Antithrombotic Agents and Cancer




Abstract
1. Introduction
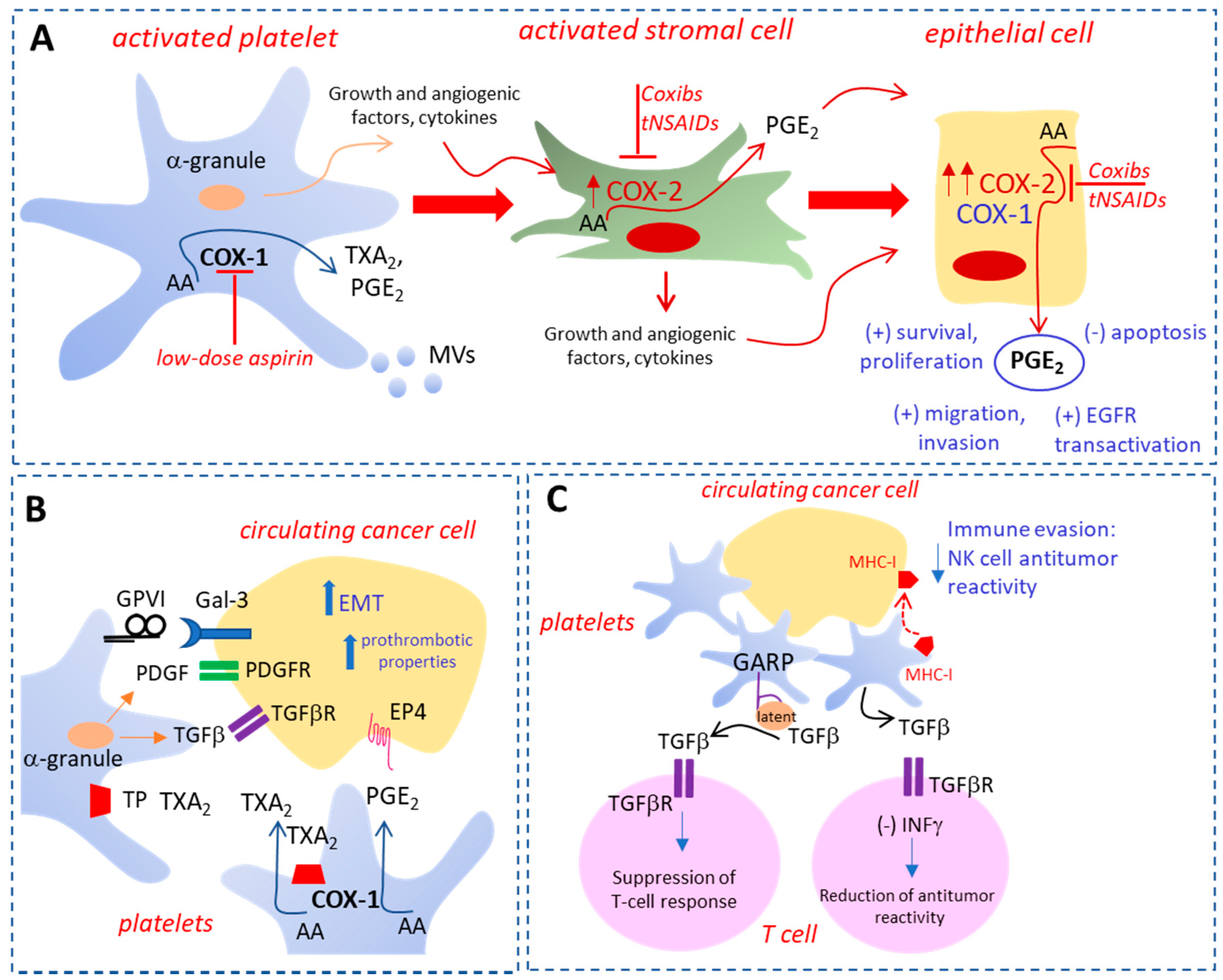
2. The Roles of Activated Platelets in Cancer
3. Effects of Antiplatelet Agents in Cancer
3.1. Low-Dose Aspirin
3.2. P2Y12 Receptor Antagonists
3.3. Thrombin Receptor Antagonists
3.4. Glycoprotein IIb/IIIa Antagonists
4. Novel Antiplatelet Agents in Clinical Development
4.1. GPVI Blockers
4.2. Antagonists of the EP3 Receptor for PGE2
4.3. GPIbα Antagonists
4.4. P-Selectin Inhibitors
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Mega, J.L.; Simon, T. Pharmacology of antithrombotic drugs: An assessment of oral antiplatelet and anticoagulant treatments. Lancet 2015, 386, 281–291. [Google Scholar] [CrossRef]
- Vandvik, P.O.; Lincoff, A.M.; Gore, J.M.; Gutterman, D.D.; Sonnenberg, F.A.; Alonso-Coello, P.; Akl, E.A.; Lansberg, M.G.; Guyatt, G.H.; Spencer, F.A. Primary and secondary prevention of cardiovascular disease: Antithrombotic Therapy and Prevention of Thrombosis: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012, 141, e637S–e668S. [Google Scholar] [CrossRef] [PubMed]
- Wojtukiewicz, M.Z.; Hempel, D.; Sierko, E.; Tucker, S.C.; Honn, K.V. Antiplatelet agents for cancer treatment: A real perspective or just an echo from the past? Cancer Metastasis Rev. 2017, 36, 305–329. [Google Scholar] [CrossRef] [PubMed]
- Khorana, A.A. Cancer and thrombosis: Implications of published guidelines for clinical practice. Ann. Oncol. 2009, 20, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Battinelli, E.M.; Markens, B.A.; Kulenthirarajan, R.A.; Machlus, K.R.; Flaumenhaft, R.; Italiano, J.E., Jr. Anticoagulation inhibits tumor cell-mediated release of platelet angiogenic proteins and diminishes platelet angiogenic response. Blood 2014, 123, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Mousa, S.A.; Petersen, L.J. Anti-cancer properties of low-molecular-weight heparin: Preclinical evidence. Thromb. Haemost. 2009, 102, 258–267. [Google Scholar] [PubMed]
- Flossmann, E.; Rothwell, P.M.; British Doctors Aspirin Trial and the UK-TIA Aspirin Trial. Effect of aspirin on long-term risk of colorectal cancer: Consistent evidence from randomised and observational studies. Lancet 2007, 369, 1603–1613. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Price, J.F.; Fowkes, F.G.; Zanchetti, A.; Roncaglioni, M.C.; Tognoni, G.; Lee, R.; Belch, J.F.; Wilson, M.; Mehta, Z.; et al. Effect of daily aspirin on long-term risk of death due to cancer: Analysis of individual patient data from randomised trials. Lancet 2011, 377, 31–41. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Wilson, M.; Price, J.F.; Belch, J.F.; Meade, T.W.; Mehta, Z. Effect of daily aspirin on risk of cancer metastasis: A study of incident cancers during randomised controlled trials. Lancet 2012, 379, 1591–1601. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Price, J.F.; Fowkes, F.G.; Zanchetti, A.; Roncaglioni, M.C.; Tognoni, G.; Lee, R.; Belch, J.F.; Wilson, M.; Mehta, Z.; et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: Analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 2012, 379, 1602–1612. [Google Scholar] [CrossRef]
- Algra, A.M.; Rothwell, P.M. Effects of regular aspirin on long-term cancer incidence and metastasis: A systemic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012, 13, 518–527. [Google Scholar] [CrossRef]
- Ruggeri, Z.M. Platelets in atherothrombosis. Nat. Med. 2002, 8, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Dovizio, M.; Bruno, A.; Contursi, A.; Grande, R.; Patrignani, P. Platelets and extracellular vesicles in cancer: Diagnostic and therapeutic implications. Cancer Metastasis Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.R.; Zhang, D.; Oswald, B.E.; Carrim, N.; Wang, X.; Hou, Y.; Zhang, Q.; Lavalle, C.; McKeown, T.; Marshall, A.H.; et al. Platelets are versatile cells: New discoveries in hemostasis, thrombosis, immune responses, tumor metastasis and beyond. Crit. Rev. Clin. Lab. Sci. 2016, 53, 409–430. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W.; Italiano, J.E., Jr.; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Dovizio, M.; Sacco, A.; Patrignani, P. Curbing tumorigenesis and malignant progression through the pharmacological control of the wound healing process. Vasc. Pharmacol. 2017, 89, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Di Francesco, L.; López Contreras, L.A.; Sacco, A.; Patrignani, P. New Insights into the Mechanism of Action of Aspirin in the Prevention of Colorectal Neoplasia. Curr. Pharm. Des. 2015, 21, 5116–5126. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Rosenberg, D.W. Multifaceted roles of PGE2 in inflammation and cancer. Semin. Immunopathol. 2013, 35, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Dixon, D.A.; Blanco, F.F.; Bruno, A.; Patrignani, P. Mechanistic aspects of COX-2 expression in colorectal neoplasia. Recent Results Cancer Res. 2013, 191, 7–37. [Google Scholar] [PubMed]
- Tsujii, M.; DuBois, R.N. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell 1995, 83, 493–501. [Google Scholar] [CrossRef]
