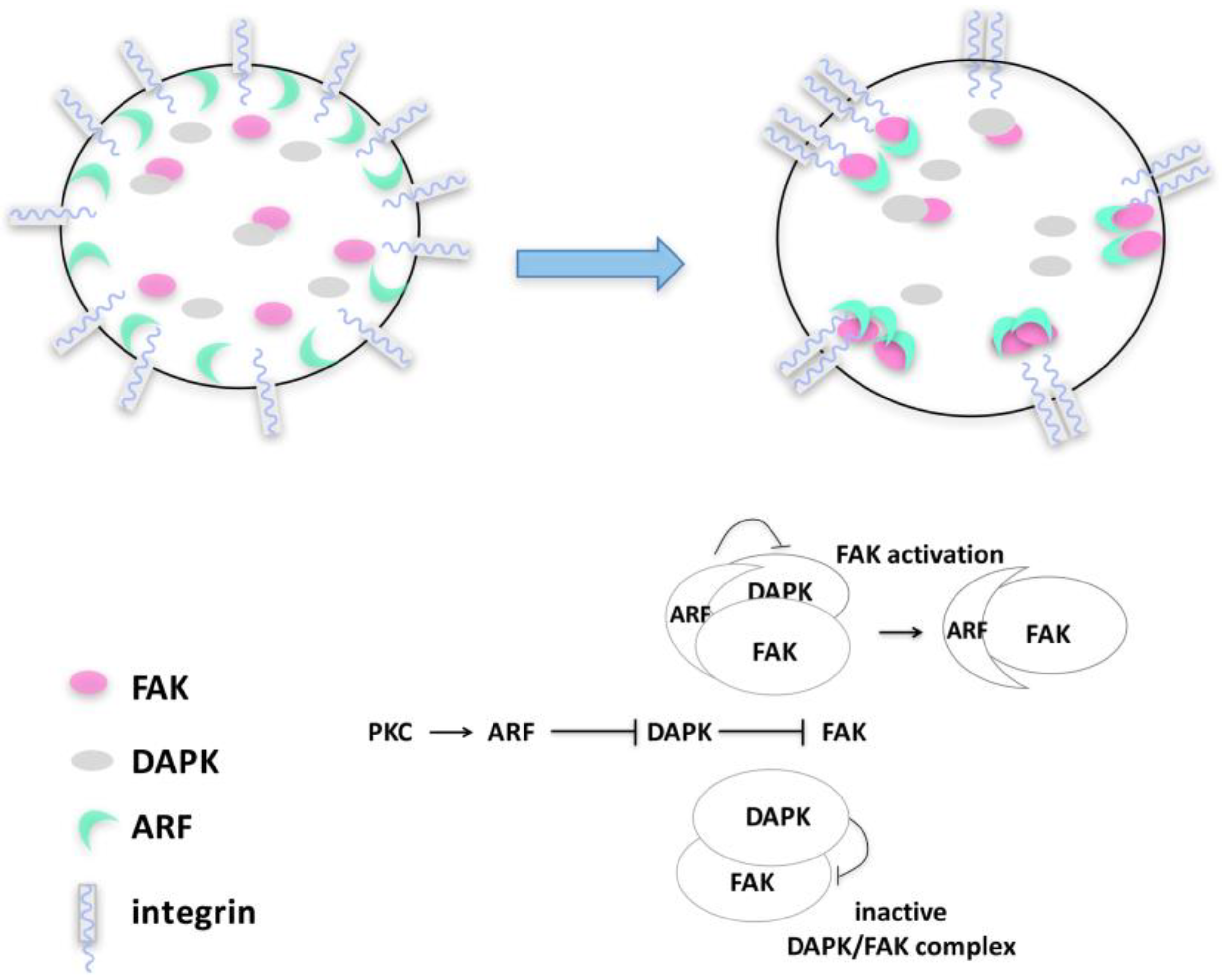
3.2. FAK Control of Autophagy: Regulation in Time and Space
4. ARF–FAK Cross-Talk
5. Conclusions: Autophagy and Cancer Evolution
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ozenne, P.; Eymin, B.; Brambilla, E.; Gazzeri, S. The arf tumor suppressor: Structure, functions and status in cancer. Int. J. Cancer 2010, 127, 2239–2247. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Tumor surveillance via the arf-p53 pathway. Genes Dev. 1998, 12, 2984–2991. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Ink4-arf locus in cancer and aging. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Pollice, A.; Vivo, M.; La Mantia, G. The promiscuity of arf interactions with the proteasome. FEBS Lett. 2008, 582, 3257–3262. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Bertwistle, D.; W, D.E.N.B.; Kuo, M.L.; Sugimoto, M.; Tago, K.; Williams, R.T.; Zindy, F.; Roussel, M.F. P53-dependent and -independent functions of the arf tumor suppressor. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Vivo, M.; Calogero, R.A.; Sansone, F.; Calabro, V.; Parisi, T.; Borrelli, L.; Saviozzi, S.; La Mantia, G. The human tumor suppressor arf interacts with spinophilin/neurabin II, a type 1 protein-phosphatase-binding protein. J. Biol. Chem. 2001, 276, 14161–14169. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, T.; Bodner, S.; van de Kamp, E.; Randle, D.H.; Sherr, C.J. Tumor spectrum in arf-deficient mice. Cancer Res. 1999, 59, 2217–2222. [Google Scholar] [PubMed]
- Ruas, M.; Peters, G. The p16ink4a/cdkn2a tumor suppressor and its relatives. Biochim. Biophys. Acta 1998, 1378, F115–F177. [Google Scholar] [CrossRef]
- Kamijo, T.; Weber, J.D.; Zambetti, G.; Zindy, F.; Roussel, M.F.; Sherr, C.J. Functional and physical interactions of the arf tumor suppressor with p53 and mdm2. Proc. Natl. Acad. Sci. USA 1998, 95, 8292–8297. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Shan, J.; Zhu, W.G.; Qin, J.; Gu, W. Transcription-independent arf regulation in oncogenic stress-mediated p53 responses. Nature 2010, 464, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Humbey, O.; Pimkina, J.; Zilfou, J.T.; Jarnik, M.; Dominguez-Brauer, C.; Burgess, D.J.; Eischen, C.M.; Murphy, M.E. The arf tumor suppressor can promote the progression of some tumors. Cancer Res. 2008, 68, 9608–9613. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Aguilera, A.; Sanchez-Beato, M.; Garcia, J.F.; Prieto, I.; Pollan, M.; Piris, M.A. P14(ARF) nuclear overexpression in aggressive b-cell lymphomas is a sensor of malfunction of the common tumor suppressor pathways. Blood 2002, 99, 1411–1418. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Carracedo, A.; Lin, H.K.; Koutcher, J.A.; Behrendt, N.; Egia, A.; Alimonti, A.; Carver, B.S.; Gerald, W.; Teruya-Feldstein, J.; et al. Differential p53-independent outcomes of p19(ARF) loss in oncogenesis. Sci. Signal. 2009, 2, ra44. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Liu, S.; Lu, W.; Yang, Q.; Williams, K.D.; Binhazim, A.A.; Carver, B.S.; Matusik, R.J.; Chen, Z. Slug regulates e-cadherin repression via p19arf in prostate tumorigenesis. Mol. Oncol. 2014, 8, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Lu, W.; Liu, S.; Yang, Q.; Goodwin, J.S.; Sathyanarayana, S.A.; Pratap, S.; Chen, Z. Mmp7 interacts with arf in nucleus to potentiate tumor microenvironments for prostate cancer progression in vivo. Oncotarget 2016, 7, 47609–47619. [Google Scholar] [CrossRef] [PubMed]
