The Osteoclast in Bone Metastasis: Player and Target


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
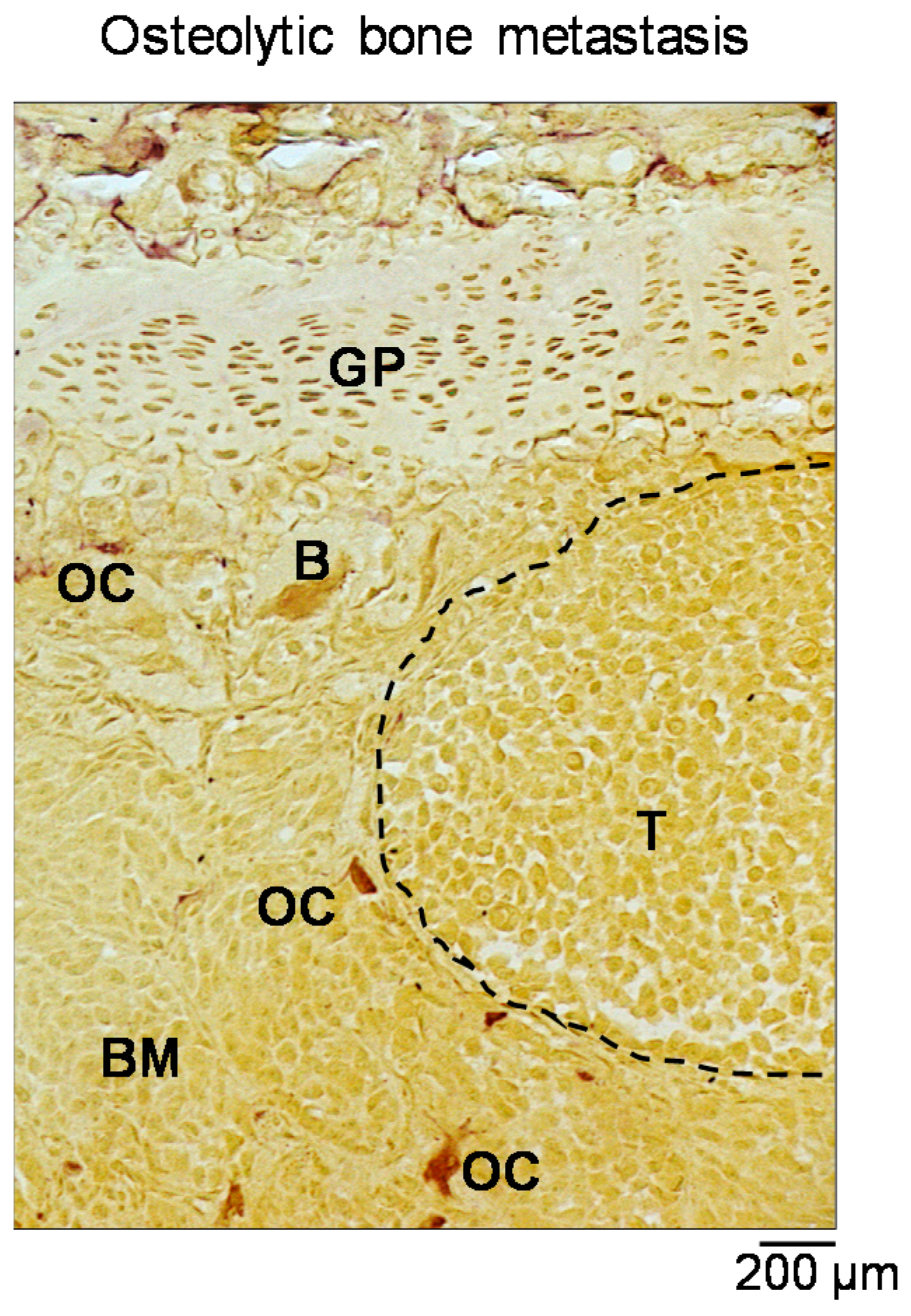
1. Bone Metastases: Pathological Features
2. Bone Physiology
2.1. Bone Remodeling: The Virtuous Cycle
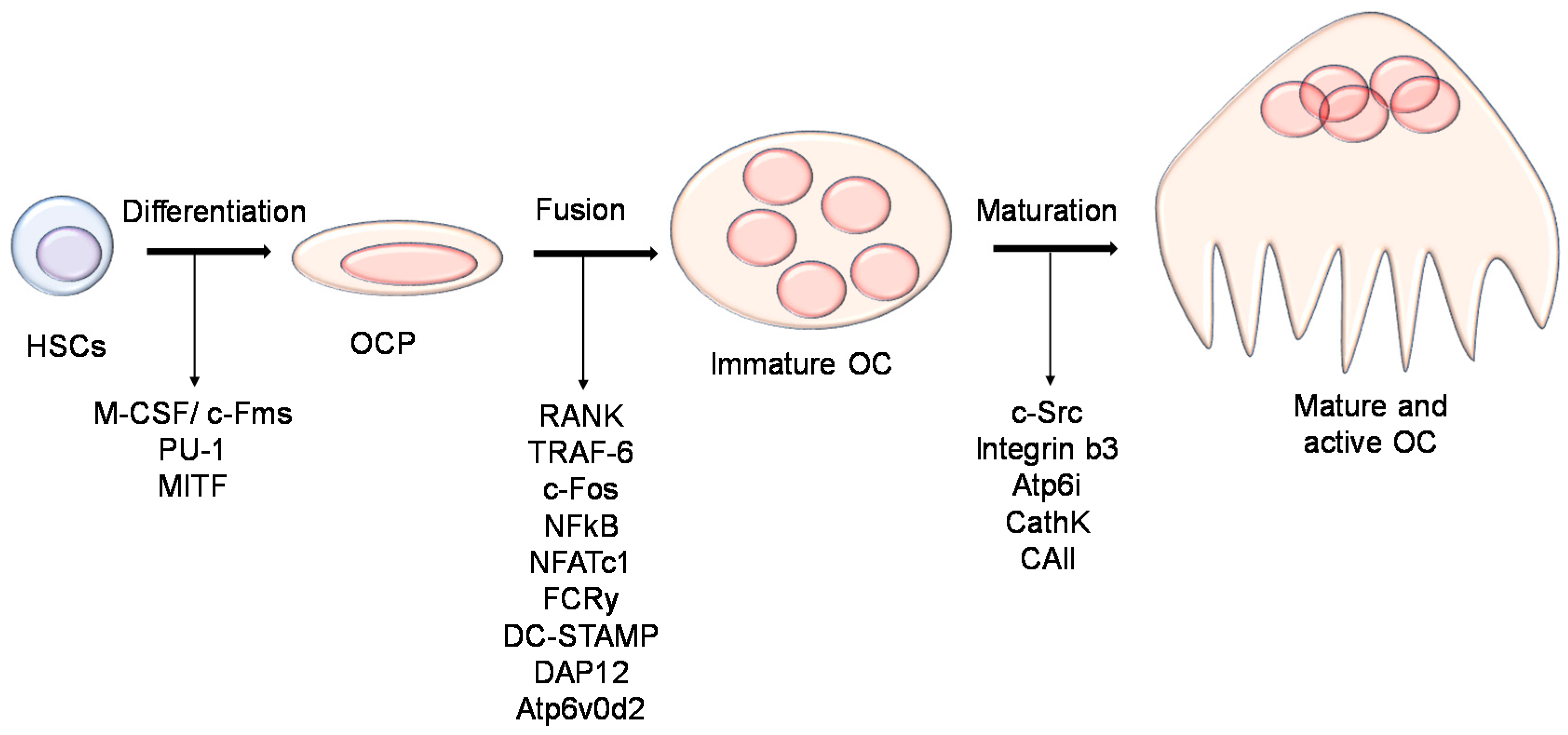
2.2. Biology of the Osteoclast
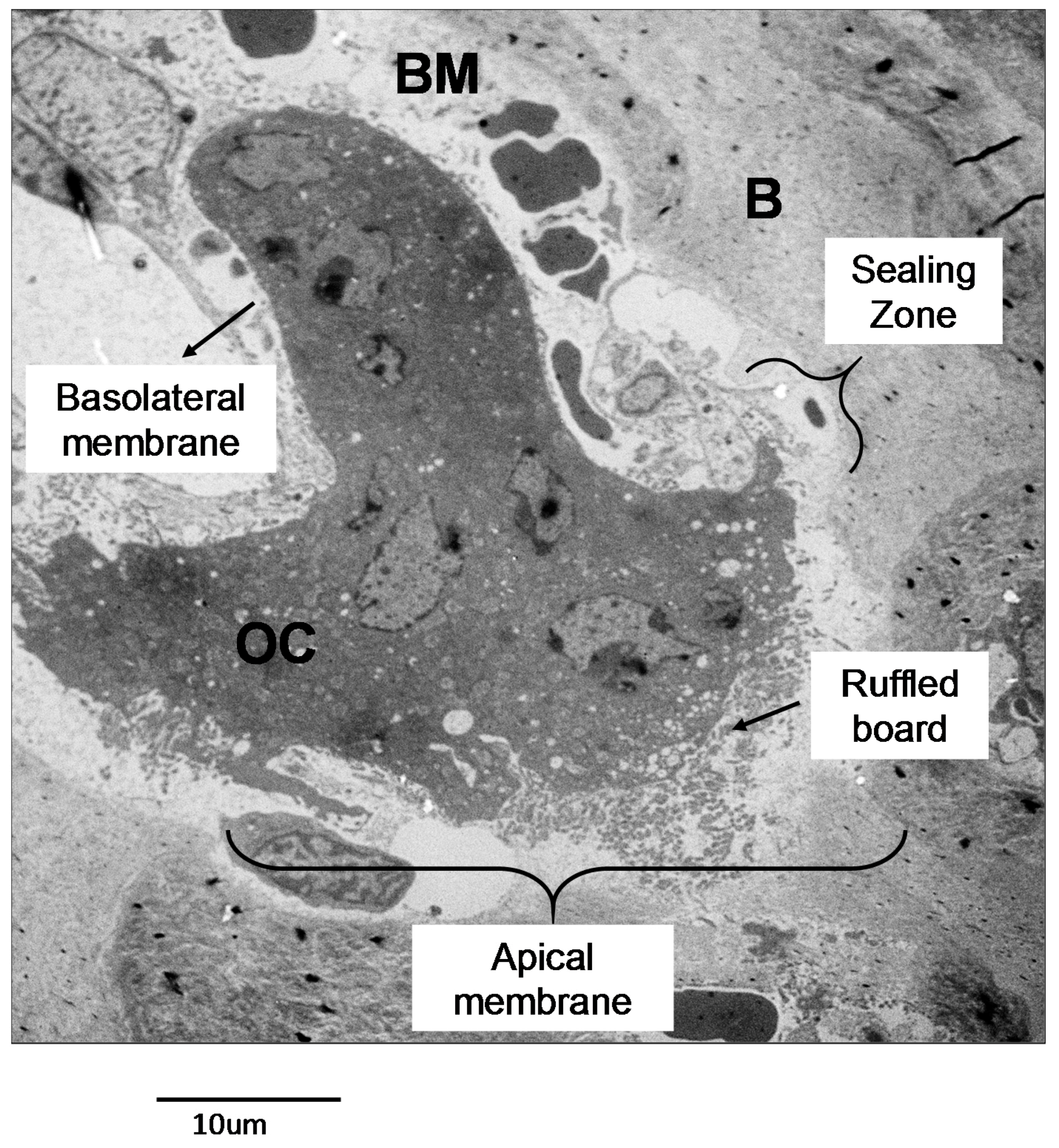
2.2.1. Osteoclast Function
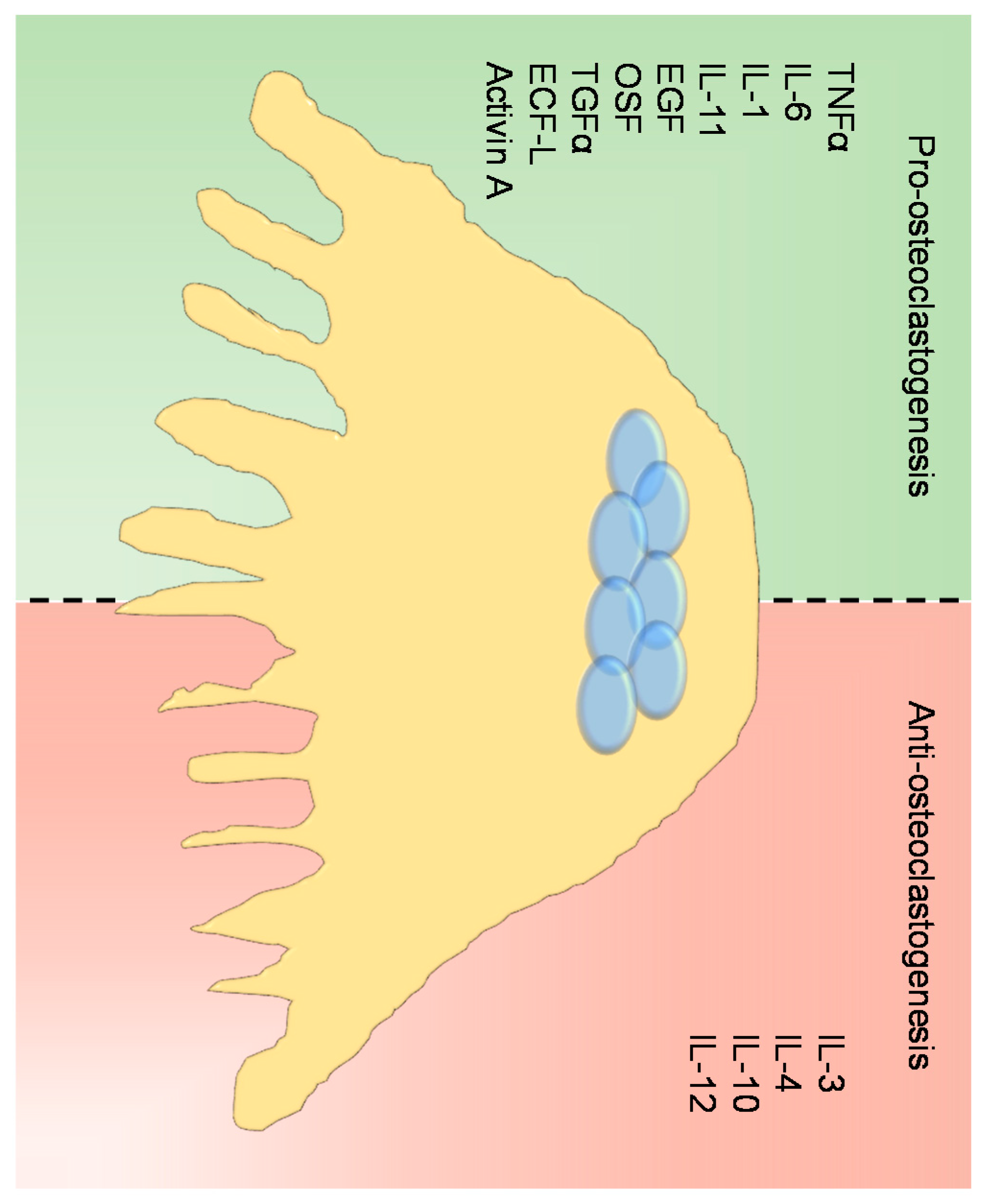
2.2.2. Regulation of Osteoclast Differentiation and Activity
3. Alterations of Bone Remodelling: The Vicious Cycle
4. The Osteoclast as Therapeutic Target
4.1. Current Antiresorptive Therapies
4.1.1. Bisphosphonates
4.1.2. Denosumab
4.2. Anti Resorptive Treatment as an Adjuvant Therapy in Breast Cancer Patients
4.3. Antitumoral Effects of Antiresorptive Therapies
4.4. New Antiresorptive Therapeutic Strategies
4.4.1. Cathepsin K Inhibitors
4.4.2. c-src Inhibitors
4.4.3. Inhibitors of Integrins
5. Anabolic Treatments
Antisclerostin Antibodies
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Coleman, R.E. Metastatic bone disease: Clinical features, pathophysiology and treatment strategies. Cancer Treat. Rev. 2001, 27, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.E.; Smith, P.; Rubens, R.D. Clinical course and prognostic factors following bone recurrence from breast cancer. Br. J. Cancer 1998, 77, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Dawson, N. How is androgen-dependent metastatic prostate cancer best treated? Hematol. Oncol. Clin. N. Am. 1996, 10, 727–747. [Google Scholar] [CrossRef]
- Koizumi, M.; Yoshimoto, M.; Kasumi, F.; Ogata, E. Comparison between solitary and multiple skeletal metastatic lesions of breast cancer patients. Ann. Oncol. 2003, 14, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.; De Boer, R.; Eidtmann, H.; Llombart, A.; Davidson, N.; Neven, P.; Von Minckwitz, G.; Sleeboom, H.P.; Forbes, J.; Barrios, C.; et al. Zoledronic acid (zoledronate) for postmenopausal women with early breast cancer receiving adjuvant letrozole (ZO-FAST study): Final 60-month results. Ann. Oncol. 2013, 24, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.; Lipton, A.; Hadji, P.; Chen, Y.M.; Kosmidis, P. Treatment of bone metastases before the onset of pain. Int. J. Clin. Oncol. 2013, 18, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Selvaggi, G.; Scagliotti, G.V. Management of bone metastases in cancer: A review. Crit. Rev. Oncol. Hematol. 2005, 56, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Clohisy, D.R.; Mantyh, P.W. Bone cancer pain. Cancer 2003, 97, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Roodman, G.D. Mechanisms of Bone Metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef]
- Kenkre, J.S.; Bassett, J.H. The Bone Remodelling Cycle. Ann. Clin. Biochem. 2018, 55, 308–327. [Google Scholar] [CrossRef] [PubMed]
- Hauge, E.M.; Qvesel, D.; Eriksen, E.F.; Mosekilde, L.; Melsen, F. Cancellous bone remodeling occurs in specialized compartments lined by cells expressing osteoblastic markers. J. Bone Miner. Res. 2001, 16, 1575–1582. [Google Scholar] [CrossRef] [PubMed]
- Everts, V.; Delaissié, J.M.; Korper, W.; Jansen, D.C.; Tigchelaar-Gutter, W.; Saftig, P.; Beertsen, W. The bone lining cell: Its role in cleaning Howship’s lacunae and initiating bone formation. J. Bone Miner. Res. 2002, 17, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Delaisse, J.-M. The reversal phase of the bone-remodeling cycle: Cellular prerequisites for coupling resorption and formation. Bonekey Rep. 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Anderson, H.C.; Garimella, R.; Tague, S.E. The role of matrix vesicles in growth plate development and biomineralization. Front. Biosci. 2005, 10, 822–837. [Google Scholar] [CrossRef] [PubMed]