- Pai, R.; Soreghan, B.; Szabo, I.L.; Pavelka, M.; Baatar, D.; Tarnawski, A.S. Prostaglandin E2 transactivates EGF receptor: A novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat. Med. 2002, 8, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R.N. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 2010, 29, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Arber, N.; Eagle, C.J.; Spicak, J.; Rácz, I.; Dite, P.; Hajer, J.; Zavoral, M.; Lechuga, M.J.; Gerletti, P.; Tang, J.; et al. Celecoxib for the prevention of colorectal adenomatous polyps. N. Engl. J. Med. 2006, 355, 885–985. [Google Scholar] [CrossRef] [PubMed]
- Bertagnolli, M.M.; Eagle, C.J.; Zauber, A.G.; Redston, M.; Solomon, S.D.; Kim, K.; Tang, J.; Rosenstein, R.B.; Wittes, J.; Corle, D.; et al. Celecoxib for the prevention of sporadic colorectal adenomas. N. Engl. J. Med. 2006, 355, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Baron, J.A.; Sandler, R.S.; Bresalier, R.S.; Quan, H.; Riddell, R.; Lanas, A.; Bolognese, J.A.; Oxenius, B.; Horgan, K.; Loftus, S.; et al. A randomized trial of rofecoxib for the chemoprevention of colorectal adenomas. Gastroenterology 2006, 131, 1674–1682. [Google Scholar] [CrossRef] [PubMed]
- Patrignani, P.; Patrono, C. Cyclooxygenase inhibitors: From pharmacology to clinical read-outs. Biochim. Biophys. Acta 2015, 1851, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Patrignani, P.; Patrono, C. Aspirin and cancer. J. Am. Coll. Cardiol. 2016, 68, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signalling between platelets and cancer cells induces an epithelial mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Guillem-Llobat, P.; Dovizio, M.; Bruno, A.; Ricciotti, E.; Cufino, V.; Sacco, A.; Grande, R.; Alberti, S.; Arena, V.; Cirillo, M.; et al. Aspirin prevents colorectal cancer metastasis in mice by splitting the crosstalk between platelets and tumor cells. Oncotarget 2016, 7, 32462–32477. [Google Scholar] [CrossRef] [PubMed]
- Dovizio, M.; Maier, T.J.; Alberti, S.; Di Francesco, L.; Marcantoni, E.; Münch, G.; John, C.M.; Suess, B.; Sgambato, A.; Steinhilber, D.; et al. Pharmacological inhibition of platelet-tumor cell cross-talk prevents platelet-induced overexpression of cyclooxygenase-2 in HT29 human colon carcinoma cells. Mol. Pharmacol. 2013, 84, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Grossi, I.M.; Fitzgerald, L.A.; Kendall, A.; Taylor, J.D.; Sloane, B.F.; Honn, K.V. Inhibition of human tumor cell induced platelet aggregation by antibodies to platelet glycoproteins Ib and IIb/IIIa. Proc. Soc. Exp. Biol. Med. 1987, 186, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, M.; Stone, R.L.; Menter, D.G.; Afshar-Kharghan, V.; Sood, A.K. The Platelet Lifeline to Cancer: Challenges and Opportunities. Cancer Cell 2018, 33, 965–983. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.R.; Yousef, G.M.; Ni, H. Cancer and platelet crosstalk: Opportunities and challenges for aspirin and other antiplatelet agents. Blood 2018, 131, 1777–1789. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Jurasz, P. The role of platelets in the tumor microenvironment: From solid tumors to leukemia. Biochim. Biophys. Acta 2016, 1863, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Kopp, H.G.; Placke, T.; Salih, H.R. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009, 69, 7775–7783. [Google Scholar] [CrossRef] [PubMed]
- Rachidi, S.; Metelli, A.; Riesenberg, B.; Wu, B.X.; Nelson, M.H.; Wallace, C.; Paulos, C.M.; Rubinstein, M.P.; Garrett-Mayer, E.; Hennig, M.; et al. Platelets subvert T cell immunity against cancer via GARP-TGFβ axis. Sci. Immunol. 2017, 2, eaai7911. [Google Scholar] [CrossRef] [PubMed]
- Tímár, J.; Tóvári, J.; Rásó, E.; Mészáros, L.; Bereczky, B.; Lapis, K. Platelet-mimicry of cancer cells: Epiphenomenon with clinical significance. Oncology 2005, 69, 185–201. [Google Scholar] [CrossRef] [PubMed]
- Placke, T.; Örgel, M.; Schaller, M.; Jung, G.; Rammensee, H.G.; Kopp, H.G.; Salih, H.R. Platelet derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res. 2012, 72, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Demers, M.; Wagner, D.D. Neutrophil extracellular traps: A new link to cancer-associated thrombosis and potential implications for tumor progression. OncoImmunology 2013, 2, e22946. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wysocki, R.W.; Amoozgar, Z.; Maiorino, L.; Fein, M.R.; Jorns, J.; Schott, A.F.; Kinugasa-Katayama, Y.; Lee, Y.; Won, N.H.; et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med. 2016, 8, 361ra138. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.K.; Rao, D.A. Platelets signal and tumors take off. Blood 2012, 120, 4667–4668. [Google Scholar] [CrossRef] [PubMed]
- Varon, D.; Shai, E. Role of platelet-derived microparticles in angiogenesis and tumor progression. Discov. Med. 2009, 8, 237–241. [Google Scholar] [PubMed]