- Ferru, A.; Fromont, G.; Gibelin, H.; Guilhot, J.; Savagner, F.; Tourani, J.M.; Kraimps, J.L.; Larsen, C.J.; Karayan-Tapon, L. The status of cdkn2a alpha (p16ink4a) and beta (p14arf) transcripts in thyroid tumour progression. Br. J. Cancer 2006, 95, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Fontana, R.; Guidone, D.; Sangermano, F.; Calabro, V.; Pollice, A.; La Mantia, G.; Vivo, M. Pkc dependent p14arf phosphorylation on threonine 8 drives cell proliferation. Sci. Rep. 2018, 8, 7056. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Zhao, X.; Sun, S.; Luo, M.; Guan, J.L. Function of focal adhesion kinase scaffolding to mediate endophilin a2 phosphorylation promotes epithelial-mesenchymal transition and mammary cancer stem cell activities in vivo. J. Biol. Chem. 2013, 288, 3322–3333. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.Q.; Liu, X.F.; Yao, L.; Chen, C.Q.; Gu, Z.D.; Ni, P.H.; Zheng, X.M.; Fan, Q.S. Somatic mutational analysis of fak in breast cancer: A novel gain-of-function mutation due to deletion of exon 33. Biochem. Biophys. Res. Commun. 2014, 443, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Goode, E.L.; Chenevix-Trench, G.; Song, H.; Ramus, S.J.; Notaridou, M.; Lawrenson, K.; Widschwendter, M.; Vierkant, R.A.; Larson, M.C.; Kjaer, S.K.; et al. A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat. Genet. 2010, 42, 874–879. [Google Scholar] [CrossRef] [PubMed]
- Vivo, M.; Fontana, R.; Ranieri, M.; Capasso, G.; Angrisano, T.; Pollice, A.; Calabro, V.; La Mantia, G. P14arf interacts with the focal adhesion kinase and protects cells from anoikis. Oncogene 2017, 36, 4913–4928. [Google Scholar] [CrossRef] [PubMed]
- Balaburski, G.M.; Hontz, R.D.; Murphy, M.E. P53 and arf: Unexpected players in autophagy. Trends Cell Biol. 2010, 20, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Pimkina, J.; Humbey, O.; Zilfou, J.T.; Jarnik, M.; Murphy, M.E. Arf induces autophagy by virtue of interaction with bcl-xl. J. Biol. Chem. 2009, 284, 2803–2810. [Google Scholar] [CrossRef] [PubMed]
- Pimkina, J.; Murphy, M.E. Arf, autophagy and tumor suppression. Autophagy 2009, 5, 397–399. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Melendez, A.; Talloczy, Z.; Seaman, M.; Eskelinen, E.L.; Hall, D.H.; Levine, B. Autophagy genes are essential for dauer development and life-span extension in c. Elegans. Science 2003, 301, 1387–1391. [Google Scholar] [CrossRef] [PubMed]
- Abada, A.; Elazar, Z. Getting ready for building: Signaling and autophagosome biogenesis. EMBO Rep. 2014, 15, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Gwangwa, M.V.; Joubert, A.M.; Visagie, M.H. Crosstalk between the warburg effect, redox regulation and autophagy induction in tumourigenesis. Cell. Mol. Biol. Lett. 2018, 23, 20. [Google Scholar] [CrossRef] [PubMed]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojkowiak, J.; Cornwell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by tumor microenvironment drives local invasion. Cancer Res. 2012, 73, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. Mtor regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Schulman, B.A. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat. Struct. Mol. Biol. 2014, 21, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Molino, D.; Nascimbeni, A.C.; Giordano, F.; Codogno, P.; Morel, E. Er-driven membrane contact sites: Evolutionary conserved machineries for stress response and autophagy regulation? Commun. Integr. Biol. 2017, 10, e1401699. [Google Scholar] [CrossRef] [PubMed]
- Shpilka, T.; Welter, E.; Borovsky, N.; Amar, N.; Mari, M.; Reggiori, F.; Elazar, Z. Lipid droplets and their component triglycerides and steryl esters regulate autophagosome biogenesis. EMBO J. 2015, 34, 2117–2131. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, C.; Yeo, S.; Liang, C.C.; Okamoto, T.; Sun, S.; Wen, J.; Guan, J.L. Distinct roles of autophagy-dependent and -independent functions of fip200 revealed by generation and analysis of a mutant knock-in mouse model. Genes Dev. 2016, 30, 856–869. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Wei, S.; Gan, B.; Peng, X.; Zou, W.; Guan, J.L. Suppression of autophagy by fip200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011, 25, 1510–1527. [Google Scholar] [CrossRef] [PubMed]
- Lazova, R.; Camp, R.L.; Klump, V.; Siddiqui, S.F.; Amaravadi, R.K.; Pawelek, J.M. Punctate lc3b expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin. Cancer Res. 2012, 18, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Bjorkoy, G.; Lamark, T.; Pankiv, S.; Overvatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/sqstm1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [PubMed]
- Seibenhener, M.L.; Geetha, T.; Wooten, M.W. Sequestosome 1/p62—More than just a scaffold. FEBS Lett. 2007, 581, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Yamada, E.; Singh, R. Mapping autophagy on to your metabolic radar. Diabetes 2012, 61, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Frohlich, L.F. P53-mediated molecular control of autophagy in tumor cells. Biomolecules 2018. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; Ryan, K.M. Dram links autophagy to p53 and programmed cell death. Autophagy 2007, 3, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mtor pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Budina-Kolomets, A.; Hontz, R.D.; Pimkina, J.; Murphy, M.E. A conserved domain in exon 2 coding for the human and murine arf tumor suppressor protein is required for autophagy induction. Autophagy 2013, 9, 1553–1565. [Google Scholar] [CrossRef] [PubMed]
- Levin-Salomon, V.; Bialik, S.; Kimchi, A. Dap-kinase and autophagy. Apoptosis 2014, 19, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Zalckvar, E.; Berissi, H.; Eisenstein, M.; Kimchi, A. Phosphorylation of beclin 1 by dap-kinase promotes autophagy by weakening its interactions with BCL-2 and BCL-xl. Autophagy 2009, 5, 720–722. [Google Scholar] [CrossRef] [PubMed]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. Dap-kinase-mediated phosphorylation on the bh3 domain of beclin 1 promotes dissociation of beclin 1 from bcl-xl and induction of autophagy. EMBO Rep. 2009, 10, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.; Ash, D.; Shaha, C. Beclin-1-p53 interaction is crucial for cell fate determination in embryonal carcinoma cells. J. Cell. Mol. Med. 2014, 18, 2275–2286. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Shen, S.; Ruckenstuhl, C.; Bauer, M.A.; Marino, G.; Galluzzi, L.; Criollo, A.; Michaud, M.; Maiuri, M.C.; Chano, T.; et al. P53 inhibits autophagy by interacting with the human ortholog of yeast ATG17, RB1CC1/FIP200. Cell Cycle 2011, 10, 2763–2769. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. Tigar, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Reef, S.; Kimchi, A. A smarf way to die: A novel short isoform of p19arf is linked to autophagic cell death. Autophagy 2006, 2, 328–330. [Google Scholar] [CrossRef] [PubMed]
- Reef, S.; Shifman, O.; Oren, M.; Kimchi, A. The autophagic inducer smarf interacts with and is stabilized by the mitochondrial p32 protein. Oncogene 2007, 26, 6677–6683. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.M.; Gu, W. P53-dependent and p53-independent activation of autophagy by ARF. Cancer Res. 2008, 68, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Reef, S.; Kimchi, A. Nucleolar p19arf, unlike mitochondrial smarf, is incapable of inducing p53-independent autophagy. Autophagy 2008, 4, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Grenier, K.; Kontogiannea, M.; Fon, E.A. Short mitochondrial arf triggers parkin/pink1-dependent mitophagy. J. Biol. Chem. 2014, 289, 29519–29530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Kuang, H.; Chen, C.; Yan, J.; Do-Umehara, H.C.; Liu, X.Y.; Dada, L.; Ridge, K.M.; Chandel, N.S.; Liu, J. The kinase jnk2 promotes stress-induced mitophagy by targeting the small mitochondrial form of the tumor suppressor arf for degradation. Nat. Immunol. 2015, 16, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Le Toumelin, G.; Criollo, A.; Rain, J.C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between BCL-x(l) and a BH3-like domain in beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef] [PubMed]
- Vivo, M.; Ranieri, M.; Sansone, F.; Santoriello, C.; Calogero, R.A.; Calabro, V.; Pollice, A.; La Mantia, G. Mimicking p14arf phosphorylation influences its ability to restrain cell proliferation. PLoS ONE 2013, 8, e53631. [Google Scholar] [CrossRef]
- Debnath, J. Detachment-induced autophagy in three-dimensional epithelial cell cultures. Methods Enzymol. 2009, 452, 423–439. [Google Scholar] [PubMed]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Kadandale, P.; Kiger, A.A. Role of selective autophagy in cellular remodeling: “Self-eating” into shape. Autophagy 2010, 6, 1194–1195. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, M.N.; Mowers, E.E.; Macleod, K.F. Autophagic degradation of focal adhesions underlies metastatic cancer dissemination. Mol. Cell. Oncol. 2017, 4, e1198299. [Google Scholar] [CrossRef] [PubMed]
- Gan, B.; Yoo, Y.; Guan, J.L. Association of focal adhesion kinase with tuberous sclerosis complex 2 in the regulation of s6 kinase activation and cell growth. J. Biol. Chem. 2006, 281, 37321–37329. [Google Scholar] [CrossRef] [PubMed]