- Cappariello, A.; Maurizi, A.; Veeriah, V.; Teti, A. Reprint of: The Great Beauty of the osteoclast. Arch. Biochem. Biophys. 2014, 561, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Colony-stimulating factor-1 receptor. Blood 1990, 75, 1–12. [Google Scholar] [PubMed]
- Wiktor-Jedrzejczak, W.; Bartocci, A.; Ferrante, A.W.; Ahmed-Ansari, A.; Sell, K.W.; Pollard, J.W.; Stanley, E.R. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (OP/OP) mouse. Proc. Natl. Acad. Sci. USA 1990, 87, 4828–4832. [Google Scholar] [CrossRef] [PubMed]
- Soysa, N.S.; Alles, N.; Aoki, K.; Ohya, K. Osteoclast formation and differentiation: An overview. J. Med. Dent. Sci. 2012, 59, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Lacey, D.L.; Timms, E.; Tan, H.L.; Kelley, M.J.; Dunstan, C.R.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA 1998, 95, 3597–3602. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Takayanagi, H. New regulation mechanisms of osteoclast differentiation. Ann. N. Y. Acad. Sci. 2011, 1240. [Google Scholar] [CrossRef] [PubMed]
- Simonet, W.; Lacey, D.; Dunstan, C.; Kelley, M.; Chang, M.-S.; Lüthy, R.; Nguyen, H.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A Novel Secreted Protein Involved in the Regulation of Bone Density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef]
- Kong, Y.Y.; Yoshida, H.; Sarosi, I.; Tan, H.L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; De Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sarosi, I.; Yan, X.Q.; Morony, S.; Capparelli, C.; Tan, H.L.; McCabe, S.; Elliott, R.; Scully, S.; Van, G.; et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc. Natl. Acad. Sci. USA 2000, 97, 1566–1571. [Google Scholar] [CrossRef] [PubMed]
- Bucay, N.; Sarosi, I.; Dunstan, C.R.; Morony, S.; Tarpley, J.; Capparelli, C.; Scully, S.; Tan, H.L.; Xu, W.; Lacey, D.L.; et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Asagiri, M.; Sato, K.; Usami, T.; Ochi, S.; Nishina, H.; Yoshida, H.; Morita, I.; Wagner, E.F.; Mak, T.W.; Serfling, E.; et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J. Exp. Med. 2005, 202, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.I.; et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef]
- Matsumoto, M.; Kogawa, M.; Wada, S.; Takayanagi, H.; Tsujimoto, M.; Katayama, S.; Hisatake, K.; Nogi, Y. Essential role of p38 mitogen-activated protein kinase in cathepsin K gene expression during osteoclastogenesis through association of NFATc1 and PU.1. J. Biol. Chem. 2004, 279, 45969–45979. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Kim, J.H.; Lee, J.; Jin, H.-M.; Lee, S.-H.; Fisher, D.E.; Kook, H.; Kim, K.K.; Choi, Y.; Kim, N. Nuclear factor of activated T cells c1 induces osteoclast-associated receptor gene expression during tumor necrosis factor-related activation-induced cytokine-mediated osteoclastogenesis. J. Biol. Chem. 2005, 280, 35209–35216. [Google Scholar] [CrossRef] [PubMed]
- Crotti, T.N.; Flannery, M.; Walsh, N.C.; Fleming, J.D.; Goldring, S.R.; McHugh, K.P. NFATc1 regulation of the human β3 integrin promoter in osteoclast differentiation. Gene 2006, 372, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Mbalaviele, G.; Chen, H.; Boyce, B.F.; Mundy, G.R.; Yoneda, T. The role of cadherin in the generation of multinucleated osteoclasts from mononuclear precursors in murine marrow. J. Clin. Investig. 1995, 95, 2757–2765. [Google Scholar] [CrossRef] [PubMed]