- Lazar, S.; Goldfinger, L.E. Platelet Microparticles and miRNA Transfer in Cancer Progression: Many Targets, Modes of Action, and Effects across Cancer Stages. Front. Cardiovasc. Med. 2018, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Jaiswal, R.; Dalla, P.; Luk, F.; Bebawy, M. Microparticles in cancer: A review of recent developments and the potential for clinical application. Semin. Cell Dev. Biol. 2015, 40, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Jiang, L.; Lin, Y.; Wu, X.; Wang, K.; He, Q.; Wang, X.; Li, W. Platelet microparticle-mediated transfer of miR-939 to epithelial ovarian cancer cells promotes epithelial to mesenchymal transition. Oncotarget 2017, 8, 97464–97475. [Google Scholar] [CrossRef] [PubMed]
- Michael, J.V.; Wurtzel, J.G.T.; Mao, G.F.; Rao, A.K.; Kolpakov, A.; Sabri, A.; Hoffman, N.E.; Rajan, S.; Tomar, D.; Madesh, M.; et al. Platelet microparticles infiltrating solid tumors transfer miRNAs that suppress tumor growth. Blood 2017, 130, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Thun, M.J.; Jacobs, E.J.; Patrono, C. The role of aspirin in cancer prevention. Nat. Rev. Clin. Oncol. 2012, 9, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, P.M.; Wilson, M.; Elwin, C.E.; Norrving, B.; Algra, A.; Warlow, C.P.; Meade, T.W. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 2010, 376, 1741–1750. [Google Scholar] [CrossRef]
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002, 324, 71–86. [Google Scholar]
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase isozymes: The biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 2004, 56, 387–437. [Google Scholar] [CrossRef] [PubMed]
- Dovizio, M.; Bruno, A.; Tacconelli, S.; Patrignani, P. Mode of action of aspirin as a chemopreventive agent. Recent Results Cancer Res. 2013, 191, 39–65. [Google Scholar] [PubMed]
- Patrignani, P.; Filabozzi, P.; Patrono, C. Selective cumulative inhibition of platelet thromboxane production by low-dose aspirin in healthy subjects. J. Clin. Investig. 1982, 69, 1366–1372. [Google Scholar] [CrossRef] [PubMed]
- Patrignani, P.; Sacco, A.; Sostres, C.; Bruno, A.; Dovizio, M.; Piazuelo, E.; Di Francesco, L.; Contursi, A.; Zucchelli, M.; Schiavone, S.; et al. Low-Dose Aspirin Acetylates Cyclooxygenase-1 in Human Colorectal Mucosa: Implications for the Chemoprevention of Colorectal Cancer. Clin. Pharmacol. Ther. 2017, 102, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Ruvinsky, I.; Sharon, N.; Lerer, T.; Cohen, H.; Stolovich-Rain, M.; Nir, T.; Dor, Y.; Zisman, P.; Meyuhas, O. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005, 19, 2199–2211. [Google Scholar] [CrossRef] [PubMed]
- Sitia, G.; Aiolfi, R.; Di Lucia, P.; Mainetti, M.; Fiocchi, A.; Mingozzi, F.; Esposito, A.; Ruggeri, Z.M.; Chisari, F.V.; Iannacone, M.; et al. Antiplatelet therapy prevents hepatocellular carcinoma and improves survival in a mouse model of chronic hepatitis B. Proc. Natl. Acad. Sci. USA 2012, 109, E2165–E2172. [Google Scholar] [CrossRef] [PubMed]
- Kohga, S.; Kinjo, M.; Tanaka, K.; Ogawa, H.; Ishimara, M.; Tanaka, N. Effects of 5-(2-chlorobenzyl)-4,5,6,7-tetrahydrothieno[3,2-C]pyridine hydrochloride (Ticlopidine), a platelet aggregation inhibitor, on blood-borne metastasis. Cancer Res. 1981, 41, 4710–4714. [Google Scholar] [PubMed]
- Gareau, A.J.; Brien, C.; Gebremeskel, S.; Liwski, R.S.; Johnston, B.; Bezuhly, M. Ticagrelor inhibits platelet-tumor cell interactions and metastasis in human and murine breast cancer. Clin. Exp. Metastasis 2018, 35, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Chanakira, A.; Westmark, P.R.; Ong, I.M.; Sheehan, J.P. Tissue factor-factor VIIa complex triggers protease activated receptor 2-dependent growth factor release and migration in ovarian cancer. Gynecol. Oncol. 2017, 145, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Kononczuk, J.; Surazynski, A.; Czyzewska, U.; Prokop, I.; Tomczyk, M.; Palka, J.; Miltyk, W. αIIbβ3-integrin Ligands: Abciximab and Eptifibatide as Proapoptotic Factors in MCF-7 Human Breast Cancer Cells. Curr. Drug Targets 2015, 16, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Parikka, M.; Nissinen, L.; Kainulainen, T.; Bruckner-Tuderman, L.; Salo, T.; Heino, J.; Tasanen, K. Collagen XVII promotes integrin-mediated squamous cell carcinoma transmigration—A novel role for alphaIIb integrin and tirofiban. Exp. Cell Res. 2006, 312, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Reheman, A.; Hou, Y.; Zhou, H.; Wang, Y.; Marshall, A.H.; Liang, C.; Dai, X.; Li, B.X.; Vanhoorelbeke, K.; et al. Anfibatide, a novel GPIb complex antagonist, inhibits platelet adhesion and thrombus formation in vitro and in vivo in murine models of thrombosis. Thromb. Haemost. 2014, 111, 279–289. [Google Scholar] [PubMed]
- Li, B.; Dai, X.; Yang, Z.; Qian, F.; Zhang, G.; Xu, Z.; Liu, J.; Liang, C.; Reheman, A.; Ni, H. First ex vivo and in vivo assessment of anfibatide, a novel glycoprotein Ib-IV-V complex antagonist, in healthy human volunteers in phase I clinical trial. J. Thromb. Haemost. 2013, 11, 23. [Google Scholar]
