Fusogenic Viruses in Oncolytic Immunotherapy



Abstract
1. Introduction
1.1. OV-Mediated Fusion as an Oncolytic Strategy
1.2. Mechanisms of Virus-Mediated Fusion
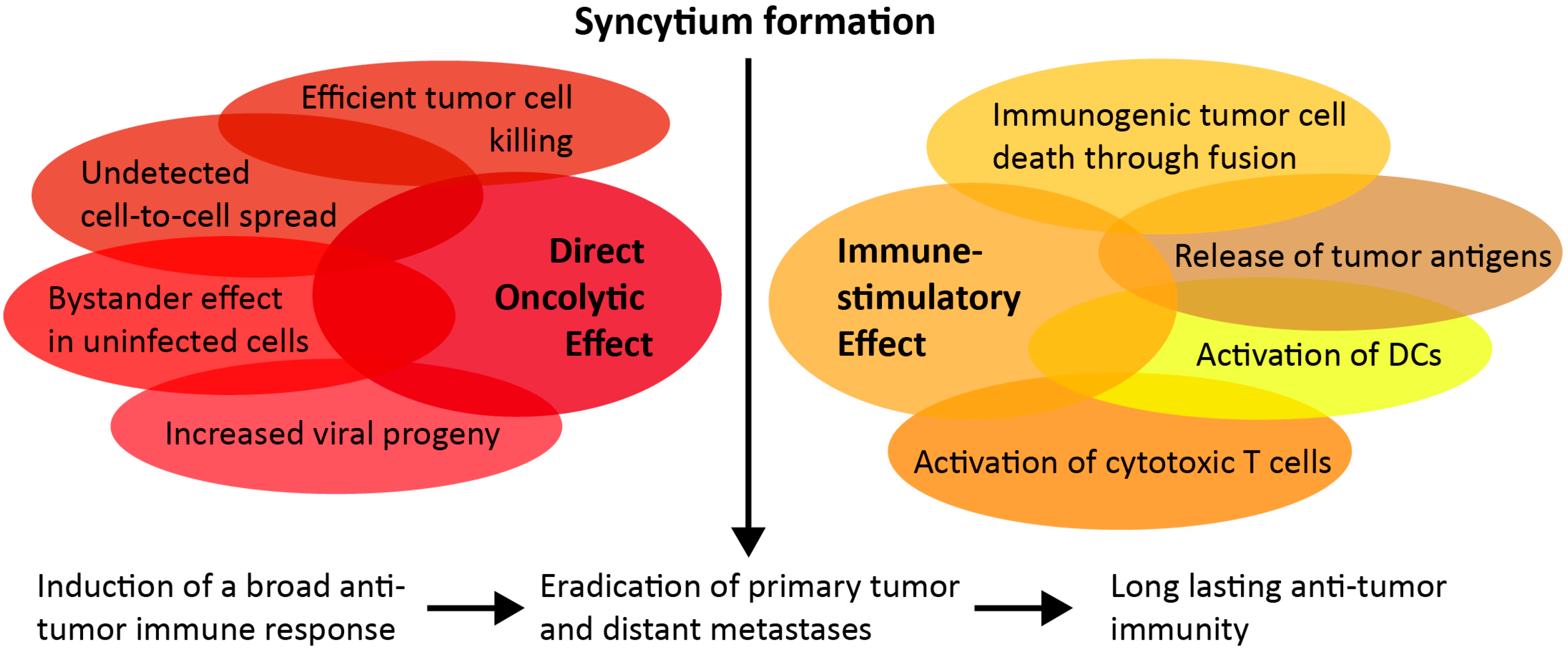
2. Cytopathic Effect of Fusogenic Proteins
3. Fusogenic Oncolytic Viruses and Their Potential in the Clinic
3.1. Viruses with Endogenous Fusion Proteins
3.2. Viruses with Engineered Fusion Proteins
3.3. Clinical Trials
4. Conclusions and Outlook
- Little or no toxicity at effective doses;
- Tumor-selective replication;
- Rapid spread throughout the tumor mass, resulting in efficient tumor debulking;
- The ability to induce potent adaptive antitumor immune responses.
