The Methylation Status of the Epigenome: Its Emerging Role in the Regulation of Tumor Angiogenesis and Tumor Growth, and Potential for Drug Targeting



Abstract
1. Introduction
2. DNA Methylation Profiles in Tumors, Metastasis and Angiogenic Genes
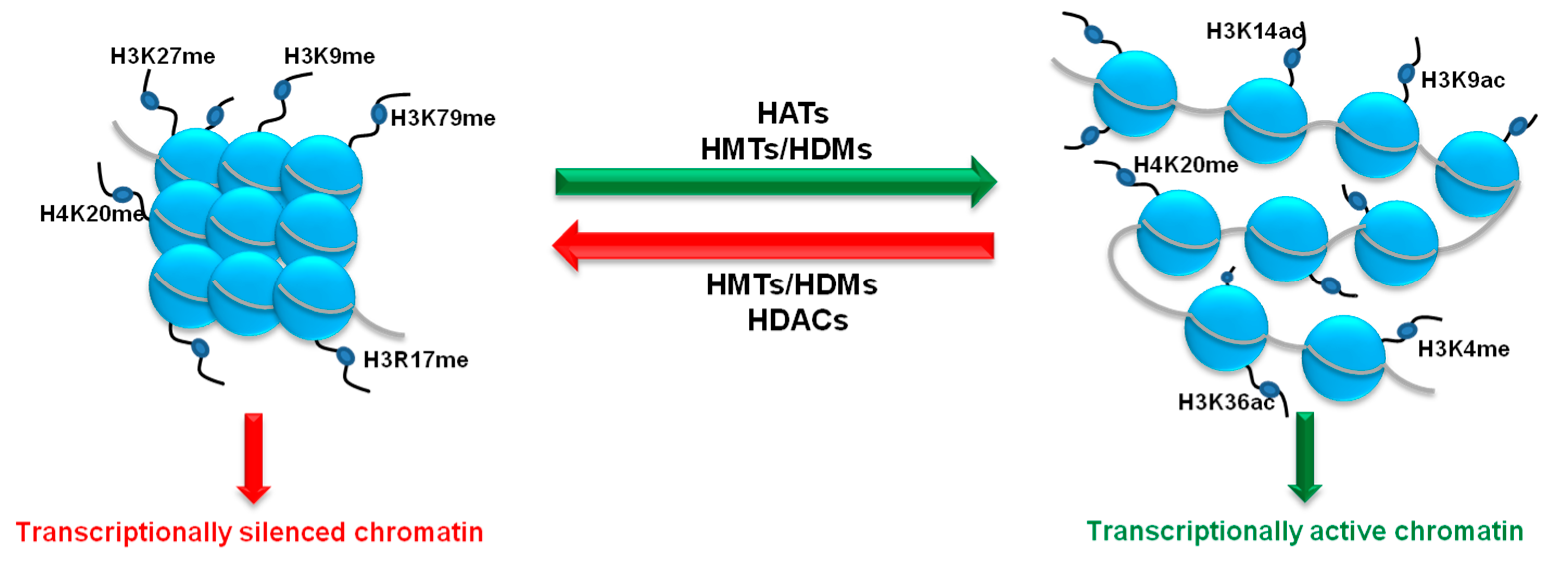
3. Histone Methylation Status as a Key Player in the Regulation of Angiogenesis Process
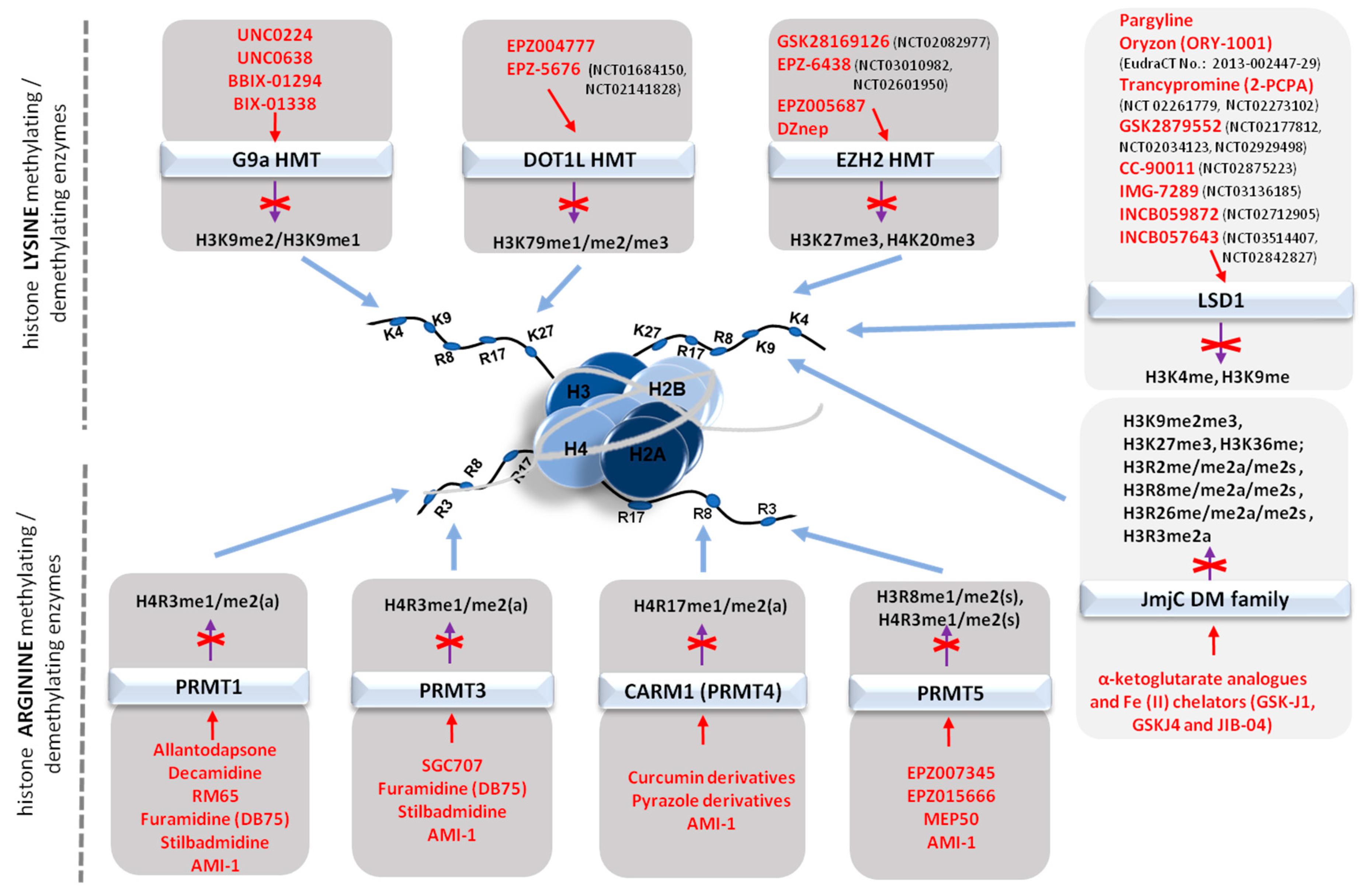
3.1. Histone Methylating Enzymes (HMTs)
3.1.1. Targeting the Polycomb Repressive Complex 2 in Cancer
3.1.2. G9a Histone Methyltransferase
3.1.3. DOT1L and Other Histone Methyltransferases
4. Histone Demethylases
5. Methylation-Focused Epidrugs in Tumor Angiogenesis Control: Drug Candidates and Ongoing Clinical Trials
5.1. DNA Methyltransferase Inhibitors (DNMTis)
5.1.1. Azacitidine and Decitabine—The Fundamental Hypomethylating Agents
5.1.2. Zebularine: A Novel DNA Methyltransferase Inhibitor
5.1.3. Antisense Oligonucleotides Inhibitors of DNMTs
5.1.4. Low Molecular Weight Inhibitors of DNMTs
5.1.5. Natural Compounds with DNMTs Inhibitory Activity
5.2. Hypomethylating Agents (DNMTis) in Co-treatment with Methyl Group Donor (SAM) in the Prevention of Tumor Progression via Tumor Angiogenesis Inhibition
5.3. Epidrugs Modulating Angiogenesis Process via the Histone Methylation Status
5.3.1. Histone Methyltransferase Inhibitors (HMTIs)
Inhibitors of histone lysine methyltransferases (HKMTs)
Inhibitors of histone arginine methyltransferases (PRMTs)
5.3.2. Histone Demethylase Inhibitors (KDMIs)
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Antiangiogenesis in cancer therapy—Endostatin and its mechanisms of action. Exp. Cell Res. 2006, 312, 594–607. [Google Scholar] [CrossRef] [PubMed]
- Varella, L.; Abraham, J.; Kruse, M. Revisiting the role of bevacizumab in the treatment of breast cancer. Semin. Oncol. 2017, 44, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. Endogenous angiogenesis inhibitors and their therapeutic implications. Int. J. Biochem. Cell Biol. 2001, 33, 357–369. [Google Scholar] [CrossRef]
- Lu, C.; Han, H.D.; Mangala, L.S.; Ali-Fehmi, R.; Newton, C.S.; Ozbun, L.; Armaiz-Pena, G.N.; Hu, W.; Stone, R.L.; Munkarah, A.; et al. Regulation of tumor angiogenesis by EZH2. Cancer Cell 2010, 18, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Wojtala, M.; Pirola, L.; Balcerczyk, A. Modulation of the vascular endothelium functioning by dietary components, the role of epigenetics. Biofactors 2017, 43, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Xu, W.S. Histone deacetylase inhibitors: Potential in cancer therapy. J. Cell. Biochem. 2009, 107, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.H.; Hao, C.L.; Liu, P.; Tian, X.; Wang, L.H.; Zhao, L.; Zhu, C.M. Valproic acid inhibits tumor angiogenesis in mice transplanted with Kasumi1 leukemia cells. Mol. Med. Rep. 2014, 9, 443–449. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mottet, D.; Castronovo, V. Histone deacetylases: Anti-angiogenic targets in cancer therapy. Curr. Cancer Drug Targets 2010, 10, 898–913. [Google Scholar] [CrossRef] [PubMed]
- Raj, K.; Mufti, G.J. Azacytidine (Vidaza(R)) in the treatment of myelodysplastic syndromes. Ther. Clin. Risk Manag. 2006, 2, 377–388. [Google Scholar] [CrossRef] [PubMed]
- De Smet, C.; Loriot, A. DNA hypomethylation in cancer: Epigenetic scars of a neoplastic journey. Epigenetics 2010, 5, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Luu, P.L.; Stirzaker, C.; Clark, S.J. Methyl-CpG-binding domain proteins: Readers of the epigenome. Epigenomics 2015, 7, 1051–1073. [Google Scholar] [CrossRef] [PubMed]
- Greger, V.; Passarge, E.; Hopping, W.; Messmer, E.; Horsthemke, B. Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum. Genet. 1989, 83, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetic gene silencing in cancer: The DNA hypermethylome. Hum. Mol. Genet. 2007, 16, R50–R59. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, H.J.; Esteller, M. CpG Islands in Cancer: Heads, Tails, and Sides. Methods Mol. Biol. 2018, 1766, 49–80. [Google Scholar] [PubMed]
- Guo, Y.L.; Zhu, T.N.; Guo, W.; Dong, Z.M.; Zhou, Z.; Cui, Y.J.; Zhao, R.J. Aberrant CpG Island Shore Region Methylation of CAV1 Is Associated with Tumor Progression and Poor Prognosis in Gastric Cardia Adenocarcinoma. Arch. Med. Res. 2016, 47, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.A.; Wu, X.; Li, A.X.; Hahn, T.; Pfeifer, G.P. Relationship between gene body DNA methylation and intragenic H3K9me3 and H3K36me3 chromatin marks. PLoS ONE 2011, 6, e18844. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, F.; Hodgson, J.G.; Eden, A.; Jackson-Grusby, L.; Dausman, J.; Gray, J.W.; Leonhardt, H.; Jaenisch, R. Induction of tumors in mice by genomic hypomethylation. Science 2003, 300, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Rainier, S.; Johnson, L.A.; Dobry, C.J.; Ping, A.J.; Grundy, P.E.; Feinberg, A.P. Relaxation of imprinted genes in human cancer. Nature 1993, 362, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, O.; Eccles, M.R.; Szeto, J.; McNoe, L.A.; Yun, K.; Maw, M.A.; Smith, P.J.; Reeve, A.E. Relaxation of insulin-like growth factor II gene imprinting implicated in Wilms’ tumour. Nature 1993, 362, 749–751. [Google Scholar] [CrossRef] [PubMed]
- Zetter, B.R. Angiogenesis and tumor metastasis. Annu. Rev. Med. 1998, 49, 407–424. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Zhou, Z.J.; Xia, G.K.; Zhang, X.H.; Wei, Z.W.; Zhu, J.T.; Yu, J.; Chen, W.; He, Y.; Schwarz, R.E.; et al. A positive crosstalk between CXCR4 and CXCR2 promotes gastric cancer metastasis. Oncogene 2017, 36, 5122–5133. [Google Scholar] [CrossRef] [PubMed]