- Fontayne, A.; Meiring, M.; Lamprecht, S.; Roodt, J.; Demarsin, E.; Barbeaux, P.; Deckmyn, H. The humanized anti-glycoprotein Ib monoclonal antibody h6B4-Fab is a potent and safe antithrombotic in a high shear arterial thrombosis model in baboons. Thromb. Haemost. 2008, 100, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Hennan, J.K.; Swillo, R.E.; Morgan, G.A.; Leik, C.E.; Brooks, J.M.; Shaw, G.D.; Schaub, R.G.; Crandall, D.L.; Vlasuk, G.P. Pharmacologic inhibition of platelet vWF-GPIb alpha interaction prevents coronary artery thrombosis. Thromb. Haemost. 2006, 95, 469–475. [Google Scholar] [PubMed]
- Erpenbeck, L.; Nieswandt, B.; Schon, M.; Pozgajova, M.; Schon, M.P. Inhibition of platelet GPIb alpha and promotion of melanoma metastasis. J. Investig. Dermatol. 2010, 130, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; He, Z.; Sai, P.; Faridi, A.; Aziz, A.; Kalavar, M.; Griciene, P.; Gintautas, J.; Steier, W. Inhibition of human CD24 binding to platelet-bound P-selectin by monoclonal antibody. Proc. West Pharmacol. Soc. 2004, 47, 28–29. [Google Scholar] [PubMed]
- Stone, J.P.; Wagner, D.D. P-selectin mediates adhesion of platelets to neuroblastoma and small cell lung cancer. J. Clin. Investig. 1993, 92, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Borsig, L.; Han, H.L.; Varki, N.M.; Varki, A. Distinct selectin ligands on colon carcinoma mucins can mediate pathological interactions among platelets, leukocytes, and endothelium. Am. J. Pathol. 1999, 155, 461–472. [Google Scholar] [CrossRef]
- Garcia, J.; Callewaert, N.; Borsig, L. P-selectin mediates metastatic progression through binding to sulfatides on tumor cells. Glycobiology 2007, 17, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.; Chen, W.X.; Zhu, J.S.; Chen, N.W.; Zhou, T.; Yao, M.; Zhang, D.Q.; Wu, Y.L. Effect of P-selectin monoclonal antibody on metastasis of gastric cancer and immune function. World J. Gastroenterol. 2003, 9, 1607–1610. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic Signalling: Therapeutic Developments. Front. Pharmacol. 2017, 8, 661. [Google Scholar] [CrossRef] [PubMed]
- Leon, C.; Ravanat, C.; Freund, M.; Cazenave, J.P.; Gachet, C. Differential involvement of the P2Y1 and P2Y12 receptors in platelet procoagulant activity. Arterioscler. Thromb. 2003, 23, 1941–1947. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Evans, R.J.; Mahaut-Smith, M.P. Ca2+ influx through P2X1 receptors amplifies P2Y1 receptor-evoked Ca2+ signaling and ADP-evoked platelet aggregation. Mol. Pharmacol. 2014, 86, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Hollopeter, G.; Jantzen, H.M.; Vincent, D.; Li, G.; England, L.; Ramakrishnan, V.; Yang, R.B.; Nurden, P.; Nurden, A.; Julius, D.; et al. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature 2001, 409, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Huang, D.; Zhou, R.; Yue, M.; Xu, T.; Yang, J.; He, L.; Tian, H.; Liu, X.; Zeng, J. Activation of dorsal horn cannabinoid CB2 receptor suppresses the expression of P2Y12 and P2Y13 receptors in neuropathic pain rats. J. Neuroinflamm. 2017, 14, 185. [Google Scholar] [CrossRef] [PubMed]
- Muniz, V.S.; Baptista-Dos-Reis, R.; Benjamim, C.F.; Mata-Santos, H.A.; Pyrrho, A.S.; Strauch, M.A.; Melo, P.A.; Vicentino, A.R.; Silva-Paiva, J.; Bandeira-Melo, C. Purinergic P2Y12 receptor activation in eosinophils and the schistosomal host response. PLoS ONE 2015, 10, e0139805. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Boeynaems, J.M. Purinergic signalling and immune cells. Purinergic Signal. 2014, 10, 529–564. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M. The platelet P2 receptors in inflammation. Hamostaseologie 2015, 3, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Ballerini, P.; Dovizio, M.; Bruno, A.; Tacconelli, S.; Patrignani, P. P2Y12 Receptors in Tumorigenesis and Metastasis. Front. Pharmacol. 2018, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Czajkowski, R.; Lei, L.; Sabała, P.; Barańska, J. ADP-evoked phospholipase C stimulation and adenylyl cyclase inhibition in glioma C6 cells occur through two distinct nucleotide receptors, P2Y(1) and P2Y(12). FEBS Lett. 2002, 513, 179–183. [Google Scholar] [CrossRef]
- Burnstock, G.; Di Virgilio, F. Purinergic signalling and cancer. Purinergic Signal. 2013, 9, 491–540. [Google Scholar] [CrossRef] [PubMed]
- Sarangi, S.; Pandey, A.; Papa, A.L.; Sengupta, P.; Kopparam, J.; Dadwal, U.; Basu, S.; Sengupta, S. P2Y12 receptor inhibition augments cytotoxic effects of cisplatin in breast cancer. Med. Oncol. 2013, 30, 567. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M. New P2Y(12) inhibitors. Circulation 2010, 121, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Schrör, K.; Siller-Matula, J.M.; Huber, K. Pharmacokinetic basis of the antiplatelet action of prasugrel. Fundam. Clin. Pharmacol. 2012, 26, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.S.; Noh, K.; Haemmerle, M.; Li, D.; Park, H.; Hu, Q.; Hisamatsu, T.; Mitamura, T.; Mak, S.L.C.; Kunapuli, S.; et al. Role of ADP receptors on platelets in the growth of ovarian cancer. Blood 2017, 130, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Y.; Li, D.; Zhang, L.; Wang, K.; Zuo, Y.; Gartner, T.K.; Liu, J. Platelet P2Y12 is involved in murine pulmonary metastasis. PLoS ONE 2013, 8, e80780. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Antman, E.M.; Gibson, C.M.; Montalescot, G.; Riesmeyer, J.; Weerakkody, G.; Winters, K.J.; Warmke, J.W.; McCabe, C.H.; Braunwald, E. TRITON-TIMI 38 Investigators. Evaluation of prasugrel compared with clopidogrel in patients with acute coronary syndromes: Design and rationale for the TRial to assess Improvement in Therapeutic Outcomes by optimizing platelet InhibitioN with prasugrel Thrombolysis in Myocardial Infarction 38 (TRITON-TIMI 38). Am. Heart J. 2006, 152, 627–635. [Google Scholar] [PubMed]