- Vignery, A. Osteoclasts and giant cells: Macrophage-macrophage fusion mechanism. Int. J. Exp. Pathol. 2000, 81, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Verrier, S.; Hogan, A.; McKie, N.; Horton, M. ADAM gene expression and regulation during human osteoclast formation. Bone 2004, 35, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Yagi, M.; Miyamoto, T.; Sawatani, Y.; Iwamoto, K.; Hosogane, N.; Fujita, N.; Morita, K.; Ninomiya, K.; Suzuki, T.; Miyamoto, K.; et al. DC-STAMP is essential for cell–cell fusion in osteoclasts and foreign body giant cells. J. Exp. Med. 2005, 202, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Rho, J.; Jeong, D.; Sul, J.Y.; Kim, T.; Kim, N.; Kang, J.S.; Miyamoto, T.; Suda, T.; Lee, S.K.; et al. V-ATPase V0subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat. Med. 2006, 12, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Marchisio, P.C.; Cirillo, D.; Naldini, L.; Primavera, M.V.; Teti, A.; Zambonin-Zallone, A. Cell-substratum interaction of cultured avian osteoclasts is mediated by specific adhesion structures. J. Cell Biol. 1984, 99, 1696–1705. [Google Scholar] [CrossRef] [PubMed]
- Schachtner, H.; Calaminus, S.D.J.; Thomas, S.G.; Machesky, L.M. Podosomes in adhesion, migration, mechanosensing and matrix remodeling. Cytoskeleton 2013, 70, 572–589. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, P.H.; Blair, H.C.; Teitelbaum, S.L.; Edwards, J.C. Characterization of the osteoclast ruffled border chloride channel and its role in bone resorption. J. Biol. Chem. 1997, 272, 18636–18643. [Google Scholar] [CrossRef] [PubMed]
- Baron, R. Polarity and membrane transport in osteoclasts. Connect. Tissue Res. 1989, 20, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Mulari, M.; Vääräniemi, J.; Väänänen, H.K. Intracellular membrane trafficking in bone resorbing osteoclasts. Microsc. Res. Tech. 2003, 61, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Hirvonen, M.J.; Fagerlund, K.; Lakkakorpi, P.; Väänänen, H.K.; Mulari, M.T.K. Novel perspectives on the transcytotic route in osteoclasts. Bonekey Rep. 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.C.; Kahn, A.J.; Crouch, E.C.; Jeffrey, J.J.; Teitelbaum, S.L. Isolated osteoclasts resorb the organic and inorganic components of bone. J. Cell Biol. 1986, 102, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Baron, R. Molecular mechanisms of bone resorption by the osteoclast. Anat. Rec. 1989, 224, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Sobacchi, C.; Schulz, A.; Coxon, F.P.; Villa, A.; Helfrich, M.H. Osteopetrosis: Genetics, treatment and new insights into osteoclast function. Nat. Rev. Endocrinol. 2013, 9, 522–536. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Guerrini, M.M.; Cassani, B.; Pangrazio, A.; Sobacchi, C. Infantile malignant, autosomal recessive osteopetrosis: The rich and the poor. Calcif. Tissue Int. 2009, 84, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Naka, K.; Kawamura, K.; Matsumoto, T.; Nakanishi, I.; Fujimoto, N.; Sato, H.; Seiki, M. Localization of matrix metalloproteinase 9 (92-kilodalton gelatinase/type IV collagenase = gelatinase B) in osteoclasts: Implications for bone resorption. Lab. Investig. 1995, 72, 311–322. [Google Scholar] [PubMed]
- Zaidi, M.; Troen, B.; Moonga, B.S.; Abe, E. Cathepsin K, osteoclastic resorption, and osteoporosis therapy. J. Bone Miner. Res. 2001, 16, 1747–1749. [Google Scholar] [CrossRef] [PubMed]
- Colnot, C. Altered fracture repair in the absence of MMP9. Development 2003, 130, 4123–4133. [Google Scholar] [CrossRef] [PubMed]