Funding
Conflicts of Interest
References
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.T.; Bell, J.C. Oncolytic virus combination therapy: Killing one bird with two stones. Mol. Ther. 2018, 26, 1414–1422. [Google Scholar] [CrossRef] [PubMed]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.Y.E.; Bell, J.C.; Nelson, B.H. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer 2018, 18, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Minn, A.J. Combination cancer therapy with immune checkpoint blockade: Mechanisms and strategies. Immunity 2018, 48, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-pd-1 immunotherapy. Cell 2017, 170, 1109.e10–1119.e10. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.; Scott, K.J.; Taggart, D.; West, E.J.; Wilson, E.; Nuovo, G.J.; Thomson, S.; Corns, R.; Mathew, R.K.; Fuller, M.J.; et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 2018, 10, eaam7577. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois-Daigneault, M.C.; Roy, D.G.; Aitken, A.S.; El Sayes, N.; Martin, N.T.; Varette, O.; Falls, T.; St-Germain, L.E.; Pelin, A.; Lichty, B.D.; et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci. Transl. Med. 2018, 10, eaao1641. [Google Scholar] [CrossRef] [PubMed]
- Altomonte, J.; Ebert, O. Replicating viral vectors for cancer therapy: Strategies to synergize with host immune responses. Microb. Biotechnol. 2012, 5, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Buijs, P.R.; Verhagen, J.H.; van Eijck, C.H.; van den Hoogen, B.G. Oncolytic viruses: From bench to bedside with a focus on safety. Hum. Vaccines Immunother. 2015, 11, 1573–1584. [Google Scholar] [CrossRef] [PubMed]
- Everts, B.; van der Poel, H.G. Replication-selective oncolytic viruses in the treatment of cancer. Cancer Gene Ther. 2005, 12, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Kratzke, R.A. Oncolytic virus therapy for cancer: The first wave of translational clinical trials. Transl. Res. 2013, 161, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Lawler, S.E.; Speranza, M.C.; Cho, C.F.; Chiocca, E.A. Oncolytic viruses in cancer treatment: A review. JAMA Oncol. 2017, 3, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Podbilewicz, B. Virus and cell fusion mechanisms. Annu. Rev. Cell Dev. Biol. 2014, 30, 111–139. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, L.D.; Hoffman, L.R.; Wolfsberg, T.G.; White, J.M. Virus-cell and cell-cell fusion. Annu. Rev. Cell Dev. Biol. 1996, 12, 627–661. [Google Scholar] [CrossRef] [PubMed]
- Salsman, J.; Top, D.; Boutilier, J.; Duncan, R. Extensive syncytium formation mediated by the reovirus fast proteins triggers apoptosis-induced membrane instability. J. Virol. 2005, 79, 8090–8100. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E. Therapeutic potential of oncolytic measles virus: Promises and challenges. Clin. Pharmacol. Ther. 2010, 88, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, H.; Bronk, S.F.; Bateman, A.; Harrington, K.; Vile, R.G.; Gores, G.J. Viral fusogenic membrane glycoprotein expression causes syncytia formation with bioenergetic cell death: Implications for gene therapy. Cancer Res. 2000, 60, 6396–6402. [Google Scholar] [PubMed]