- Serebruany, V.; Floyd, J.; Chew, D. Excess of solid cancers after prasugrel: The Food and Drug Administration outlook. Am. J. Ther. 2010. [Google Scholar] [CrossRef] [PubMed]
- Mauri, L.; Kereiakes, D.J.; Yeh, R.W.; Driscoll-Shempp, P.; Cutlip, D.E.; Steg, P.G.; Normand, S.L.; Braunwald, E.; Wiviott, S.D.; Cohen, D.J.; et al. Twelve or 30 months of dual antiplatelet therapy after drug-eluting stents. N. Engl. J. Med. 2014, 371, 2155–2166. [Google Scholar] [CrossRef] [PubMed]
- Kotronias, R.A.; Kwok, C.S.; Wong, C.W.; Kinnaird, T.; Zaman, A.; Mamas, M.A. Cancer event rate and mortality with thienopyridines: A systematic review and meta-analysis. Drug Saf. 2017, 40, 229–240. [Google Scholar] [CrossRef] [PubMed]
- FDA. Plavix (Clopidogrel): Drug Safety Communication—Long-Term Treatment Does Not Change Risk of Death. 2015. Available online: http://www.fda.gov/Drugs/DrugSafety/ucm471286.htm (accessed on 19 September 2018).
- Andersen, H.; Greenberg, D.L.; Fujikawa, K.; Xu, W.; Chung, D.W.; Davie, E.W. Protease-activated receptor 1 is the primary mediator of thrombin-stimulated platelet procoagulant activity. Proc. Natl. Acad. Sci. USA 1999, 96, 11189–11193. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, S.R. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J. Thromb. Haemost. 2005, 3, 1800–1814. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, S.R. Thrombin signalling and protease-activated receptors. Nature 2000, 407, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Boire, A.; Covic, L.; Agarwal, A.; Jacques, S.; Sherifi, S.; Kuliopulos, A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 2005, 120, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Grisaru-Granovsky, S.; Salah, Z.; Maoz, M.; Pruss, D.; Beller, U.; Bar-Shavit, R. Differential expression of protease activated receptor 1 (Par1) and pY397FAK in benign and malignant human ovarian tissue samples. Int. J. Cancer 2005, 113, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Massi, D.; Naldini, A.; Ardinghi, C.; Carraro, F.; Franchi, A.; Paglierani, M.; Tarantini, F.; Ketabchi, S.; Cirino, G.; Hollenberg, M.D.; et al. Expression of protease-activated receptors 1 and 2 in melanocytic nevi and malignant melanoma. Hum. Pathol. 2005, 36, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Black, P.C.; Mize, G.J.; Karlin, P.; Greenberg, D.L.; Hawley, S.J.; True, L.D.; Vessella, R.L.; Takayama, T.K. Overexpression of protease-activated receptors-1,-2, and-4 (PAR-1, -2, and -4) in prostate cancer. Prostate 2007, 67, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, D.; Hirono, Y.; Goi, T.; Katayama, K.; Yamaguchi, A. Prognostic value of protease-activated receptor-1 (PAR-1) and matrix metalloproteinase-1 (MMP-1) in gastric cancer. Anticancer Res. 2008, 28, 847–854. [Google Scholar] [PubMed]
- Song, J.S.; Kang, C.M.; Park, C.K.; Yoon, H.K. Thrombin induces epithelial-mesenchymal transition via PAR-1, PKC, and ERK1/2 pathways in A549 cells. Exp. Lung Res. 2013, 39, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Chen, S.; Qin, Y.; Zhang, H.; Wang, H.; Meng, J.; Huai, L.; Zhang, Q.; Yin, T.; Lei, Y.; et al. Doxycycline inhibits breast cancer EMT and metastasis through PAR-1/NF-κB/miR-17/E-cadherin pathway. Oncotarget 2017, 8, 104855–104866. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Shin, J.Y.; Lee, K.D.; Bae, Y.K.; Choi, I.J.; Park, S.H.; Chun, K.H. Galectin-3 facilitates cell motility in gastric cancer by up-regulating protease-activated receptor-1 (PAR-1) and matrix metalloproteinase-1 (MMP-1). PLoS ONE 2011, 6, e25103. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Majoor, C.J.; Roelofs, J.J.; de Kruif, M.D.; Horlings, H.M.; Borensztajn, K.; Spek, C.A. Potential importance of protease activated receptor (PAR)-1 expression in the tumor stroma of non-small-cell lung cancer. BMC Cancer 2017, 17, 113. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.N.; Rosenfeldt, L.; Frederick, M.; Miller, W.; Waltz, D.; Kombrinck, K.; McElhinney, K.E.; Flick, M.J.; Monia, B.P.; Revenko, A.S. Colon Cancer Growth and Dissemination Relies upon Thrombin, Stromal PAR-1, and Fibrinogen. Cancer Res. 2015, 75, 4235–4243. [Google Scholar] [CrossRef] [PubMed]
- Wang, A. Review of vorapaxar for the prevention of atherothrombotic events. Expert Opin. Pharmacother. 2015, 16, 2509–2522. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Boucher, M.; Kaunelis, D. PAR-1 Antagonists: An Emerging Antiplatelet Drug Class. CADTH Issues in Emerging Health Technologies; Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, Canada, 2016. [Google Scholar]