- Del Fattore, A.; Teti, A.; Rucci, N. Osteoclast receptors and signaling. Arch. Biochem. Biophys. 2008, 473, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Takahashi, N.; Jimi, E.; Udagawa, N.; Takami, M.; Kotake, S.; Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; et al. Tumor Necrosis Factor α Stimulates Osteoclast Differentiation by a Mechanism Independent of the ODF/RANKL-RANK Interaction. J. Exp. Med. 2000, 191, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.; Takeshita, S.; Barker, J.E.; Kanagawa, O.; Ross, F.P.; Teitelbaum, S.L. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Investig. 2000, 106, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, P.; Persson, E.; Conaway, H.H.; Lerner, U.H. IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF-kappa B ligand, osteoprotegerin, and receptor activator of NF-kappa B in mouse calvariae. J. Immunol. 2002, 169, 3353–3362. [Google Scholar] [CrossRef] [PubMed]
- Kudo, O.; Sabokbar, A.; Pocock, A.; Itonaga, I.; Fujikawa, Y.; Athanasou, N.A. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone 2003, 32, 1–7. [Google Scholar] [CrossRef]
- Cronstein, B.N. Interleukin-6 a key mediator of systemic and local symptoms in rheumatoid arthritis. Bull. NYU Hosp. Jt. Dis. 2007, 65 (Suppl. 1), S11–S15. [Google Scholar] [PubMed]
- Yao, Z.; Xing, L.; Qin, C.; Schwarz, E.M.; Boyce, B.F. Osteoclast precursor interaction with bone matrix induces osteoclast formation directly by an interleukin-1-mediated autocrine mechanism. J. Biol. Chem. 2008, 283, 9917–9924. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; MacDonald, B.R.; Hon, J.; Winkler, M.E.; Derynck, R.; Mundy, G.R.; Roodman, G.D. Recombinant human transforming growth factor-α stimulates the formation of osteoclast-like cells in long-term human marrow cultures. J. Clin. Investig. 1986, 78, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Fuller, K.; Bayley, K.E.; Chambers, T.J. Activin A is an essential cofactor for osteoclast induction. Biochem. Biophys. Res. Commun. 2000, 268, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, N.; Menaa, C.; Maeda, H.; Haile, D.J.; Reddy, S.V. Osteoclast-stimulating Factor Interacts with the Spinal Muscular Atrophy Gene Product to Stimulate Osteoclast Formation. J. Biol. Chem. 2001, 276, 41035–41039. [Google Scholar] [CrossRef] [PubMed]
- Owens, J.M.; Gallagher, A.C.; Chambers, T.J. IL-10 modulates formation of osteoclasts in murine hemopoietic cultures. J. Immunol. 1996, 157, 936–940. [Google Scholar] [PubMed]
- Horwood, N.J.; Elliott, J.; Martin, T.J.; Gillespie, M.T. IL-12 alone and in synergy with IL-18 inhibits osteoclast formation in vitro. J. Immunol. 2001, 166, 4915–4921. [Google Scholar] [CrossRef] [PubMed]
- Bendixen, A.C.; Shevde, N.K.; Dienger, K.M.; Willson, T.M.; Funk, C.D.; Pike, J.W. IL-4 inhibits osteoclast formation through a direct action on osteoclast precursors via peroxisome proliferator-activated receptor 1. Proc. Natl. Acad. Sci. USA 2001, 98, 2443–2448. [Google Scholar] [CrossRef] [PubMed]
- Khapli, S.M.; Mangashetti, L.S.; Yogesha, S.D.; Wani, M.R. IL-3 acts directly on osteoclast precursors and irreversibly inhibits receptor activator of NF-κB ligand-induced osteoclast differentiation by diverting the cells to macrophage lineage. J. Immunol. 2003, 171, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, J.A.; Partridge, N.C. Physiological bone remodeling: Systemic regulation and growth factor involvement. Physiology 2016, 31, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Manolagas, S.C.; O’Brien, C.A.; Almeida, M. The role of estrogen and androgen receptors in bone health and disease. Nat. Rev. Endocrinol. 