- Inoue, H.; Tani, K. Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ. 2014, 21, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N.; et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Showalter, A.; Limaye, A.; Oyer, J.L.; Igarashi, R.; Kittipatarin, C.; Copik, A.J.; Khaled, A.R. Cytokines in immunogenic cell death: Applications for cancer immunotherapy. Cytokine 2017, 97, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Herschke, F.; Plumet, S.; Duhen, T.; Azocar, O.; Druelle, J.; Laine, D.; Wild, T.F.; Rabourdin-Combe, C.; Gerlier, D.; Valentin, H. Cell-cell fusion induced by measles virus amplifies the type I interferon response. J. Virol. 2007, 81, 12859–12871. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.R.; Harrington, K.J.; Kottke, T.; Ahmed, A.; Melcher, A.A.; Gough, M.J.; Linardakis, E.; Riddle, D.; Dietz, A.; Lohse, C.M.; et al. Viral fusogenic membrane glycoproteins kill solid tumor cells by nonapoptotic mechanisms that promote cross presentation of tumor antigens by dendritic cells. Cancer Res. 2002, 62, 6566–6578. [Google Scholar] [PubMed]
- Errington, F.; Jones, J.; Merrick, A.; Bateman, A.; Harrington, K.; Gough, M.; O’Donnell, D.; Selby, P.; Vile, R.; Melcher, A. Fusogenic membrane glycoprotein-mediated tumour cell fusion activates human dendritic cells for enhanced IL-12 production and T-cell priming. Gene Ther. 2006, 13, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Delpeut, S.; Rudd, P.A.; Labonte, P.; von Messling, V. Membrane fusion-mediated autophagy induction enhances morbillivirus cell-to-cell spread. J. Virol. 2012, 86, 8527–8535. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Delos, S.E.; Brecher, M.; Schornberg, K. Structures and mechanisms of viral membrane fusion proteins: Multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189–219. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479–480, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.; Chen, Z.; Walsh, S.; Wu, S. Avian reovirus-induced syncytium formation is independent of infectious progeny virus production and enhances the rate, but is not essential, for virus-induced cytopathology and virus egress. Virology 1996, 224, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Ciechonska, M.; Duncan, R. Reovirus fast proteins: Virus-encoded cellular fusogens. Trends Microbiol. 2014, 22, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, J.A.; Salsman, J.; de Antueno, R.; Touhami, A.; Jericho, M.H.; Clancy, E.K.; Duncan, R. The p14 fusion-associated small transmembrane (fast) protein effects membrane fusion from a subset of membrane microdomains. J. Biol. Chem. 2006, 281, 31778–31789. [Google Scholar] [CrossRef] [PubMed]
- Perfettini, J.L.; Castedo, M.; Roumier, T.; Andreau, K.; Nardacci, R.; Piacentini, M.; Kroemer, G. Mechanisms of apoptosis induction by the HIV-1 envelope. Cell Death Differ. 2005, 12 (Suppl. 1), 916–923. [Google Scholar] [CrossRef] [PubMed]
- Nardacci, R.; Perfettini, J.L.; Grieco, L.; Thieffry, D.; Kroemer, G.; Piacentini, M. Syncytial apoptosis signaling network induced by the HIV-1 envelope glycoprotein complex: An overview. Cell Death Dis. 2015, 6, e1846. [Google Scholar] [CrossRef] [PubMed]
- Scheller, C.; Jassoy, C. Syncytium formation amplifies apoptotic signals: A new view on apoptosis in HIV infection in vitro. Virology 2001, 282, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Esolen, L.M.; Park, S.W.; Hardwick, J.M.; Griffin, D.E. Apoptosis as a cause of death in measles virus-infected cells. J. Virol. 1995, 69, 3955–3958. [Google Scholar] [PubMed]
- Galanis, E.; Bateman, A.; Johnson, K.; Diaz, R.M.; James, C.D.; Vile, R.; Russell, S.J. Use of viral fusogenic membrane glycoproteins as novel therapeutic transgenes in gliomas. Hum. Gene Ther. 2001, 12, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.H.; Salon, C.; Brambilla, E.; Lavillette, D.; Szecsi, J.; Cosset, F.L.; Coll, J.L. Fusogenic membrane glycoproteins induce syncytia formation and death in vitro and in vivo: A potential therapy agent for lung cancer. Cancer Gene Ther. 2010, 17, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Jia, H.; Liu, R.; Wu, J.; Han, H.; Zuo, Y.; Yang, S.; Huang, W. Inhibition