- Wang, J.; Kulkarni, A.; Chintala, M.; Fink, L.M.; Hauer-Jensen, M. Inhibition of protease-activated receptor 1 ameliorates intestinal radiation mucositis in a preclinical rat model. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Hashemzadeh, M.; Furukawa, M.; Goldsberry, S.; Movahed, M.R. Chemical structures and mode of action of intravenous glycoprotein IIb/IIIa receptor blockers: A review. Exp. Clin. Cardiol. 2008, 13, 192–197. [Google Scholar] [PubMed]
- Wagner, C.L.; Mascelli, M.A.; Neblock, D.S.; Weisman, H.F.; Coller, B.S.; Jordan, R.E. Analysis of GPIIb/IIIa receptor number by quantification of 7E3 binding to human platelets. Blood 1996, 88, 907–914. [Google Scholar] [PubMed]
- Rivera, J.; Lozano, M.L.; Navarro-Nunez, L.; Vicente, V. Platelet receptors and signaling in the dynamics of thrombus formation. Haematologica 2009, 700–711, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Kloczewiak, M.; Timmons, S.; Hawiger, J. Recognition site for the platelet receptor is present on the 15-residue carboxy-terminal fragment of the gamma chain of human fibrinogen and is not involved in the fibrin polymerization reaction. Thromb. Res. 1983, 29, 249–255. [Google Scholar] [CrossRef]
- Ni, H.; Denis, C.V.; Subbarao, S.; Degen, J.L.; Sato, T.N.; Hynes, R.O.; Wagner, D.D. Persistence of platelet thrombus formation in arterioles of mice lacking both von Willebrand factor and fibrinogen. J. Clin. Investig. 2000, 106, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Reheman, A.; Chen, P.; Zhu, G.; Hynes, R.O.; Freedman, J.; Wagner, D.D.; Ni, H. Fibrinogen and von Willebrand factor-independent platelet aggregation in vitro and in vivo. J. Thromb. Haemost. 2006, 4, 2230–2237. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gallant, R.C.; Ni, H. Extracellular matrix proteins in the regulation of thrombus formation. Curr. Opin. Hematol. 2016, 23, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Amirkhosravi, A.; Mousa, S.A.; Amaya, M.; Blaydes, S.; Desai, H.; Meyer, T.; Francis, J.L. Inhibition of tumor cell-induced platelet aggregation and lung metastasis by the oral GpIIb/IIIa antagonist XV454. Thromb. Haemost. 2003, 90, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Dang, S.; Hong, T.; Tang, J.; Fan, J.; Bu, D.; Sun, Y.; Wang, Z.; Wisniewski, T. A humanized single-chain antibody against beta 3 integrin inhibits pulmonary metastasis by preferentially fragmenting activated platelets in the tumor microenvironment. Blood 2012, 120, 2889–2898. [Google Scholar] [CrossRef] [PubMed]
- Bakewell, S.J.; Nestor, P.; Prasad, S.; Tomasson, M.H.; Dowland, N.; Mehrotra, M.; Scarborough, R.; Kanter, J.; Abe, K.; Phillips, D.; et al. Platelet and osteoclast beta 3 integrins are critical for bone metastasis. Proc. Nat. Acad. Sci. USA 2003, 100, 14205–14210. [Google Scholar] [CrossRef] [PubMed]
- Lonsdorf, A.S.; Krämer, B.F.; Fahrleitner, M.; Schönberger, T.; Gnerlich, S.; Ring, S.; Gehring, S.; Schneider, S.W.; Kruhlak, M.J.; Meuth, S.G.; et al. Engagement of αIIbβ3 (GPIIb/IIIa) with ανβ3 integrin mediates interaction of melanoma cells with platelets: A connection to hematogenous metastasis. J. Biol. Chem. 2012, 287, 2168–2178. [Google Scholar] [CrossRef] [PubMed]
- Boukerche, H.; Berthier-Vergnes, O.; Bailly, M.; Doré, J.F.; Leung, L.L.; McGregor, J.L. A monoclonal antibody (LYP18) directed against the blood platelet glycoprotein IIb/IIIa complex inhibits human melanoma growth in vivo. Blood 1989, 74, 909–912. [Google Scholar] [PubMed]
- Schneider, D.J. Anti-platelet therapy: Glycoprotein IIb-IIIa antagonists. Br. J. Clin. Pharmacol. 2011, 72, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Kalantzi, K.I.; Tsoumani, M.E.; Goudevenos, I.A.; Tselepis, A.D. Pharmacodynamic properties of antiplatelet agents: Current knowledge and future perspectives. Expert Rev. Clin. Pharmacol. 2012, 5, 319–336. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Li, L.; Guan, L.; Yang, H.; Wu, C.; Liu, Y. Roles for GP IIb/IIIa and αvβ3 integrins in MDA-MB-231 cell invasion and shear flow-induced cancer cell mechanotransduction. Cancer Lett. 2014, 344, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Lincoff, A.M.; Califf, R.M.; Topol, E.J. Platelet glycoprotein IIb/IIIa receptor blockade in coronary artery disease. J. Am. Coll. Cardiol. 2000, 35, 1103–1115. [Google Scholar] [CrossRef]
- Chew, D.P.; Bhatt, D.L.; Sapp, S.; Topol, E.J. Increased mortality with oral platelet glycoprotein IIb/IIIa antagonists: A meta-analysis of phase III multicenter randomized trials. Circulation 2001, 103, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, M.; Meade, G.; Stoll, P.; Ylanne, J.; Bassler, N.; Chen, Y.C.; Hagemeyer, C.E.; Ahrens, I.; Moran, N.; Kenny, D.; et al. Conformation-specific blockade of the integrin GPIIb/IIIa: A novel antiplatelet strategy that selectively targets activated platelets. Circ. Res. 2006, 99, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Yap, M.L.; McFadyen, J.D.; Wang, X.; Zia, N.A.; Hohmann, J.D.; Ziegler, M.; Yao, Y.; Pham, A.; Harris, M.; Donnelly, P.S.; et al. Targeting Activated Platelets: A Unique and Potentially Universal Approach for Cancer Imaging. Theranostics 2017, 7, 2565–2574. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Watson, S.P. Platelet-collagen interaction: Is GPVI the central receptor? Blood 2003, 102, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Ungerer, M.; Münch, G. Novel antiplatelet drugs in clinical development. Thromb. Haemost. 2013, 110, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Ungerer, M.; Rosport, K.; Bültmann, A.; Piechatzek, R.; Uhland, K.; Schlieper, P.; Gawaz, M.; Münch, G. Novel antiplatelet drug revacept (dimeric glycoprotein VI-Fc) specifically and efficiently inhibited collagen-induced platelet aggregation without affecting general hemostasis in humans. Circulation 2011, 123, 1891–1899. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Russell, S.; Ware, J. Platelet glycoprotein VI facilitates experimental lung metastasis in syngenic mouse models. J. Thromb. Haemost. 2009, 7, 1713–1717. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Hara, A.; Xiao, C.Y.; Okada, Y.; Takahata, O.; Nakaya, K.; Sugimoto, Y.; Ichikawa, A.; Narumiya, S.; Ushikubi, F. Increased bleeding tendency and decreased susceptibility to thromboembolism in mice lacking the prostaglandin E receptor subtype EP(3). Circulation 2001, 104, 1176–1180. [Google Scholar] [CrossRef] [PubMed]
- Fabre, J.E.; Nguyen, M.; Athirakul, K.; Coggins, K.; McNeish, J.D.; Austin, S.; Parise, L.K.; FitzGerald, G.A.; Coffman, T.M.; Koller, B.H. Activation of the murine EP3 receptor for PGE2 inhibits cAMP production and promotes platelet aggregation. J. Clin. Investig. 2001, 107, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; Tilly, P.; Hentsch, D.; Vonesch, J.L.; Fabre, J.E. Vascular wall-produced prostaglandin E2 exacerbates arterial thrombosis and atherothrombosis through platelet EP3 receptors. J. Exp. Med. 2007, 204, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Tilly, P.; Charles, A.L.; Ludwig, S.; Slimani, F.; Gross, S.; Meilhac, O.; Geny, B.; Stefansson, K.; Gurney, M.E.; Fabre, J.E. Blocking the EP3 receptor for PGE2 with DG-041 decreases thrombosis without impairing haemostatic competence. Cardiovasc. Res. 2014, 101, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.R.; Carrim, N.; Neves, M.A.; McKeown, T.; Stratton, T.W.; Coelho, R.M.; Lei, X.; Chen, P.; Xu, J.; Dai, X.; et al. Platelets and platelet adhesion molecules: Novel mechanisms of thrombosis and anti-thrombotic therapies. Thromb. J. 2016, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Erpenbeck, L.; Schön, M.P. Deadly allies: The fatal interplay between platelets and metastasizing cancer cells. Blood 2010, 29, 3427–3436. [Google Scholar] [CrossRef] [PubMed]
- Koedam, J.A.; Cramer, E.M.; Briend, E.; Furie, B.; Furie, B.C.; Wagner, D.D. P-selectin, a granule membrane protein of platelets and endothelial cells, follows the regulated secretory pathway in AtT-20 cells. J. Cell Biol. 1992, 116, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Geng, J.G. P-selectin mediates adhesion of leukocytes, platelets, and cancer cells in inflammation, thrombosis, and cancer growth and metastasis. Arch. Immunol. Ther. Exp. 2006, 54, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Radziwon-Balicka, A.; Santos-Martinez, M.J.; Corbalan, J.J.; O’Sullivan, S.; Treumann, A.; Gilmer, J.F.; Radomski, M.W.; Medina, C. Mechanisms of platelet-stimulated colon cancer invasion: Role of clusterin and thrombospondin 1 in regulation of the P38MAPK-MMP-9 pathway. Carcinogenesis 2014, 35, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Egan, K.; Crowley, D.; Smyth, P.; O’Toole, S.; Spillane, C.; Martin, C.; Gallagher, M.; Canney, A.; Norris, L.; Conlon, N.; et al. Platelet adhesion and degranulation induce pro-survival and pro-angiogenic signalling in ovarian cancer cells. PLoS ONE 2011, 6, e26125. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Borsig, L.; Varki, N.M.; Varki, A. P-selectin deficiency attenuates tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 1998, 95, 9325–9330. [Google Scholar] [CrossRef] [PubMed]
- Dovizio, M.; Alberti, S.; Guillem-Llobat, P.; Patrignani, P. Role of platelets in inflammation and cancer: Novel therapeutic strategies. Basic Clin. Pharmacol. Toxicol. 2014, 114, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Patrono, C. The Multifaceted Clinical Readouts of Platelet Inhibition by Low-Dose Aspirin. J. Am. Coll. Cardiol. 2015, 66, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Bibbins-Domingo, K.; U.S. Preventive Services Task Force. Aspirin use for the primary prevention of cardiovascular disease and colorectal cancer: U.S. Preventive Services Task Force Recommendation Statement. Ann. Intern. Med. 2016, 164, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Best, M.G.; Sol, N.; Kooi, I.; Tannous, J.; Westerman, B.A.; Rustenburg, F.; Schellen, P.; Verschueren, H.; Post, E.; Koster, J.; et al. RNA-Seq of tumoreducated platelets enables blood-based pan-cancer, multiclass, and molecular pathway cancer diagnostics. Cancer Cell 2015, 28, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Best, M.G.; Sol, N.; GJG, S.; Vancura, A.; Muller, M.; Niemeijer, A.N.; Fejes, A.V.; Fat, L.A.T.K.; Huis, A.E.; Leurs, C.; et al. Swarm intelligence-enhanced detection of nonsmall cell lung cancer using tumor-educated platelets. Cancer Cell 2017, 32, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Best, M.G.; Vancura, A.; Wurdinger, T. Platelet RNA as a circulating biomarker trove for cancer diagnostics. J. Thromb. Haemost. 2017, 15, 1295–1306. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Drug Class | Drug Target | Agents | In Vivo and in Vitro Studies | Reported Effects |