2013, 9, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Imai, Y.; Matsumoto, T.; Sato, S.; Takeuchi, K.; Igarashi, K.; Harada, Y.; Azuma, Y.; Krust, A.; Yamamoto, Y.; et al. Estrogen prevents bone loss via estrogen receptor α and induction of Fas ligand in osteoclasts. Cell 2007, 130, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Rucci, N.; Teti, A. Osteomimicry: How the Seed Grows in the Soil. Calcif. Tissue Int. 2018, 102, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Curatolo, C.; Ludovico, G.M.; Correale, M.; Pagliarulo, A.; Abbate, I.; Cirrillo Marucco, E.; Barletta, A. Advanced prostate cancer follow-up with prostate-specific antigen, prostatic acid phosphatase, osteocalcin and bone isoenzyme of alkaline phosphatase. Eur. Urol. 1992, 21 (Suppl. 1), 105–107. [Google Scholar] [CrossRef] [PubMed]
- Koeneman, K.S.; Yeung, F.; Chung, L.W.K. Osteomimetic properties of prostate cancer cells: A hypothesis supporting the predilection of prostate cancer metastasis and growth in the bone environment. Prostate 1999, 39, 246–261. [Google Scholar] [CrossRef]
- Bellahcène, A.; Bachelier, R.; Detry, C.; Lidereau, R.; Clézardin, P.; Castronovo, V. Transcriptome analysis reveals an osteoblast-like phenotype for human osteotropic breast cancer cells. Breast Cancer Res. Treat. 2007, 101, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, M.; Kawakami, A.; Nakashima, T.; Matsuoka, N.; Urayama, S.; Kawabe, Y.; Fujiyama, K.; Kiriyama, T.; Aoyagi, T.; Maeda, K.; et al. Tumor necrosis factor-α and interleukin-1β increase the Fas-mediated apoptosis of human osteoblasts. J. Lab. Clin. Med. 1999, 134, 222–231. [Google Scholar] [CrossRef]
- Guise, T.A.; Yin, J.J.; Taylor, S.D.; Kumagai, Y.; Dallas, M.; Boyce, B.F.; Yoneda, T.; Mundy, G.R. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J. Clin. Investig. 1996, 98, 1544–1549. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Takyar, F.M.; Swan, K.; Jeong, J.; Vanhouten, J.; Sullivan, C.; Dann, P.; Yu, H.; Fiaschi-Taesch, N.; Chang, W.; et al. Calcium-sensing receptor promotes breast cancer by stimulating intracrine actions of parathyroid hormone-related protein. Cancer Res. 2016, 76, 5348–5360. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, T.; Flamini, E.; Mercatali, L.; Sacanna, E.; Serra, P.; Amadori, D. Pathogenesis of osteoblastic bone metastases from prostate cancer. Cancer 2010, 116, 1406–1418. [Google Scholar] [CrossRef] [PubMed]
- Guise, T.A.; Mohammad, K.S.; Clines, G.; Stebbins, E.G.; Wong, D.H.; Higgins, L.S.; Vessella, R.; Corey, E.; Padalecki, S.; Suva, L.; et al. Basic mechanisms responsible for osteolytic and osteoblastic bone metastases. Clin. Cancer Res. 2006, 12. [Google Scholar] [CrossRef] [PubMed]
- Clines, G.; Mohammad, K.S.; Bao, Y.; Stephens, O.W.; Suva, L.J.; Shaughnessy, J.D.; Fox, J.W.; Chirgwin, J.M.; Guise, T. A Dickkopf homolog 1 mediates endothelin-1-stimulated new bone formation. Mol. Endocrinol. 2007, 21, 486–498. [Google Scholar] [CrossRef] [PubMed]
- Reddi, H.; Roodman, D.; Freeman, C.; Mohla, S. Mechanisms of tumor metastasis to the bone: Challenges and opportunities. J. Bone Miner. Res. 2003, 18, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Kynaston, H.G.; Jiang, W.G. Bone metastasis in prostate cancer: Molecular and cellular mechanisms (Review). Int. J. Mol. Med. 2007, 20, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Cook, L.M.; Shay, G.; Aruajo, A.; Lynch, C.C. Integrating new discoveries into the “vicious cycle” paradigm of prostate to bone metastases. Cancer Metastasis Rev. 2014, 33, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.J.; Crockett, J.C.; Coxon, F.P.; Mönkkönen, J. Biochemical and molecular mechanisms of action of bisphosphonates. Bone 2011, 49, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Coxon, F.P.; Thompson, K.; Rogers, M.J. Recent advances in understanding the mechanism of action of bisphosphonates. Curr. Opin. Pharmacol. 