of NF-kappab in fusogenic membrane glycoprotein causing HL-60 cell death: Implications for acute myeloid leukemia. Cancer Lett. 2009, 273, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Grases, D.; Rojas, J.J.; Gros, A.; Vilardell, F.; Vile, R.; Mercade, E.; Cascallo, M.; Alemany, R. GALV expression enhances the therapeutic efficacy of an oncolytic adenovirus by inducing cell fusion and enhancing virus distribution. Gene Ther. 2012, 19, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Liu, Z.; Bartlett, D.L. Oncolytic immunotherapy: Dying the right way is a key to eliciting potent antitumor immunity. Front. Oncol. 2014, 4, 74. [Google Scholar] [CrossRef] [PubMed]
- Workenhe, S.T.; Mossman, K.L. Oncolytic virotherapy and immunogenic cancer cell death: Sharpening the sword for improved cancer treatment strategies. Mol. Ther. 2014, 22, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado-Castano, S.; Sanchez-Aparicio, M.T.; Garcia-Sastre, A.; Villar, E. The therapeutic effect of death: Newcastle disease virus and its antitumor potential. Virus Res. 2015, 209, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, O.G.; Errington-Mais, F.; Steele, L.; Hadac, E.; Jennings, V.; Scott, K.; Peach, H.; Phillips, R.M.; Bond, J.; Pandha, H.; et al. Measles virus causes immunogenic cell death in human melanoma. Gene Ther. 2013, 20, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Matveeva, O.V.; Guo, Z.S.; Shabalina, S.A.; Chumakov, P.M. Oncolysis by paramyxoviruses: Multiple mechanisms contribute to therapeutic efficiency. Mol. Ther. Oncolytics 2015, 2, 15011. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus accessory aspects of cell death: Recommendations of the nccd 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Melcher, A.; Todryk, S.; Hardwick, N.; Ford, M.; Jacobson, M.; Vile, R.G. Tumor immunogenicity is determined by the mechanism of cell death via induction of heat shock protein expression. Nat. Med. 1998, 4, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Bullough, F.; Murphy, S.; Emiliusen, L.; Lavillette, D.; Cosset, F.L.; Cattaneo, R.; Russell, S.J.; Vile, R.G. Fusogenic membrane glycoproteins as a novel class of genes for the local and immune-mediated control of tumor growth. Cancer Res. 2000, 60, 1492–1497. [Google Scholar] [PubMed]
- Holm, C.K.; Jensen, S.B.; Jakobsen, M.R.; Cheshenko, N.; Horan, K.A.; Moeller, H.B.; Gonzalez-Dosal, R.; Rasmussen, S.B.; Christensen, M.H.; Yarovinsky, T.O.; et al. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor sting. Nat. Immunol. 2012, 13, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Linardakis, E.; Bateman, A.; Phan, V.; Ahmed, A.; Gough, M.; Olivier, K.; Kennedy, R.; Errington, F.; Harrington, K.J.; Melcher, A.; et al. Enhancing the efficacy of a weak allogeneic melanoma vaccine by viral fusogenic membrane glycoprotein-mediated tumor cell-tumor cell fusion. Cancer Res. 2002, 62, 5495–5504. [Google Scholar] [PubMed]
- Lanzrein, M.; Schlegel, A.; Kempf, C. Entry and uncoating of enveloped viruses. Biochem. J. 1994, 302 Pt 2, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Gaudin, Y.; Ruigrok, R.W.; Brunner, J. Low-pH induced conformational changes in viral fusion proteins: Implications for the fusion mechanism. J. Gen. Virol. 1995, 76 Pt 7, 1541–1556. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.T.; Richardson, C.D. The host cell receptors for measles virus and their interaction with the viral hemagglutinin (H) protein. Viruses 2016, 8, 250. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A. Paramyxovirus fusion: A hypothesis for changes. Virology 1993, 197, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A.; Kolakofsky, D. Paramyxoviridae. Fields Virol. 2001, 1, 1305–1340. [Google Scholar]