- Sobolik, T.; Su, Y.J.; Wells, S.; Ayers, G.D.; Cook, R.S.; Richmond, A. CXCR4 drives the metastatic phenotype in breast cancer through induction of CXCR2 and activation of MEK and PI3K pathways. Mol. Biol. Cell 2014, 25, 566–582. [Google Scholar] [CrossRef] [PubMed]
- Pulukuri, S.M.; Patibandla, S.; Patel, J.; Estes, N.; Rao, J.S. Epigenetic inactivation of the tissue inhibitor of metalloproteinase-2 (TIMP-2) gene in human prostate tumors. Oncogene 2007, 26, 5229–5237. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.C.; Eberhart, C.G.; Grossman, S.A.; Herman, J.G. Epigenetic silencing of multiple genes in primary CNS lymphoma. Int. J. Cancer 2006, 119, 2487–2491. [Google Scholar] [CrossRef] [PubMed]
- Maleva Kostovska, I.; Jakimovska, M.; Popovska-Jankovic, K.; Kubelka-Sabit, K.; Karagjozov, M.; Plaseska-Karanfilska, D. TIMP3 Promoter Methylation Represents an Epigenetic Marker of BRCA1ness Breast Cancer Tumours. Pathol. Oncol. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Saraiva-Esperon, U.; Ruibal, A.; Herranz, M. The contrasting epigenetic role of RUNX3 when compared with that of MGMT and TIMP3 in glioblastoma multiforme clinical outcomes. J. Neurol. Sci. 2014, 347, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Das, A.M.; Bolkestein, M.; van der Klok, T.; Oude Ophuis, C.M.; Vermeulen, C.E.; Rens, J.A.; Dinjens, W.N.; Atmodimedjo, P.N.; Verhoef, C.; Koljenovic, S.; et al. Tissue inhibitor of metalloproteinase-3 (TIMP3) expression decreases during melanoma progression and inhibits melanoma cell migration. Eur. J. Cancer 2016, 66, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.L.; Lv, P.; Han, J.; Zhu, X.; Hong, L.L.; Zhu, W.Y.; Wang, X.B.; Wu, Y.C.; Li, P.; Ling, Z.Q. Methylated TIMP-3 DNA in body fluids is an independent prognostic factor for gastric cancer. Arch. Pathol. Lab. Med. 2014, 138, 1466–1473. [Google Scholar] [CrossRef] [PubMed]
- Oue, N.; Matsumura, S.; Nakayama, H.; Kitadai, Y.; Taniyama, K.; Matsusaki, K.; Yasui, W. Reduced expression of the TSP1 gene and its association with promoter hypermethylation in gastric carcinoma. Oncology 2003, 64, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Hellebrekers, D.M.; Jair, K.W.; Vire, E.; Eguchi, S.; Hoebers, N.T.; Fraga, M.F.; Esteller, M.; Fuks, F.; Baylin, S.B.; van Engeland, M.; et al. Angiostatic activity of DNA methyltransferase inhibitors. Mol. Cancer Ther. 2006, 5, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Lindner, D.J.; Wu, Y.; Haney, R.; Jacobs, B.S.; Fruehauf, J.P.; Tuthill, R.; Borden, E.C. Thrombospondin-1 expression in melanoma is blocked by methylation and targeted reversal by 5-aza-deoxycytidine suppresses angiogenesis. Matrix Biol. 2013, 32, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Greco, M.; D’Alo, F.; Scardocci, A.; Criscuolo, M.; Fabiani, E.; Guidi, F.; Di Ruscio, A.; Migliara, G.; Pagano, L.; Fianchi, L.; et al. Promoter methylation of DAPK1, E-cadherin and thrombospondin-1 in de novo and therapy-related myeloid neoplasms. Blood Cells Mol. Dis. 2010, 45, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Dang, S.; Hou, P. Gene methylation in gastric cancer. Clin. Chim. Acta 2013, 424, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Pakneshan, P.; Tetu, B.; Rabbani, S.A. Demethylation of urokinase promoter as a prognostic marker in patients with breast carcinoma. Clin. Cancer Res. 2004, 10, 3035–3041. [Google Scholar] [CrossRef] [PubMed]
- Quentmeier, H.; Eberth, S.; Romani, J.; Weich, H.A.; Zaborski, M.; Drexler, H.G. DNA methylation regulates expression of VEGF-R2 (KDR) and VEGF-R3 (FLT4). BMC Cancer 2012, 12, 19. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.; Zhong, J.; Zhang, S.; Zhang, Y.; Yu, Q.; Yang, P.; Wang, M.H.; Fulton, D.J.; Shi, H.; Dong, Z.; et al. Loss of methyl-CpG-binding domain protein 2 enhances endothelial angiogenesis and protects mice against hind-limb ischemic injury. Circulation 2011, 123, 2964–2974. [Google Scholar] [CrossRef] [PubMed]
- Ping, S.Y.; Shen, K.H.; Yu, D.S. Epigenetic regulation of vascular endothelial growth factor a dynamic expression in transitional cell carcinoma. Mol. Carcinog. 2013, 52, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.; Kwabi-Addo, B.; Ittmann, M.; Jelinek, J.; Shen, L.; Yu, Y.; Issa, J.P. Identification of novel tumor markers in prostate, colon and breast cancer by unbiased methylation profiling. PLoS ONE 2008, 3, e2079. [Google Scholar] [CrossRef]
- Choi, B.; Han, T.S.; Min, J.; Hur, K.; Lee, S.M.; Lee, H.J.; Kim, Y.J.; Yang, H.K. MAL and TMEM220 are novel DNA methylation markers in human gastric cancer. Biomarkers 2017, 22, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Matsui, S.; Kagara, N.; Mishima, C.; Naoi, Y.; Shimoda, M.; Shimomura, A.; Shimazu, K.; Kim, S.J.; Noguchi, S. Methylation of the SEPT9_v2 promoter as a novel marker for the detection of circulating tumor DNA in breast cancer patients. Oncol. Rep. 2016, 36, 2225–2235. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.M.; Guzzetta, A.A.; Bailey, V.J.; Downing, S.R.; Van Neste, L.; Chiappinelli, K.B.; Keeley, B.P.; Stark, A.; Herrera, A.; Wolfgang, C.; et al. Novel methylation biomarker panel for the early detection of pancreatic cancer. Clin. Cancer Res. 2013, 19, 6544–6555. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Histone methylation in transcriptional control. Curr. Opin. Genet. Dev. 2002, 12, 198–209. [Google Scholar] [CrossRef]