2006, 6, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Mönkkönen, H.; Auriola, S.; Lehenkari, P.; Kellinsalmi, M.; Hassinen, I.E.; Vepsäläinen, J.; Mönkkönen, J. A new endogenous ATP analog (ApppI) inhibits the mitochondrial adenine nucleotide translocase (ANT) and is responsible for the apoptosis induced by nitrogen-containing bisphosphonates. Br. J. Pharmacol. 2006, 147, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Rodan, G.A.; Reszka, A.A. Bisphosphonate mechanism of action. Curr. Mol. Med. 2002, 2, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L.S.; Gordon, D.; Tchekmedyian, S.; Yanagihara, R.; Hirsh, V.; Krzakowski, M.; Pawlicki, M.; de Souza, P.; Zheng, M.; Urbanowitz, G.; et al. Zoledronic acid versus placebo in the treatment of skeletal metastases in patients with lung cancer and other solid tumors: A phase III, double-blind, randomized trial--the Zoledronic Acid Lung Cancer and Other Solid Tumors Study Group. J. Clin. Oncol. 2003, 21, 3150–3157. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.; Clézardin, P. Bone-Targeted Therapies in Cancer-Induced Bone Disease. Calcif. Tissue Int. 2018, 102, 227–250. [Google Scholar] [CrossRef] [PubMed]
- Reyes, C.; Hitz, M.; Prieto-Alhambra, D.; Abrahamsen, B. Risks and Benefits of Bisphosphonate Therapies. J. Cell. Biochem. 2016, 117, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Body, J.J.; Bone, H.G.; de Boer, R.H.; Stopeck, A.; Van Poznak, C.; Damião, R.; Fizazi, K.; Henry, D.H.; Ibrahim, T.; Lipton, A.; et al. Hypocalcaemia in patients with metastatic bone disease treated with denosumab. Eur. J. Cancer 2015, 51, 1812–1821. [Google Scholar] [CrossRef] [PubMed]
- Lüftner, D.; Niepel, D.; Steger, G.G. Therapeutic approaches for protecting bone health in patients with breast cancer. Breast 2018, 37, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Suarez, E.; Jacob, A.P.; Jones, J.; Miller, R.; Roudier-Meyer, M.P.; Erwert, R.; Pinkas, J.; Branstetter, D.; Dougall, W.C. RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature 2010, 468, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Schramek, D.; Leibbrandt, A.; Sigl, V.; Kenner, L.; Pospisilik, J.A.; Lee, H.J.; Hanada, R.; Joshi, P.A.; Aliprantis, A.; Glimcher, L.; et al. Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature 2010, 468, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Pfitzner, B.M.; Branstetter, D.; Loibl, S.; Denkert, C.; Lederer, B.; Schmitt, W.D.; Dombrowski, F.; Werner, M.; Rüdiger, T.; Dougall, W.C.; et al. RANK expression as a prognostic and predictive marker in breast cancer. Breast Cancer Res. Treat. 2014, 145, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Ullen, A.; Schwarz, S.; Lennartsson, L.; Kalkner, K.-M.; Sandstrom, P.; Costa, F.; Lennernas, B.; Linder, S.; Nilsson, S. Zoledronic acid induces caspase-dependent apoptosis in renal cancer cell lines. Scand. J. Urol. Nephrol. 2009, 43, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhang, Q.; Shi, S.; Yen, Y.; Li, X.; Zhang, Y.; Zhou, K.; Le, A.D. Bisphosphonates suppress insulin-like growth factor 1-induced angiogenesis via the HIF-1α/VEGF signaling pathways in human breast cancer cells. Int. J. Cancer 2010, 126, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Zekria, J.; Mansour, M.; Karim, S.M. The anti-tumour effects of zoledronic acid. J. Bone Oncol. 2014, 3, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Prentice, R.L.; Anderson, G.L. The women’s health initiative: Lessons learned. Annu. Rev. Public Health 2008, 29, 131–150. [Google Scholar] [CrossRef] [PubMed]