- Frame, M.C.; Patel, H.; Serrels, B.; Lietha, D.; Eck, M.J. The ferm domain: Organizing the structure and function of fak. Nat. Rev. Mol. Cell Biol. 2010, 11, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Kleinschmidt, E.G.; Schlaepfer, D.D. Focal adhesion kinase signaling in unexpected places. Curr. Opin. Cell. Biol 2017, 45, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Tuloup-Minguez, V.; Greffard, A.; Codogno, P.; Botti, J. Regulation of autophagy by extracellular matrix glycoproteins in hela cells. Autophagy 2001, 7, 27–39. [Google Scholar]
- Owen, K.A.; Meyer, C.B.; Bouton, A.H.; Casanova, J.E. Activation of focal adhesion kinase by salmonella suppresses autophagy via an AKT/MTOR signaling pathway and promotes bacterial survival in macrophages. PLoS Pathog. 2014, 10, e1004159. [Google Scholar] [CrossRef] [PubMed]
- Sandilands, E.; Schoenherr, C.; Frame, M.C. P70s6k is regulated by focal adhesion kinase and is required for src-selective autophagy. Cell. Signal. 2015, 27, 1816–1823. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Zhu, Q.; Dee, R.; Opheim, Z.; Mack, C.P.; Cyr, D.M.; Taylor, J.M. Focal adhesion kinase-mediated phosphorylation of beclin1 protein suppresses cardiomyocyte autophagy and initiates hypertrophic growth. J. Biol. Chem. 2017, 292, 2065–2079. [Google Scholar] [CrossRef] [PubMed]
- Leidal, A.M.; Debnath, J. ‘Doubling down’ on the autophagy pathway to suppress tumor growth. Genes Dev. 2014, 28, 1137–1139. [Google Scholar] [CrossRef] [PubMed]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Autophagy in cancer metastasis. Oncogene 2017, 36, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Gan, B.; Melkoumian, Z.K.; Wu, X.; Guan, K.L.; Guan, J.L. Identification of fip200 interaction with the tsc1-tsc2 complex and its role in regulation of cell size control. J. Cell Biol. 2005, 170, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.T.; Chen, X.L.; Lim, Y.; Hanson, D.A.; Vo, T.T.; Howerton, K.; Larocque, N.; Fisher, S.J.; Schlaepfer, D.D.; Ilic, D. Nuclear fak promotes cell proliferation and survival through ferm-enhanced p53 degradation. Mol. Cell 2008, 29, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Ou, W.B.; Lu, M.; Eilers, G.; Li, H.; Ding, J.; Meng, X.; Wu, Y.; He, Q.; Sheng, Q.; Zhou, H.M.; et al. Co-targeting of fak and mdm2 triggers additive anti-proliferative effects in mesothelioma via a coordinated reactivation of p53. Br. J. Cancer 2016, 115, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Sandilands, E.; Serrels, B.; McEwan, D.G.; Morton, J.P.; Macagno, J.P.; McLeod, K.; Stevens, C.; Brunton, V.G.; Langdon, W.Y.; Vidal, M.; et al. Autophagic targeting of src promotes cancer cell survival following reduced fak signalling. Nat. Cell Biol. 2011, 14, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Sandilands, E.; Serrels, B.; Wilkinson, S.; Frame, M.C. Src-dependent autophagic degradation of ret in fak-signalling-defective cancer cells. EMBO Rep. 2012, 13, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Troiano, A.; Lomoriello, I.S.; di Martino, O.; Fusco, S.; Pollice, A.; Vivo, M.; La Mantia, G.; Calabro, V. Y-box binding protein-1 is part of a complex molecular network linking deltanp63alpha to the PI3K/AKT pathway in cutaneous squamous cell carcinoma. J. Cell. Physiol. 2015, 230, 2067–2074. [Google Scholar] [CrossRef] [PubMed]