- Di Lorenzo, A.; Bedford, M.T. Histone arginine methylation. FEBS Lett. 2011, 585, 2024–2031. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.; Bannister, A.J.; Kouzarides, T. Unsafe SETs: Histone lysine methyltransferases and cancer. Trends Biochem. Sci. 2002, 27, 396–402. [Google Scholar] [CrossRef]
- Paro, R.; Strutt, H.; Cavalli, G. Heritable chromatin states induced by the Polycomb and Trithorax group genes. Novartis Found. Symp. 1998, 214, 51–61. [Google Scholar] [PubMed]
- Dreger, H.; Ludwig, A.; Weller, A.; Stangl, V.; Baumann, G.; Meiners, S.; Stangl, K. Epigenetic regulation of cell adhesion and communication by enhancer of zeste homolog 2 in human endothelial cells. Hypertension 2012, 60, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Fornaro, L.; Bocci, G.; Sun, L.; Farrar, W.L.; Falcone, A.; Danesi, R. EZH2 inhibition: Targeting the crossroad of tumor invasion and angiogenesis. Cancer Metastasis Rev. 2012, 31, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Richter, G.H.; Plehm, S.; Fasan, A.; Rossler, S.; Unland, R.; Bennani-Baiti, I.M.; Hotfilder, M.; Lowel, D.; von Luettichau, I.; Mossbrugger, I.; et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 5324–5329. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [PubMed]
- Van Leenders, G.J.; Dukers, D.; Hessels, D.; van den Kieboom, S.W.; Hulsbergen, C.A.; Witjes, J.A.; Otte, A.P.; Meijer, C.J.; Raaphorst, F.M. Polycomb-group oncogenes EZH2, BMI1, and RING1 are overexpressed in prostate cancer with adverse pathologic and clinical features. Eur. Urol. 2007, 52, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Kim, L.J.Y.; Wu, Q.; Wallace, L.C.; Prager, B.C.; Sanvoranart, T.; Gimple, R.C.; Wang, X.; Mack, S.C.; Miller, T.E.; et al. Targeting glioma stem cells through combined BMI1 and EZH2 inhibition. Nat. Med. 2017, 23, 1352–1361. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Park, J.W.; Chun, Y.S. Jumonji histone demethylases as emerging therapeutic targets. Pharmacol. Res. 2016, 105, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Osawa, T.; Muramatsu, M.; Wang, F.; Tsuchida, R.; Kodama, T.; Minami, T.; Shibuya, M. Increased expression of histone demethylase JHDM1D under nutrient starvation suppresses tumor growth via down-regulating angiogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 20725–20729. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yan, Y.; Davidson, T.L.; Shinkai, Y.; Costa, M. Hypoxic stress induces dimethylated histone H3 lysine 9 through histone methyltransferase G9a in mammalian cells. Cancer Res. 2006, 66, 9009–9016. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.W.; Hua, K.T.; Kao, H.J.; Chi, C.C.; Wei, L.H.; Johansson, G.; Shiah, S.G.; Chen, P.S.; Jeng, Y.M.; Cheng, T.Y.; et al. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 2010, 70, 7830–7840. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, J.; Kim, W.H.; Lee, Y.M. Hypoxic silencing of tumor suppressor RUNX3 by histone modification in gastric cancer cells. Oncogene 2009, 28, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ye, D.; Guo, W.; Yu, W.; He, Y.; Hu, J.; Wang, Y.; Zhang, L.; Liao, Y.; Song, H.; et al. G9a is essential for EMT-mediated metastasis and maintenance of cancer stem cell-like characters in head and neck squamous cell carcinoma. Oncotarget 2015, 6, 6887–6901. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Li, T.; Wang, X.; Zhao, E.; Choi, J.H.; Yang, L.; Zha, Y.; Dong, Z.; Huang, S.; Asara, J.M.; et al. The histone H3 methyltransferase G9A epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab. 2013, 18, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, M.; Chiba, T.; Zen, Y.; Oshima, M.; Kusakabe, Y.; Noguchi, Y.; Yuki, K.; Koide, S.; Tara, S.; Saraya, A.; et al. Histone lysine methyltransferase G9a is a novel epigenetic target for the treatment of hepatocellular carcinoma. Oncotarget 2017, 8, 21315–21326. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.J.; Shun, C.T.; Yen, M.L.; Chou, C.H.; Lin, M.C. Methyltransferase G9a promotes cervical cancer angiogenesis and decreases patient survival. Oncotarget 2017, 8, 62081–62098. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.Y.; Seok, J.Y.; Choi, Y.S.; Lee, S.H.; Bae, J.S.; Lee, Y.M. The Histone Methyltransferase Inhibitor BIX01294 Inhibits HIF-1alpha Stability and Angiogenesis. Mol. Cells 2015, 38, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Wojtala, M.; Macierzynska-Piotrowska, E.; Rybaczek, D.; Pirola, L.; Balcerczyk, A. Pharmacological and transcriptional inhibition of the G9a histone methyltransferase suppresses proliferation and modulates redox homeostasis in human microvascular endothelial cells. Pharmacol. Res. 2018, 128, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Wu, X.; Zhao, Q.; Gao, J.; Huo, D.; Liu, X.; Ye, Z.; Dong, X.; Fu, Z.; Shang, Y.; et al. DOT1L promotes angiogenesis through cooperative regulation of VEGFR2 with ETS-1. Oncotarget 2016, 7, 69674–69687. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, J.; Lin, J.; Zhou, L.; Song, Y.; Wei, B.; Luo, X.; Chen, Z.; Chen, Y.; Xiong, J.; et al. The transcription factor GATA1 and the histone methyltransferase SET7 interact to promote VEGF-mediated angiogenesis and tumor growth and predict clinical outcome of breast cancer. Oncotarget 2016, 7, 9859–9875. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Shi, L.; Gui, B.; Yu, W.; Wang, J.; Zhang, D.; Han, X.; Yao, Z.; Shang, Y. Binding of the JmjC demethylase JARID1B to LSD1/NuRD suppresses angiogenesis and metastasis in breast cancer cells by repressing chemokine CCL14. Cancer Res. 2011, 71, 6899–6908. [Google Scholar] [CrossRef] [PubMed]