- Chlebowski, R.T.; Chen, Z.; Cauley, J.A.; Anderson, G.; Rodabough, R.J.; McTiernan, A.; Lane, D.S.; Manson, J.E.; Snetselaar, L.; Yasmeen, S.; et al. Oral bisphosphonate use and breast cancer incidence in postmenopausal women. J. Clin. Oncol. 2010, 28, 3582–3590. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.; Powles, T.; Paterson, A.; Gnant, M.; Anderson, S.; Diel, I.; Gralow, J.; von Minckwitz, G.; Moebus, V.; Bergh, J.; et al. Adjuvant bisphosphonate treatment in early breast cancer: Meta-analyses of individual patient data from randomised trials. Lancet 2015, 386, 1353–1361. [Google Scholar] [CrossRef]
- Biskup, E.; Cai, F.; Vetter, M. Bone targeted therapies in advanced breast cancer. Swiss Med. Wkly. 2017, 147, 1–11. [Google Scholar] [CrossRef]
- Littlewood-Evans, A.J.; Bilbe, G.; Bowler, W.B.; Farley, D.; Wlodarski, B.; Kokubo, T.; Inaoka, T.; Sloane, J.; Evans, D.B.; Gallagher, J.A. The osteoclast-associated protease cathepsin K is expressed in human breast carcinoma. Cancer Res. 1997, 57, 5386–5390. [Google Scholar] [PubMed]
- Le Gall, C.; Bellahcene, A.; Bonnelye, E.; Gasser, J.A.; Castronovo, V.; Green, J.; Zimmermann, J.; Clezardin, P. A Cathepsin K Inhibitor Reduces Breast Cancer Induced Osteolysis and Skeletal Tumor Burden. Cancer Res. 2007, 67, 9894–9902. [Google Scholar] [CrossRef] [PubMed]
- Podgorski, I. Future of anticathepsin K drugs: Dual therapy for skeletal disease and atherosclerosis? Future Med. Chem. 2009, 1, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A.B.; Wynne, C.; Ramirez, G.; He, W.; Song, Y.; Berd, Y.; Wang, H.; Mehta, A.; Lombardi, A. The cathepsin k inhibitor odanacatib suppresses bone resorption in women with breast cancer and established bone metastases: Results of a 4-week, double-blind, randomized, controlled trial. Clin. Breast Cancer 2010, 10, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Marzia, M.; Sims, N.A.; Voit, S.; Migliaccio, S.; Taranta, A.; Bernardini, S.; Faraggiana, T.; Yoneda, T.; Mundy, G.R.; Boyce, B.F.; et al. Decreased C-Src Expression Enhances Osteoblast Differentiation and Bone Formation. J. Cell Biol. 2000, 151, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Myoui, A.; Nishimura, R.; Williams, P.J.; Hiraga, T.; Tamura, D.; Michigami, T.; Mundy, G.R.; Yoneda, T. C-Src tyrosine kinase activity is associated with tumor colonization in bone and lung in an animal model of human breast cancer metastasis. Cancer Res. 2003, 63, 5028–5033. [Google Scholar] [PubMed]
- Rucci, N.; Recchia, I.; Angelucci, A.; Alamanou, M.; Del Fattore, A.; Fortunati, D.; Susa, M.; Fabbro, D.; Bologna, M.; Teti, A. Inhibition of protein kinase c-Src reduces the incidence of breast cancer metastases and increases survival in mice: Implications for therapy. J. Pharmacol. Exp. Ther. 2006, 318, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Huang, C.F.; Murshed, M.; Chu, K.; Araujo, J.C.; Ye, X.; Decrombrugghe, B.; Yu-Lee, L.Y.; Gallick, G.E.; Lin, S.H. Src family kinase/ABl inhibitor dasatinib suppresses proliferation and enhances differentiation of osteoblasts. Oncogene 2010, 29, 3196–3207. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, S.A.; Valentino, M.L.; Arakelian, A.; Ali, S.; Boschelli, F. SKI-606 (Bosutinib) blocks prostate cancer invasion, growth, and metastasis in vitro and in vivo through regulation of genes involved in cancer growth and skeletal metastasis. Mol. Cancer Ther. 2010, 9, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Bai, L.; Yap, S.; Gao, A.C.; Kung, H.