- Scheid, A.; Choppin, P.W. Two disulfide-linked polypeptide chains constitute the active F protein of paramyxoviruses. Virology 1977, 80, 54–66. [Google Scholar] [CrossRef]
- Morrison, T.G. Structure, function, and intracellular processing of paramyxovirus membrane proteins. Virus Res. 1988, 10, 113–135. [Google Scholar] [CrossRef]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Katsoulidis, E.; Kaur, S.; Platanias, L.C. Deregulation of interferon signaling in malignant cells. Pharmaceuticals 2010, 3, 406–418. [Google Scholar] [CrossRef] [PubMed]
- Stojdl, D.F.; Lichty, B.; Knowles, S.; Marius, R.; Atkins, H.; Sonenberg, N.; Bell, J.C. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 2000, 6, 821–825. [Google Scholar] [PubMed]
- Bull, C.; den Brok, M.H.; Adema, G.J. Sweet escape: Sialic acids in tumor immune evasion. Biochim. Biophys. Acta 2014, 1846, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Matrosovich, M.; Herrler, G.; Klenk, H.D. Sialic acid receptors of viruses. Top. Curr. Chem. 2015, 367, 1–28. [Google Scholar] [PubMed]
- Bossart, K.N.; Fusco, D.L.; Broder, C.C. Paramyxovirus entry. Adv. Exp. Med. Biol. 2013, 790, 95–127. [Google Scholar] [PubMed]
- Matveeva, O.V.; Guo, Z.S.; Senin, V.M.; Senina, A.V.; Shabalina, S.A.; Chumakov, P.M. Oncolysis by paramyxoviruses: Preclinical and clinical studies. Mol. Ther. Oncolytics 2015, 2, 15017. [Google Scholar] [CrossRef] [PubMed]
- Zaitsev, V.; von Itzstein, M.; Groves, D.; Kiefel, M.; Takimoto, T.; Portner, A.; Taylor, G. Second sialic acid binding site in newcastle disease virus hemagglutinin-neuraminidase: Implications for fusion. J. Virol. 2004, 78, 3733–3741. [Google Scholar] [CrossRef] [PubMed]
- Pearlstein, E.; Salk, P.L.; Yogeeswaran, G.; Karpatkin, S. Correlation between spontaneous metastatic potential, platelet-aggregating activity of cell surface extracts, and cell surface sialylation in 10 metastatic-variant derivatives of a rat renal sarcoma cell line. Proc. Natl. Acad. Sci. USA 1980, 77, 4336–4339. [Google Scholar] [CrossRef] [PubMed]
- Yogeeswaran, G.; Salk, P.L. Metastatic potential is positively correlated with cell surface sialylation of cultured murine tumor cell lines. Science 1981, 212, 1514–1516. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.; Elkabets, M.; Perlmutter, M.; Porgador, A.; Voronov, E.; Apte, R.N.; Lichtenstein, R.G. Sialylation of 3-methylcholanthrene-induced fibrosarcoma determines antitumor immune responses during immunoediting. J. Immunol. 2010, 185, 5869–5878. [Google Scholar] [CrossRef] [PubMed]
- Powell, L.D.; Whiteheart, S.W.; Hart, G.W. Cell surface sialic acid influences tumor cell recognition in the mixed lymphocyte reaction. J. Immunol. 1987, 139, 262–270. [Google Scholar] [PubMed]
- Kinoh, H.; Inoue, M. New cancer therapy using genetically-engineered oncolytic sendai virus vector. Front. Biosci. 2008, 13, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Tatsuta, K.; Tanaka, S.; Tajiri, T.; Shibata, S.; Komaru, A.; Ueda, Y.; Inoue, M.; Hasegawa, M.; Suita, S.; Sueishi, K.; et al. Complete elimination of established neuroblastoma by synergistic action of γ-irradiation and DCs treated with rSeV expressing interferon-β gene. Gene Ther. 2009, 16, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Yonemitsu, Y.; Ueda, Y.; Kinoh, H.; Hasegawa, M. Immunostimulatory virotherapy using recombinant sendai virus as a new cancer therapeutic regimen. Front. Biosci. 2008, 13, 1892–1898. [Google Scholar] [CrossRef] [PubMed]