- Kwa, F.A.; Balcerczyk, A.; Licciardi, P.; El-Osta, A.; Karagiannis, T.C. Chromatin modifying agents—The cutting edge of anticancer therapy. Drug Discov. Today 2011, 16, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Sorm, F.; Piskala, A.; Cihak, A.; Vesely, J. 5-Azacytidine, a new, highly effective cancerostatic. Experientia 1964, 20, 202–203. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.clinicaltrials.gov/ (accessed on 7 June 2018).
- Available online: https://pharmaintelligence.informa.com/products-and-services/data-and-analysis/trialtrove (accessed on 7 June 2018).
- Palii, S.S.; Van Emburgh, B.O.; Sankpal, U.T.; Brown, K.D.; Robertson, K.D. DNA methylation inhibitor 5-Aza-2′-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol. Cell. Biol. 2008, 28, 752–771. [Google Scholar] [CrossRef] [PubMed]
- Szyf, M. Epigenetics, DNA methylation, and chromatin modifying drugs. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 243–263. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Issa, J.P.; Garcia-Manero, G.; Kantarjian, H. Evolution of decitabine development: Accomplishments, ongoing investigations, and future strategies. Cancer 2008, 112, 2341–2351. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Baylin, S.B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003, 349, 2042–2054. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Carafa, V.; Benedetti, R.; Altucci, L. Trials with ‘epigenetic’ drugs: An update. Mol. Oncol. 2012, 6, 657–682. [Google Scholar] [CrossRef] [PubMed]
- Derissen, E.J.; Beijnen, J.H.; Schellens, J.H. Concise drug review: Azacitidine and decitabine. Oncologist 2013, 18, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Miller-Kasprzak, E.; Jagodzinski, P.P. 5-Aza-2′-deoxycytidine increases the expression of anti-angiogenic vascular endothelial growth factor 189b variant in human lung microvascular endothelial cells. Biomed. Pharmacother. 2008, 62, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Lyu, T.; Jia, N.; Wang, J.; Yan, X.; Yu, Y.; Lu, Z.; Bast, R.C., Jr.; Hua, K.; Feng, W. Expression and epigenetic regulation of angiogenesis-related factors during dormancy and recurrent growth of ovarian carcinoma. Epigenetics 2013, 8, 1330–1346. [Google Scholar] [CrossRef] [PubMed]
- Jueliger, S.; Lyons, J.; Cannito, S.; Pata, I.; Pata, P.; Shkolnaya, M.; Lo Re, O.; Peyrou, M.; Villarroya, F.; Pazienza, V.; et al. Efficacy and epigenetic interactions of novel DNA hypomethylating agent guadecitabine (SGI-110) in preclinical models of hepatocellular carcinoma. Epigenetics 2016, 11, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Matei, D.; Ghamande, S.; Roman, L.; Alvarez Secord, A.; Nemunaitis, J.; Markham, M.J.; Nephew, K.P.; Jueliger, S.; Oganesian, A.; Naim, S.; et al. A phase I clinical trial of Guadecitabine and Carboplatin in platinum-resistant, recurrent ovarian cancer: Clinical, pharmacokinetic, and pharmacodynamic analyses. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hellebrekers, D.M.; Castermans, K.; Vire, E.; Dings, R.P.; Hoebers, N.T.; Mayo, K.H.; Oude Egbrink, M.G.; Molema, G.; Fuks, F.; van Engeland, M.; et al. Epigenetic regulation of tumor endothelial cell anergy: Silencing of intercellular adhesion molecule-1 by histone modifications. Cancer Res. 2006, 66, 10770–10777. [Google Scholar] [CrossRef] [PubMed]
- Amato, R.J.; Stephenson, J.; Hotte, S.; Nemunaitis, J.; Belanger, K.; Reid, G.; Martell, R.E. MG98, a second-generation DNMT1 inhibitor, in the treatment of advanced renal cell carcinoma. Cancer Investig. 2012, 30, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Winquist, E.; Knox, J.; Ayoub, J.P.; Wood, L.; Wainman, N.; Reid, G.K.; Pearce, L.; Shah, A.; Eisenhauer, E. Phase II trial of DNA methyltransferase 1 inhibition with the antisense oligonucleotide MG98 in patients with metastatic renal carcinoma: A National Cancer Institute of Canada Clinical Trials Group investigational new drug study. Investig. New Drugs 2006, 24, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.; Boittiaux, I.; Tilborg, A.; Lambert, D.; Wouters, J. The dicyclo-hexyl-amine salt of RG108 (N-phthalyl-L-tryptophan), a potential epigenetic modulator. Acta Crystallogr. Sect. E Struct. Rep. Online 2010, 66, o3175-6. [Google Scholar] [CrossRef] [PubMed]
- Karakus, G.; Akin Polat, Z.; Sahin Yaglioglu, A.; Karahan, M.; Yenidunya, A.F. Synthesis, characterization, and assessment of cytotoxic, antiproliferative, and antiangiogenic effects of a novel procainamide hydrochloride-poly(maleic anhydride-co-styrene) conjugate. J. Biomater. Sci. Polym. Ed. 2013, 24, 1260–1276. [Google Scholar] [CrossRef] [PubMed]
- Morissette, G.; Moreau, E.; Rene, C.G.; Marceau, F. N-substituted 4-aminobenzamides (procainamide analogs): An assessment of multiple cellular effects concerning ion trapping. Mol. Pharmacol. 2005, 68, 1576–1589. [Google Scholar] [PubMed]
- Shian, S.G.; Kao, Y.R.; Wu, F.Y.; Wu, C.W. Inhibition of invasion and angiogenesis by zinc-chelating agent disulfiram. Mol. Pharmacol. 2003, 64, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lin, Z.; Yin, X.; Tang, L.; Luo, H.; Li, H.; Zhang, Y.; Luo, W. In Vitro and in vivo study of hydralazine, a potential anti-angiogenic agent. Eur. J. Pharmacol. 2016, 779, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Arbiser, J.L.; Klauber, N.; Rohan, R.; van Leeuwen, R.; Huang, M.T.; Fisher, C.; Flynn, E.; Byers, H.R. Curcumin is an in vivo inhibitor of angiogenesis. Mol. Med. 1998, 4, 376–383. [Google Scholar] [PubMed]