- Schoenherr, C.; Byron, A.; Sandilands, E.; Paliashvili, K.; Baillie, G.S.; Garcia-Munoz, A.; Valacca, C.; Cecconi, F.; Serrels, B.; Frame, M.C. Ambra1 spatially regulates src activity and Src/FAK-mediated cancer cell invasion via trafficking networks. Elife 2017. [Google Scholar] [CrossRef] [PubMed]
- Schoenherr, C.; Serrels, B.; Proby, C.; Cunningham, D.L.; Findlay, J.E.; Baillie, G.S.; Heath, J.K.; Frame, M.C. Eps8 controls Src- and FAK-dependent phenotypes in squamous carcinoma cells. J. Cell Sci. 2014, 127, 5303–5316. [Google Scholar] [CrossRef] [PubMed]
- Hsia, D.A.; Mitra, S.K.; Hauck, C.R.; Streblow, D.N.; Nelson, J.A.; Ilic, D.; Huang, S.; Li, E.; Nemerow, G.R.; Leng, J.; et al. Differential regulation of cell motility and invasion by fak. J. Cell Biol. 2003, 160, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Yuan, H.X.; Guan, K.L. Autophagy regulation by nutrient signaling. Cell Res. 2014, 24, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.H.; Wang, W.J.; Kuo, J.C. The tumor suppressor dap-kinase links cell adhesion and cytoskeleton reorganization to cell death regulation. J. Biomed. Sci. 2006, 13, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.C.; Wang, W.J.; Yao, C.C.; Wu, P.R.; Chen, R.H. The tumor suppressor dapk inhibits cell motility by blocking the integrin-mediated polarity pathway. J. Cell Biol. 2006, 172, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Kuo, J.C.; Yao, C.C.; Chen, R.H. Dap-kinase induces apoptosis by suppressing integrin activity and disrupting matrix survival signals. J. Cell Biol. 2002, 159, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, M.; Vivo, M.; De Simone, M.; Guerrini, L.; Pollice, A.; La Mantia, G.; Calabro, V. Sumoylation and ubiquitylation crosstalk in the control of deltanp63alpha protein stability. Gene 2018, 645, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Kadare, G.; Toutant, M.; Formstecher, E.; Corvol, J.C.; Carnaud, M.; Boutterin, M.C.; Girault, J.A. Pias1-mediated sumoylation of focal adhesion kinase activates its autophosphorylation. J. Biol. Chem. 2003, 278, 47434–47440. [Google Scholar] [CrossRef] [PubMed]
- Llanos, S.; Clark, P.A.; Rowe, J.; Peters, G. Stabilization of p53 by p14arf without relocation of mdm2 to the nucleolus. Nat. Cell Biol. 2001, 3, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Vivo, M.; Matarese, M.; Sepe, M.; Di Martino, R.; Festa, L.; Calabro, V.; La Mantia, G.; Pollice, A. Mdm2-mediated degradation of p14ARF: A novel mechanism to control arf levels in cancer cells. PLoS ONE 2015, 10, e0117252. [Google Scholar] [CrossRef] [PubMed]
- Tancioni, I.; Miller, N.L.; Uryu, S.; Lawson, C.; Jean, C.; Chen, X.L.; Kleinschmidt, E.G.; Schlaepfer, D.D. Fak activity protects nucleostemin in facilitating breast cancer spheroid and tumor growth. Breast Cancer Res. 2015, 17, 47. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.; Kuo, M.L.; Roussel, M.F.; Sherr, C.J. Nucleolar arf tumor suppressor inhibits ribosomal rna processing. Mol. Cell 2003, 11, 415–424. [Google Scholar] [CrossRef]