- Kurooka, M.; Kaneda, Y. Inactivated sendai virus particles eradicate tumors by inducing immune responses through blocking regulatory t cells. Cancer Res. 2007, 67, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, A.; Kurooka, M.; Miki, T.; Kaneda, Y. Intratumoral injection of inactivated sendai virus particles elicits strong antitumor activity by enhancing local CXCL10 expression and systemic NK cell activation. Cancer Immunol. Immunother. 2008, 57, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Saga, K.; Tamai, K.; Yamazaki, T.; Kaneda, Y. Systemic administration of a novel immune-stimulatory pseudovirion suppresses lung metastatic melanoma by regionally enhancing IFN-γ production. Clin. Cancer Res. 2013, 19, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Beard, C.W.; Hanson, R.P. Diseases of Poultry, 7th ed.; Iowa State University Press: Ames, IA, USA, 1981. [Google Scholar]
- Huang, Z.; Panda, A.; Elankumaran, S.; Govindarajan, D.; Rockemann, D.D.; Samal, S.K. The hemagglutinin-neuraminidase protein of newcastle disease virus determines tropism and virulence. J. Virol. 2004, 78, 4176–4184. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, O.S.; Koch, G.; Hartog, L.; Ravenshorst, N.; Peeters, B.P. Virulence of newcastle disease virus is determined by the cleavage site of the fusion protein and by both the stem region and globular head of the haemagglutinin-neuraminidase protein. J. Gen. Virol. 2005, 86, 1759–1769. [Google Scholar] [CrossRef] [PubMed]
- Park, M.S.; Garcia-Sastre, A.; Cros, J.F.; Basler, C.F.; Palese, P. Newcastle disease virus V protein is a determinant of host range restriction. J. Virol. 2003, 77, 9522–9532. [Google Scholar] [CrossRef] [PubMed]
- Fabian, Z.; Torocsik, B.; Kiss, K.; Csatary, L.K.; Bodey, B.; Tigyi, J.; Csatary, C.; Szeberenyi, J. Induction of apoptosis by a newcastle disease virus vaccine (MTH-68/H) in PC12 rat phaeochromocytoma cells. Anticancer Res. 2001, 21, 125–135. [Google Scholar] [PubMed]
- Bar-Eli, N.; Giloh, H.; Schlesinger, M.; Zakay-Rones, Z. Preferential cytotoxic effect of newcastle disease virus on lymphoma cells. J. Cancer Res. Clin. Oncol. 1996, 122, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V.; Griesbach, A.; Ahlert, T. Antitumor effects of newcastle disease virus in vivo: Local versus systemic effects. Int. J. Oncol. 2001, 18, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Lorence, R.M.; Katubig, B.B.; Reichard, K.W.; Reyes, H.M.; Phuangsab, A.; Sassetti, M.D.; Walter, R.J.; Peeples, M.E. Complete regression of human fibrosarcoma xenografts after local newcastle disease virus therapy. Cancer Res. 1994, 54, 6017–6021. [Google Scholar] [PubMed]
- Phuangsab, A.; Lorence, R.M.; Reichard, K.W.; Peeples, M.E.; Walter, R.J. Newcastle disease virus therapy of human tumor xenografts: Antitumor effects of local or systemic administration. Cancer Lett. 2001, 172, 27–36. [Google Scholar] [CrossRef]
- Bai, L.; Koopmann, J.; Fiola, C.; Fournier, P.; Schirrmacher, V. Dendritic cells pulsed with viral oncolysates potently stimulate autologous t cells from cancer patients. Int. J. Oncol. 2002, 21, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Vigil, A.; Park, M.S.; Martinez, O.; Chua, M.A.; Xiao, S.; Cros, J.F.; Martinez-Sobrido, L.; Woo, S.L.; Garcia-Sastre, A. Use of reverse genetics to enhance the oncolytic properties of newcastle disease virus. Cancer Res. 2007, 67, 8285–8292. [Google Scholar] [CrossRef] [PubMed]
- Altomonte, J.; Marozin, S.; Schmid, R.M.; Ebert, O. Engineered newcastle disease virus as an improved oncolytic agent against hepatocellular carcinoma. Mol. Ther. 2010, 18, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Select Agents and Toxins. Available online: https://www.selectagents.gov/selectagentsandtoxinslist.html (accessed on 25 June 2018).