- Fu, Z.; Chen, X.; Guan, S.; Yan, Y.; Lin, H.; Hua, Z.C. Curcumin inhibits angiogenesis and improves defective hematopoiesis induced by tumor-derived VEGF in tumor model through modulating VEGF-VEGFR2 signaling pathway. Oncotarget 2015, 6, 19469–19482. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.D.; Ellis, L.M. Inhibition of tumour invasion and angiogenesis by epigallocatechin gallate (EGCG), a major component of green tea. Int. J. Exp. Pathol. 2001, 82, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Ganapathy, S.; Hingorani, S.R.; Srivastava, R.K. EGCG inhibits growth, invasion, angiogenesis and metastasis of pancreatic cancer. Front. Biosci. 2008, 13, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.W.; Makey, K.L.; Tucker, K.B.; Chinchar, E.; Mao, X.; Pei, I.; Thomas, E.Y.; Miele, L. EGCG, a major green tea catechin suppresses breast tumor angiogenesis and growth via inhibiting the activation of HIF-1alpha and NFkappaB, and VEGF expression. Vasc. Cell 2013, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.S.; Lee, H.S.; Shin, J.; Kwon, H.J. Psammaplin, A.; a marine natural product, inhibits aminopeptidase N and suppresses angiogenesis in vitro. Cancer Lett. 2004, 203, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Cheng, X.; Connolly, B.A.; Dickman, M.J.; Hurd, P.J.; Hornby, D.P. Zebularine: A novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases. J. Mol. Biol. 2002, 321, 591–599. [Google Scholar] [CrossRef]
- Nakamura, K.; Nakabayashi, K.; Htet Aung, K.; Aizawa, K.; Hori, N.; Yamauchi, J.; Hata, K.; Tanoue, A. DNA methyltransferase inhibitor zebularine induces human cholangiocarcinoma cell death through alteration of DNA methylation status. PLoS ONE 2015, 10, e0120545. [Google Scholar] [CrossRef] [PubMed]
- Ben-Kasus, T.; Ben-Zvi, Z.; Marquez, V.E.; Kelley, J.A.; Agbaria, R. Metabolic activation of zebularine, a novel DNA methylation inhibitor, in human bladder carcinoma cells. Biochem. Pharmacol. 2005, 70, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Champion, C.; Guianvarc’h, D.; Senamaud-Beaufort, C.; Jurkowska, R.Z.; Jeltsch, A.; Ponger, L.; Arimondo, P.B.; Guieysse-Peugeot, A.L. Mechanistic insights on the inhibition of c5 DNA methyltransferases by zebularine. PLoS ONE 2010, 5, e12388. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.C.; Weisenberger, D.J.; Gonzales, F.A.; Liang, G.; Xu, G.L.; Hu, Y.G.; Marquez, V.E.; Jones, P.A. Continuous zebularine treatment effectively sustains demethylation in human bladder cancer cells. Mol. Cell. Biol. 2004, 24, 1270–1278. [Google Scholar] [CrossRef] [PubMed]
- Brueckner, B.; Garcia Boy, R.; Siedlecki, P.; Musch, T.; Kliem, H.C.; Zielenkiewicz, P.; Suhai, S.; Wiessler, M.; Lyko, F. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 2005, 65, 6305–6311. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Liu, Y.; Lau, J.L.; Min, J. Epigenetic targets and drug discovery Part 2: Histone demethylation and DNA methylation. Pharmacol. Ther. 2015, 151, 121–140. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xie, Z.; Jones, W.; Pavlovicz, R.E.; Liu, S.; Yu, J.; Li, P.K.; Lin, J.; Fuchs, J.R.; Marcucci, G.; et al. Curcumin is a potent DNA hypomethylation agent. Bioorg. Med. Chem. Lett. 2009, 19, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.Z.; Wang, Y.; Ai, N.; Hou, Z.; Sun, Y.; Lu, H.; Welsh, W.; Yang, C.S. Tea polyphenol (−)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003, 63, 7563–7570. [Google Scholar] [PubMed]
- Kumar, A.; Dhawan, S.; Hardegen, N.J.; Aggarwal, B.B. Curcumin (Diferuloylmethane) inhibition of tumor necrosis factor (TNF)-mediated adhesion of monocytes to endothelial cells by suppression of cell surface expression of adhesion molecules and of nuclear factor-kappaB activation. Biochem. Pharmacol. 1998, 55, 775–783. [Google Scholar] [CrossRef]
- Yoysungnoen, P.; Wirachwong, P.; Bhattarakosol, P.; Niimi, H.; Patumraj, S. Effects of curcumin on tumor angiogenesis and biomarkers, COX-2 and VEGF, in hepatocellular carcinoma cell-implanted nude mice. Clin. Hemorheol. Microcirc. 2006, 34, 109–115. [Google Scholar] [PubMed]
- Aggarwal, B.B.; Kumar, A.; Bharti, A.C. Anticancer potential of curcumin: Preclinical and clinical studies. Anticancer Res. 2003, 23, 363–398. [Google Scholar] [PubMed]
- Kunnumakkara, A.B.; Bordoloi, D.; Harsha, C.; Banik, K.; Gupta, S.C.; Aggarwal, B.B. Curcumin mediates anticancer effects by modulating multiple cell signaling pathways. Clin. Sci. 2017, 131, 1781–1799. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Patchva, S.; Aggarwal, B.B. Therapeutic roles of curcumin: Lessons learned from clinical trials. AAPS J. 2013, 15, 195–218. [Google Scholar] [CrossRef] [PubMed]
- Niemann, H.; Marmann, A.; Lin, W.; Proksch, P. Sponge derived bromotyrosines: Structural diversity through natural combinatorial chemistry. Nat. Prod. Commun. 2015, 10, 219–231. [Google Scholar] [PubMed]