|---|---|---|---|---|
| NSAIDs | Platelet COX-1 | Low-dose aspirin | In vitro co-culture of platelets and human colon adenocarcinoma cell line HT29 [29] HT29-induced hematogenous metastasis in vivo [29] In vivo mouse model of chronic hepatitis B [58] | Prevention of platelet-induced EMT and migration (disruption of cancer cell metastatic potential) [29] Prevention of platelet-induced metastatic and prothrombotic phenotype [29] Prevention of immune-mediated liver injury and fibrosis and HCC development (in combination with clopidogrel) [58] |
| Thienopyridines | ADP receptor P2Y12 | TiclopidineTicagrerol | In vivo model of spontaneous lung metastasis [59] In vitro co-culture of platelets and HT29 colon cancer cells [29] In vitro co-culture of platelets human breast cancer cell lines (MCF-7, MDA-MB-468, and MDA-MB-231) [60] Orthotopic 4T1 breast cancer model [60] | Suppression of metastasis dissemination [59] Prevention of platelet-induced EMT and migration (disruption of cancer cell metastatic potential) [29] Prevention of platelet–cancer cell crosstalk [60] Reduction of metastasis formation and number of tumor cell-platelet aggregates and improvement of survival [60] |
| PAR-1 antagonists | Protease-activated receptor PAR-1 | Vorapaxar | In vitro studies with human ovarian cancer cells (SKOV-3, OVCAR-3 and CaOV-3) [61] | Reduction of PAR-1 agonist-mediated effects including cell proliferation [61] |
| Glycoprotein IIb/IIIa antagonists | Glycoprotein (GP) IIb/IIIa | AbciximabEpitifabideTirofiban | MCF-7 breast cancer cells [62] In vitro co-culture of thrombin-activated platelets and human breast carcinoma MDA-MB-231 cells [62] MCF-7 breast cancer cells [62] In vitro studies with highly invasive human tongue squamous carcinoma cell line HSC-3 [63] | Tumorigenesis and metastasis control [62] Constriction of tumor cell invasive potential [62] Tumorigenesis and metastasis control [62] Inhibition of promigratory effect induced by Col15 [63] |
| GPIb inhibitors | Platelet GPIb | Anfibatide anti-GPIbα antibody (h6B4-Fab, GPG-290, and anti-GPIbα) | In vitro and in vivo murine models of thrombosis [64] and phase II human clinical trials [65] High shear arterial thrombosis model in baboons [66] Canine model of artery thrombosis [67] In vivo metastasis model B16F10 melanoma cells [68] | Inhibition of platelet adhesion, aggregation and thrombus formation, without increasing bleeding time [64,65] Reduction of thrombus formation at an injured femoral artery site [66] Prevention of coronary artery thrombosis [67] Promotion of melanoma metastasis [68] |
| P-selectin (CD62P) inhibitors | Platelet P-selectin and tumor P-selectin ligands | Anti-P-selectin antibody (GA-6), P-selectin Mab, anti-CD24 (P-selectin ligand) antibody FL80 | Prostate cancer cell line DU145 [69] Mucin-type ligands bearingsialyl-Lewis X small-celllung cancers, colon cancer and neuroblastoma [70,71] Colon cancer cells MC-38 expressing sulfatedgalactosylceramide-typeligands [72] Murine model of gastric cancer [73] | Prevention of platelet binding to prostate cancer cells [69] Prevention of P-selectin adhesion of platelets to cancer cells [70,71] Prevention of P-selectin-mediated metastasis progression [72] Reduction of gastric cancer metastasis [73] |
| GPVI antagonists | Platelet GPVI | Revacept | In vitro co-culture of platelets and human colon adenocarcinoma cell line HT29 [30] | Prevention of platelet-induced COX-2 upregulation and EMT [30] |
| EP3 antagonist | PGE2 receptor EP3 | DG041 | In vitro co-culture of platelets and human colon adenocarcinoma cell line HT29 [29] | Prevention of platelet-induced EMT and migration (disruption of cancer cell metastatic potential) [29] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruno, A.; Dovizio, M.; Tacconelli, S.; Contursi, A.; Ballerini, P.; Patrignani, P. Antithrombotic Agents and Cancer. Cancers 2018, 10, 253. https://doi.org/10.3390/cancers10080253
Bruno A, Dovizio M, Tacconelli S, Contursi A, Ballerini P, Patrignani P. Antithrombotic Agents and Cancer. Cancers. 2018; 10(8):253. https://doi.org/10.3390/cancers10080253
Chicago/Turabian StyleBruno, Annalisa, Melania Dovizio, Stefania Tacconelli, Annalisa Contursi, Patrizia Ballerini, and Paola Patrignani. 2018. "Antithrombotic Agents and Cancer" Cancers 10, no. 8: 253. https://doi.org/10.3390/cancers10080253
APA StyleBruno, A., Dovizio, M., Tacconelli, S., Contursi, A., Ballerini, P., & Patrignani, P. (2018). Antithrombotic Agents and Cancer. Cancers, 10(8), 253. https://doi.org/10.3390/cancers10080253