- Blechacz, B.; Russell, S.J. Measles virus as an oncolytic vector platform. Curr. Gene Ther. 2008, 8, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Opyrchal, M.; Aderca, I.; Schroeder, M.A.; Sarkaria, J.N.; Domingo, E.; Federspiel, M.J.; Galanis, E. Oncolytic measles virus strains have significant antitumor activity against glioma stem cells. Gene Ther. 2013, 20, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Iankov, I.D.; Dispenzieri, A.; Galanis, E. Attenuated oncolytic measles virus strains as cancer therapeutics. Curr. Pharm. Biotechnol. 2012, 13, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, K.; Hanauer, J.R.; Prufer, S.; Munch, R.C.; Volker, I.; Filippis, C.; Jost, C.; Hanschmann, K.M.; Cattaneo, R.; Peng, K.W.; et al. Darpin-targeting of measles virus: Unique bispecificity, effective oncolysis, and enhanced safety. Mol. Ther. 2013, 21, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Peng, K.W.; Harvey, M.; Greiner, S.; Lorimer, I.A.; James, C.D.; Russell, S.J. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat. Biotechnol. 2005, 23, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Iankov, I.D.; Allen, C.; Russell, S.J.; Galanis, E. Oncolytic measles virus retargeting by ligand display. Methods Mol. Biol. 2012, 797, 141–162. [Google Scholar] [PubMed]
- Galanis, E.; Atherton, P.J.; Maurer, M.J.; Knutson, K.L.; Dowdy, S.C.; Cliby, W.A.; Haluska, P., Jr.; Long, H.J.; Oberg, A.; Aderca, I.; et al. Oncolytic measles virus expressing the sodium iodide symporter to treat drug-resistant ovarian cancer. Cancer Res. 2015, 75, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Blechacz, B.; Splinter, P.L.; Greiner, S.; Myers, R.; Peng, K.W.; Federspiel, M.J.; Russell, S.J.; LaRusso, N.F. Engineered measles virus as a novel oncolytic viral therapy system for hepatocellular carcinoma. Hepatology 2006, 44, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.; Lampe, J.; Bossow, S.; Zimmermann, M.; Neubert, W.; Bitzer, M.; Lauer, U.M. A novel armed oncolytic measles vaccine virus for the treatment of cholangiocarcinoma. Hum. Gene Ther. 2013, 24, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Asada, T. Treatment of human cancer with mumps virus. Cancer 1974, 34, 1907–1928. [Google Scholar] [CrossRef]
- Okuno, Y.; Asada, T.; Yamanishi, K.; Otsuka, T.; Takahashi, M.; Tanioka, T.; Aoyama, H.; Fukui, O.; Matsumoto, K.; Uemura, F.; et al. [mumps virus therapy of neoplasms (2)]. Nihon Rinsho 1977, 35, 3820–3825. [Google Scholar] [PubMed]
- Minton, J.P. Mumps virus and BCG vaccine in metastatic melanoma. Arch. Surg. 1973, 106, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Ammayappan, A.; Russell, S.J.; Federspiel, M.J. Recombinant mumps virus as a cancer therapeutic agent. Mol. Ther. Oncolytics 2016, 3, 16019. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Haviv, Y.S.; Derdeyn, C.A.; Lam, J.; Coolidge, C.; Hunter, E.; Curiel, D.T.; Blackwell, J.L. Human immunodeficiency virus type 1-mediated syncytium formation is compatible with adenovirus replication and facilitates efficient dispersion of viral gene products and de novo-synthesized virus particles. Hum. Gene Ther. 2001, 12, 2155–2165. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Breton, C.; Russell, L.O.; Russell, S.J.; Peng, K.W. Faster replication and higher expression levels of viral glycoproteins give the vesicular stomatitis virus/measles virus hybrid VSV-FH a growth advantage over measles virus. J. Virol. 2014, 88, 8332–8339. [Google Scholar] [CrossRef] [PubMed]