- Baud, M.G.; Leiser, T.; Haus, P.; Samlal, S.; Wong, A.C.; Wood, R.J.; Petrucci, V.; Gunaratnam, M.; Hughes, S.M.; Buluwela, L.; et al. Defining the mechanism of action and enzymatic selectivity of psammaplin A against its epigenetic targets. J. Med. Chem. 2012, 55, 1731–1750. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Kim, H.S.; Kang, Y.J.; Yoon, S.; Lee, J.; Choi, W.S.; Jung, J.H.; Kim, H.S. Psammaplin A induces Sirtuin 1-dependent autophagic cell death in doxorubicin-resistant MCF-7/ADR human breast cancer cells and xenografts. Biochim. Biophys. Acta 2015, 1850, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Man, G.C.W.; Chan, T.H.; Kwong, J.; Wang, C.C. A prodrug of green tea polyphenol (−)-epigallocatechin-3-gallate (Pro-EGCG) serves as a novel angiogenesis inhibitor in endometrial cancer. Cancer Lett. 2018, 412, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.; Han, L.; Zhou, Y.; Sun, S. Green tea polyphenol EGCG reverse cisplatin resistance of A549/DDP cell line through candidate genes demethylation. Biomed. Pharmacother. 2015, 69, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Hussain, A.; Sundaram, M.K.; Alalami, U.; Gunasekera, D.; Ramesh, L.; Hamza, A.; Quraishi, U. (−)-Epigallocatechin-3-gallate reverses the expression of various tumor-suppressor genes by inhibiting DNA methyltransferases and histone deacetylases in human cervical cancer cells. Oncol. Rep. 2015, 33, 1976–1984. [Google Scholar] [CrossRef] [PubMed]
- Rashidi, B.; Malekzadeh, M.; Goodarzi, M.; Masoudifar, A.; Mirzaei, H. Green tea and its anti-angiogenesis effects. Biomed. Pharmacother. 2017, 89, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C.; Mato, J.M. S-adenosylmethionine in liver health, injury, and cancer. Physiol. Rev. 2012, 92, 1515–1542. [Google Scholar] [CrossRef] [PubMed]
- Detich, N.; Hamm, S.; Just, G.; Knox, J.D.; Szyf, M. The methyl donor S-Adenosylmethionine inhibits active demethylation of DNA: A candidate novel mechanism for the pharmacological effects of S-Adenosylmethionine. J. Biol. Chem. 2003, 278, 20812–20820. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Li, Y.N.; Wang, F.; Zhang, W.M.; Geng, X. S-adenosylmethionine inhibits the growth of cancer cells by reversing the hypomethylation status of c-myc and H-Ras in human gastric cancer and colon cancer. Int. J. Biol. Sci. 2010, 6, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Pulukuri, S.M.; Estes, N.; Patel, J.; Rao, J.S. Demethylation-linked activation of urokinase plasminogen activator is involved in progression of prostate cancer. Cancer Res. 2007, 67, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Parashar, S.; Cheishvili, D.; Arakelian, A.; Hussain, Z.; Tanvir, I.; Khan, H.A.; Szyf, M.; Rabbani, S.A. S-adenosylmethionine blocks osteosarcoma cells proliferation and invasion in vitro and tumor metastasis in vivo: Therapeutic and diagnostic clinical applications. Cancer Med. 2015, 4, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Stefanska, B.; Huang, J.; Bhattacharyya, B.; Suderman, M.; Hallett, M.; Han, Z.G.; Szyf, M. Definition of the landscape of promoter DNA hypomethylation in liver cancer. Cancer Res. 2011, 71, 5891–5903. [Google Scholar] [CrossRef] [PubMed]
- Chik, F.; Machnes, Z.; Szyf, M. Synergistic anti-breast cancer effect of a combined treatment with the methyl donor S-adenosyl methionine and the DNA methylation inhibitor 5-aza-2′-deoxycytidine. Carcinogenesis 2014, 35, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Sahin, M.; Sahin, E.; Gumuslu, S.; Erdogan, A.; Gultekin, M. DNA methylation or histone modification status in metastasis and angiogenesis-related genes: A new hypothesis on usage of DNMT inhibitors and S-adenosylmethionine for genome stability. Cancer Metastasis Rev. 2010, 29, 655–676. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.; Helin, K. Histone methyltransferases in cancer. Semin. Cell Dev. Biol. 2010, 21, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Raposo, A.E.; Piller, S.C. Protein arginine methylation: An emerging regulator of the cell cycle. Cell Div. 2018, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Balcerczyk, A.; Rybaczek, D.; Wojtala, M.; Pirola, L.; Okabe, J.; El-Osta, A. Pharmacological inhibition of arginine and lysine methyltransferases induces nuclear abnormalities and suppresses angiogenesis in human endothelial cells. Biochem. Pharmacol. 2016, 121, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Madonna, R.; Van Linthout, S.; Thum, T.; Schulz, R.; Ferdinandy, P.; Perrino, C. Epigenetic modulation of vascular diseases: Assessing the evidence and exploring the opportunities. Vascul. Pharmacol. 2018, 107, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Morera, L.; Lubbert, M.; Jung, M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenetics 2016, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Pang, B.; Wang, Q.; Yang, S.; Gao, T.; Ding, Q.; Liu, H.; Yang, Y.; Fan, H.; Zhang, R.; et al. EZH2 upregulation correlates with tumor invasiveness, proliferation, and angiogenesis in human pituitary adenomas. Hum. Pathol. 2017, 66, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, M.; Uchimaru, K. Targeting EZH2 in cancer therapy. Curr. Opin. Oncol. 2017, 29, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Wang, Y.; Sreekumar, A.; Buckley, K.M.; Shi, H.; Jillella, A.; Ustun, C.; Rao, R.; Fernandez, P.; Chen, J.; et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood 2009, 114, 2733–2743. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Takashina, T.; Kinoshita, I.; Kikuchi, E.; Shimizu, Y.; Sakakibara-Konishi, J.; Oizumi, S.; Marquez, V.E.; Nishimura, M.; Dosaka-Akita, H. Epigenetic therapy with 3-deazaneplanocin A, an inhibitor of the histone methyltransferase EZH2, inhibits growth of non-small cell lung cancer cells. Lung Cancer 2012, 78, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Wigle, T.J.; Warholic, N.M.; Sneeringer, C.J.; Allain, C.J.; Klaus, C.R.; Sacks, J.D.; Raimondi, A.; Majer, C.R.; Song, J.; et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 2012, 8, 890–896. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A., 3rd; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wu, F.; Wu, J. Targeting histone methylation for cancer therapy: Enzymes, inhibitors, biological activity and perspectives. J. Hematol. Oncol. 2016, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Poulard, C.; Corbo, L.; Le Romancer, M. Protein arginine methylation/demethylation and cancer. Oncotarget 2016, 7, 67532–67550. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Qian, K.; Ho, M.C.; Zheng, Y.G. Small Molecule Inhibitors of Protein Arginine Methyltransferases. Expert Opin. Investig. Drugs 2016, 25, 335–358. [Google Scholar] [CrossRef] [PubMed]