-J.; Evans, C.P. Effect of the specific Src family kinase inhibitor saracatinib on osteolytic lesions using the PC-3 bone model. Mol. Cancer Ther. 2010, 9, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Maroni, P.; Bendinelli, P.; Matteucci, E.; Locatelli, A.; Nakamura, T.; Scita, G.; Desiderio, M.A. Osteolytic bone metastasis is hampered by impinging on the interplay among autophagy, anoikis and ossification. Cell Death Dis. 2014, 5, e1005. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.Z.; Micocci, K.C.; Natoli, A.; Redvers, R.P.; Paquet-Fifield, S.; Martin, A.C.B.M.; Denoyer, D.; Ling, X.; Kim, S.H.; Tomasin, R.; et al. Tumor but not stromal expression of β3 integrin is essential, and is required early, for spontaneous dissemination of bone-metastatic breast cancer. J. Pathol. 2015, 235, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Gramoun, A.; Shorey, S.; Bashutski, J.D.; Dixon, S.J.; Sims, S.M.; Heersche, J.N.M.; Manolson, M.F. Effects of Vitaxin®, a novel therapeutic in trial for metastatic bone tumors, on osteoclast functions in vitro. J. Cell. Biochem. 2007, 102, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bachelier, R.; Treilleux, I.; Pujuguet, P.; Peyruchaud, O.; Baron, R.; Clément-Lacroix, P.; Clézardin, P. Tumor αvβ3 integrin is a therapeutic target for breast cancer bone metastases. Cancer Res. 2007, 67, 5821–5830. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Suen, P.K.; Qin, L. Sclerostin, an emerging therapeutic target for treating osteoporosis and osteoporotic fracture: A general review. J. Orthop. Transl. 2016, 4, 1–13. [Google Scholar] [CrossRef]
- Mendoza-Villanueva, D.; Zeef, L.; Shore, P. Metastatic breast cancer cells inhibit osteoblast differentiation through the Runx2/CBFβ-dependent expression of the Wnt antagonist, sclerostin. Breast Cancer Res. 2011, 13, R106. [Google Scholar] [CrossRef] [PubMed]
- Kyvernitakis, I.; Rachner, T.D.; Urbschat, A.; Hars, O.; Hofbauer, L.C.; Hadji, P. Effect of aromatase inhibition on serum levels of sclerostin and dickkopf-1, bone turnover markers and bone mineral density in women with breast cancer. J. Cancer Res. Clin. Oncol. 2014, 140, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- McClung, M.R.; Grauer, A.; Boonen, S.; Bolognese, M.A.; Brown, J.P.; Diez-Perez, A.; Langdahl, B.L.; Reginster, J.-Y.; Zanchetta, J.R.; Wasserman, S.M.; et al. Romosozumab in postmenopausal women with low bone mineral density. N. Engl. J. Med. 2014, 370, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Recker, R.R.; Benson, C.T.; Matsumoto, T.; Bolognese, M.A.; Robins, D.A.; Alam, J.; Chiang, A.Y.; Hu, L.; Krege, J.H.; Sowa, H.; et al. A randomized, double-blind phase 2 clinical trial of blosozumab, a sclerostin antibody, in postmenopausal women with low bone mineral density. J. Bone Miner. Res. 2015, 30, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Reagan, M.R.; McDonald, M.; Terry, R.; Pettitt, J.; Le, L.; Mohanty, S.; Kneissel, M.; Kramer, I.; Brooks, D.; Bouxsein, M.; et al. Anti-sclerostin treatment prevents multiple myeloma induced bone loss and reduces tumor burden. Blood 2015, 126, 119. [Google Scholar]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maurizi, A.; Rucci, N. The Osteoclast in Bone Metastasis: Player and Target. Cancers 2018, 10, 218. https://doi.org/10.3390/cancers10070218
Maurizi A, Rucci N. The Osteoclast in Bone Metastasis: Player and Target. Cancers. 2018; 10(7):218. https://doi.org/10.3390/cancers10070218
Chicago/Turabian StyleMaurizi, Antonio, and Nadia Rucci. 2018. "The Osteoclast in Bone Metastasis: Player and Target" Cancers 10, no. 7: 218. https://doi.org/10.3390/cancers10070218
APA StyleMaurizi, A., & Rucci, N. (2018). The Osteoclast in Bone Metastasis: Player and Target. Cancers, 10(7), 218. https://doi.org/10.3390/cancers10070218