- Abbi, S.; Ueda, H.; Zheng, C.; Cooper, L.A.; Zhao, J.; Christopher, R.; Guan, J.L. Regulation of focal adhesion kinase by a novel protein inhibitor FIP200. Mol. Biol. Cell 2002, 13, 3178–3191. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Takamura, A.; Kishi, C.; Iemura, S.; Natsume, T.; Guan, J.L.; Mizushima, N. Fip200, a ulk-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 2008, 181, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Caino, M.C.; Chae, Y.C.; Vaira, V.; Ferrero, S.; Nosotti, M.; Martin, N.M.; Weeraratna, A.; O’Connell, M.; Jernigan, D.; Fatatis, A.; et al. Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J. Clin. Investig. 2013, 123, 2907–2920. [Google Scholar] [CrossRef] [PubMed]
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; McEvoy, A.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M.; et al. The autophagy-associated factors dram1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2013, 32, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Yao, Y.; Xu, G.; Zhou, C.; Zhang, Y.; Sun, J.; Jiang, R.; Shao, Q.; Chen, Y. Cd24 regulates sorafenib resistance via activating autophagy in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 646. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, metabolism, and cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, M.N.; Mowers, E.E.; Drake, L.E.; Collier, C.; Chen, H.; Zamora, M.; Mui, S.; Macleod, K.F. Autophagy promotes focal adhesion disassembly and cell motility of metastatic tumor cells through the direct interaction of paxillin with LC3. Cell. Rep. 2016, 15, 1660–1672. [Google Scholar] [CrossRef] [PubMed]
- Bustelo, X.R. Intratumoral stages of metastatic cells: A synthesis of ontogeny, Rho/Rac gtpases, epithelial-mesenchymal transitions, and more. Bioessays 2012, 34, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Boeckeler, K.; Rosse, C.; Howell, M.; Parker, P.J. Manipulating signal delivery—Plasma-membrane Erk activation in apkc-dependent migration. J. Cell Sci. 2010, 123, 2725–2732. [Google Scholar] [CrossRef] [PubMed]
- Rosse, C.; Boeckeler, K.; Linch, M.; Radtke, S.; Frith, D.; Barnouin, K.; Morsi, A.S.; Hafezparast, M.; Howell, M.; Parker, P.J. Binding of dynein intermediate chain 2 to paxillin controls focal adhesion dynamics and migration. J. Cell Sci. 2012, 125, 3733–3738. [Google Scholar] [CrossRef] [PubMed]
- Rosse, C.; Linch, M.; Kermorgant, S.; Cameron, A.J.; Boeckeler, K.; Parker, P.J. Pkc and the control of localized signal dynamics. Nat. Rev. Mol. Cell Biol. 2010, 11, 103–112. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fontana, R.; Vivo, M. Dynamics of p14ARF and Focal Adhesion Kinase-Mediated Autophagy in Cancer. Cancers 2018, 10, 221. https://doi.org/10.3390/cancers10070221
Fontana R, Vivo M. Dynamics of p14ARF and Focal Adhesion Kinase-Mediated Autophagy in Cancer. Cancers. 2018; 10(7):221. https://doi.org/10.3390/cancers10070221
Chicago/Turabian StyleFontana, Rosa, and Maria Vivo. 2018. "Dynamics of p14ARF and Focal Adhesion Kinase-Mediated Autophagy in Cancer" Cancers 10, no. 7: 221. https://doi.org/10.3390/cancers10070221
APA StyleFontana, R., & Vivo, M. (2018). Dynamics of p14ARF and Focal Adhesion Kinase-Mediated Autophagy in Cancer. Cancers, 10(7), 221. https://doi.org/10.3390/cancers10070221