- Ratovitski, T.; Arbez, N.; Stewart, J.C.; Chighladze, E.; Ross, C.A. PRMT5-mediated symmetric arginine dimethylation is attenuated by mutant huntingtin and is impaired in Huntington’s disease (HD). Cell Cycle 2015, 14, 1716–1729. [Google Scholar] [CrossRef] [PubMed]
- Quan, X.; Yue, W.; Luo, Y.; Cao, J.; Wang, H.; Wang, Y.; Lu, Z. The protein arginine methyltransferase PRMT5 regulates Abeta-induced toxicity in human cells and Caenorhabditis elegans models of Alzheimer’s disease. J. Neurochem. 2015, 134, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Castellano, S.; Milite, C.; Ragno, R.; Simeoni, S.; Mai, A.; Limongelli, V.; Novellino, E.; Bauer, I.; Brosch, G.; Spannhoff, A.; et al. Design, synthesis and biological evaluation of carboxy analogues of arginine methyltransferase inhibitor 1 (AMI-1). ChemMedChem 2010, 5, 398–414. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.H.; Wang, X.; Tian, S.C.; Ma, N.Q.; Zhang, X.Y.; Liu, Y.; Zhang, B.L.; Wu, Y.J. Arginine methyltransferase inhibitor 1 exhibits antitumor effects against cervical cancer in vitro and in vivo. Pharmazie 2018, 73, 269–273. [Google Scholar] [PubMed]
- Bissinger, E.M.; Heinke, R.; Spannhoff, A.; Eberlin, A.; Metzger, E.; Cura, V.; Hassenboehler, P.; Cavarelli, J.; Schule, R.; Bedford, M.T.; et al. Acyl derivatives of p-aminosulfonamides and dapsone as new inhibitors of the arginine methyltransferase hPRMT1. Bioorg. Med. Chem. 2011, 19, 3717–3731. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Yan, C.; Qian, K.; Su, H.; Kofsky-Wofford, S.A.; Lee, W.C.; Zhao, X.; Ho, M.C.; Ivanov, I.; Zheng, Y.G. Diamidine compounds for selective inhibition of protein arginine methyltransferase 1. J. Med. Chem. 2014, 57, 2611–2622. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Li, M.; Wang, B.; Zheng, Y.G. Discovery and mechanistic study of a class of protein arginine methylation inhibitors. J. Med. Chem. 2010, 53, 6028–6039. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Qian, K.; Yan, C.; He, M.; Jassim, B.A.; Ivanov, I.; Zheng, Y.G. Discovery of Decamidine as a New and Potent PRMT1 Inhibitor. MedChemComm 2017, 8, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Heinke, R.; Spannhoff, A.; Meier, R.; Trojer, P.; Bauer, I.; Jung, M.; Sippl, W. Virtual screening and biological characterization of novel histone arginine methyltransferase PRMT1 inhibitors. ChemMedChem 2009, 4, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Kaniskan, H.U.; Szewczyk, M.M.; Yu, Z.; Eram, M.S.; Yang, X.; Schmidt, K.; Luo, X.; Dai, M.; He, F.; Zang, I.; et al. A potent, selective and cell-active allosteric inhibitor of protein arginine methyltransferase 3 (PRMT3). Angew. Chem. Int. Ed. Engl. 2015, 54, 5166–5170. [Google Scholar] [CrossRef] [PubMed]
- Kaniskan, H.U.; Eram, M.S.; Zhao, K.; Szewczyk, M.M.; Yang, X.; Schmidt, K.; Luo, X.; Xiao, S.; Dai, M.; He, F.; et al. Discovery of Potent and Selective Allosteric Inhibitors of Protein Arginine Methyltransferase 3 (PRMT3). J. Med. Chem. 2018, 61, 1204–1217. [Google Scholar] [CrossRef] [PubMed]
- Huynh, T.; Chen, Z.; Pang, S.; Geng, J.; Bandiera, T.; Bindi, S.; Vianello, P.; Roletto, F.; Thieffine, S.; Galvani, A.; et al. Optimization of pyrazole inhibitors of Coactivator Associated Arginine Methyltransferase 1 (CARM1). Bioorg. Med. Chem. Lett. 2009, 19, 2924–2927. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Zhang, B.; Mao, R.; Zhang, C.; Wang, Y.; Xing, J.; Liu, Y.C.; Luo, X.; Ding, H.; Yang, Y.; et al. Discovery and optimization of selective inhibitors of protein arginine methyltransferase 5 by docking-based virtual screening. Org. Biomol. Chem. 2017, 15, 3648–3661. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Jiang, C.; Tao, H.; Liu, J.; Zhang, H.; Luo, C. Identification of a novel selective small-molecule inhibitor of protein arginine methyltransferase 5 (PRMT5) by virtual screening, resynthesis and biological evaluations. Bioorg. Med. Chem. Lett. 2018, 28, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Chan-Penebre, E.; Kuplast, K.G.; Majer, C.R.; Boriack-Sjodin, P.A.; Wigle, T.J.; Johnston, L.D.; Rioux, N.; Munchhof, M.J.; Jin, L.; Jacques, S.L.; et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015, 11, 432–437. [Google Scholar] [CrossRef] [PubMed]
- D’Oto, A.; Tian, Q.W.; Davidoff, A.M.; Yang, J. Histone demethylases and their roles in cancer epigenetics. J. Med. Oncol. Ther. 2016, 1, 34–40. [Google Scholar] [PubMed]
- Hoffmann, I.; Roatsch, M.; Schmitt, M.L.; Carlino, L.; Pippel, M.; Sippl, W.; Jung, M. The role of histone demethylases in cancer therapy. Mol. Oncol. 2012, 6, 683–703. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.C.; Chang, J.; Zhang, T.; Suo, F.Z.; Chen, X.B.; Liu, Y.; Zhao, B.; Yu, B.; Liu, H.M. An Overview on Screening Methods for Lysine Specific Demethylase 1 (LSD1) Inhibitors. Curr. Med. Chem. 2017, 24, 2496–2504. [Google Scholar] [CrossRef] [PubMed]
- Schenk, T.; Chen, W.C.; Gollner, S.; Howell, L.; Jin, L.; Hebestreit, K.; Klein, H.U.; Popescu, A.C.; Burnett, A.; Mills, K.; et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 2012, 18, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chang, J.; Varghese, D.; Dellinger, M.; Kumar, S.; Best, A.M.; Ruiz, J.; Bruick, R.; Pena-Llopis, S.; Xu, J.; et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat. Commun. 2013, 4, 2035. [Google Scholar] [CrossRef] [PubMed]
- Kruidenier, L.; Chung, C.W.; Cheng, Z.; Liddle, J.; Che, K.; Joberty, G.; Bantscheff, M.; Bountra, C.; Bridges, A.; Diallo, H.; et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 2012, 488, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Thinnes, C.C.; England, K.S.; Kawamura, A.; Chowdhury, R.; Schofield, C.J.; Hopkinson, R.J. Targeting histone lysine demethylases—Progress, challenges, and the future. Biochim. Biophys. Acta 2014, 1839, 1416–1432. [Google Scholar] [CrossRef] [PubMed]
- Cha, T.L.; Zhou, B.P.; Xia, W.; Wu, Y.; Yang, C.C.; Chen, C.T.; Ping, B.; Otte, A.P.; Hung, M.C. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 2005, 310, 306–310. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA Modification | Genetic Action | Biological Effects | References |
|---|---|---|---|
| Hypermethylation of DNA | Hypermethylation of promoter CpG islands | Silencing of tumor suppressor genes; Inhibition of transcription factors; Inactivation of metasasis inhibitors | [13,14,15] |
| CpG shore methylation | Abnormal transcriptional inactivation | [16,17] | |
| Hypomethylation of DNA | Decreased methylation in gene promoter regions | Activation of metastasis and tumor promoting genes | [18] |
| Hypomethylation of gene bodies | Altered and incorrect gene expression due to activation of alternative transcription start sites (TSSs) regulatory sequences | [19] | |
| Global hypomethylation of genome | Chromosomal instability and reactivation of repetitive genomic sequences | [20] | |
| Loss of imprinting | Activation of imprinted genes (IGF-2, H19) | [21,22] |
| Drug Name | Epigenetic Action | Leading Center | Approval Date | Clinically Approved Indications |
|---|---|---|---|---|
| Azacitidine (Vidaza®) | DNMTs inhibiton | Celgene | May 2004 | Acute Myelogenous Leukemia, AML Chronic Myelogenous Leukemia, CML Myelodysplastic Syndromes, MDS |
| Decitabine (Dacogen®) | DNMTs inhibiton | Astex Pharmaceuticals (Otsuka) | May 2006 | Acute Myelogenous Leukemia, AML Chronic Myelogenous Leukemia, CML Myelodysplastic Syndromes, MDS |
| Class | Epidrug | Epigenetic Target | Molecular Target | Biological Effects of Treatment | References |
|---|---|---|---|---|---|
| Nucleoside analogs | 5-azacytidine (Aza) | DNMT1 | Decreased level of VEGFs: pro-angiogenic (121a, 165a) and anti-angiogenic (121b, 165b); Increased expression of TSP1, TIMP3 and CDH1 and anti-angiogenic VEGF(189b) | Decreased ECs proliferation; Decreased tumor vessel development in vivo | [34,81,82] |
| Decitabine (DAC) | DNMT1 | Increased expression of: EGFL7, JUNB, IGFBP3, miR126, TSP1, WIF | Decreased ECs proliferation; Decreased tumor vessel development in vivo | [80] | |
| Guadecitabine (SGI-110; antimetabolite of DAC) | DNMT1 | Increased expression of: CDKN2A, DLEC1, RUNX3 | Decreased microvessel density in vivo | [83,84] | |
| Zebularine (Zeb) | DNMT1 | Increased level of: ICAM1, TSP1, JUNB, IGFBP3 | Increased leukocyte adhesion to ECs | [33,85] | |
| Antisense oligonucleotides | MG98 | DNMT1 | Re-expression of p16 | Decreased cell proliferation | [86,87] |
| Low molecular weight molecules | RG108 | DNMT1 | Re-expression of p16, SRFP1, TIMP-3 | Decreased cell proliferation | [88] |
| Procainamide | DNMT1 | Inhibition of NF-κB | Decreased cell proliferation, capillary network formation | [89,90] | |
| Disulfiram | DNMT1 | Increased expression of: RECK | Decreased activity of MMP2 and MMP9 | [91] | |
| Hydralazine (HYD) | DNMT1 DNMT3a DNMT3b | Re-expression of: p16, RAR-β | Decreased ability of ECs for: tube network formation, migration and proliferation; Decreased level of VEGF and microvessel density in vivo | [92] | |
| Natural compounds (Epi-nutrients) | Curcumin | DNMT1 | Decreased expression of: STAT3 | Decreased ECs proliferation | [93,94] |
| (−)-Epigallo-catechin-3-gallate (EGCG) | DNMT1 | Increased expression of: RECK. Inhibition the activation of: HIF-α, NF-κB and VEGF expression | Decreased ability of ECs for capillary network formation; Decreased microcapillary density in vivo | [95,96,97] | |
| Psammaplin A (PsA) | DNMT1 HDACs | Suppression of invasion and tube formation of ECs | [98] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pirola, L.; Ciesielski, O.; Balcerczyk, A. The Methylation Status of the Epigenome: Its Emerging Role in the Regulation of Tumor Angiogenesis and Tumor Growth, and Potential for Drug Targeting. Cancers 2018, 10, 268. https://doi.org/10.3390/cancers10080268
Pirola L, Ciesielski O, Balcerczyk A. The Methylation Status of the Epigenome: Its Emerging Role in the Regulation of Tumor Angiogenesis and Tumor Growth, and Potential for Drug Targeting. Cancers. 2018; 10(8):268. https://doi.org/10.3390/cancers10080268
Chicago/Turabian StylePirola, Luciano, Oskar Ciesielski, and Aneta Balcerczyk. 2018. "The Methylation Status of the Epigenome: Its Emerging Role in the Regulation of Tumor Angiogenesis and Tumor Growth, and Potential for Drug Targeting" Cancers 10, no. 8: 268. https://doi.org/10.3390/cancers10080268
APA StylePirola, L., Ciesielski, O., & Balcerczyk, A. (2018). The Methylation Status of the Epigenome: Its Emerging Role in the Regulation of Tumor Angiogenesis and Tumor Growth, and Potential for Drug Targeting. Cancers, 10(8), 268. https://doi.org/10.3390/cancers10080268