- Altomonte, J.; Braren, R.; Schulz, S.; Marozin, S.; Rummeny, E.J.; Schmid, R.M.; Ebert, O. Synergistic antitumor effects of transarterial viroembolization for multifocal hepatocellular carcinoma in rats. Hepatology 2008, 48, 1864–1873. [Google Scholar] [CrossRef] [PubMed]
- Ebert, O.; Shinozaki, K.; Kournioti, C.; Park, M.S.; Garcia-Sastre, A.; Woo, S.L. Syncytia induction enhances the oncolytic potential of vesicular stomatitis virus in virotherapy for cancer. Cancer Res. 2004, 64, 3265–3270. [Google Scholar] [CrossRef] [PubMed]
- Le Boeuf, F.; Gebremeskel, S.; McMullen, N.; He, H.; Greenshields, A.L.; Hoskin, D.W.; Bell, J.C.; Johnston, B.; Pan, C.; Duncan, R. Reovirus fast protein enhances vesicular stomatitis virus oncolytic virotherapy in primary and metastatic tumor models. Mol. Ther. Oncolytics 2017, 6, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Tao, L.; Jin, A.; Vile, R.; Brenner, M.K.; Zhang, X. Expression of a fusogenic membrane glycoprotein by an oncolytic herpes simplex virus potentiates the viral antitumor effect. Mol. Ther. 2003, 7, 748–754. [Google Scholar] [CrossRef]
- Nakamori, M.; Fu, X.; Pettaway, C.A.; Zhang, X. Potent antitumor activity after systemic delivery of a doubly fusogenic oncolytic herpes simplex virus against metastatic prostate cancer. Prostate 2004, 60, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Simpson, G.R.; Han, Z.; Liu, B.; Wang, Y.; Campbell, G.; Coffin, R.S. Combination of a fusogenic glycoprotein, prodrug activation, and oncolytic herpes simplex virus for enhanced local tumor control. Cancer Res. 2006, 66, 4835–4842. [Google Scholar] [CrossRef] [PubMed]
- Price, D.L.; Lin, S.F.; Han, Z.; Simpson, G.; Coffin, R.S.; Wong, J.; Li, S.; Fong, Y.; Wong, R.J. Oncolysis using herpes simplex virus type 1 engineered to express cytosine deaminase and a fusogenic glycoprotein for head and neck squamous cell carcinoma. Arch. Otolaryngol. Head Neck Surg. 2010, 136, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Kelly, K.; Mittra, A.; Gonzalez, S.J.; Song, K.Y.; Simpson, G.; Coffin, R.; Fong, Y. A third-generation herpesvirus is effective against gastroesophageal cancer. J. Surg. Res. 2010, 163, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Simpson, G.R.; Horvath, A.; Annels, N.E.; Pencavel, T.; Metcalf, S.; Seth, R.; Peschard, P.; Price, T.; Coffin, R.S.; Mostafid, H.; et al. Combination of a fusogenic glycoprotein, pro-drug activation and oncolytic HSV as an intravesical therapy for superficial bladder cancer. Br. J. Cancer 2012, 106, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; McDonald, C.; Giannini, C.; Peng, K.W.; Rosales, G.; Russell, S.J.; Galanis, E. Adenoviral vectors expressing fusogenic membrane glycoproteins activated via matrix metalloproteinase cleavable linkers have significant antitumor potential in the gene therapy of gliomas. J. Gene Med. 2004, 6, 1216–1227. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Gros, A.; Cascallo, M.; Vile, R.; Mercade, E.; Alemany, R. Syncytia formation affects the yield and cytotoxicity of an adenovirus expressing a fusogenic glycoprotein at a late stage of replication. Gene Ther. 2008, 15, 1240–1245. [Google Scholar] [CrossRef] [PubMed]
- Diaz, R.M.; Bateman, A.; Emiliusen, L.; Fielding, A.; Trono, D.; Russell, S.J.; Vile, R.G. A lentiviral vector expressing a fusogenic glycoprotein for cancer gene therapy. Gene Ther. 2000, 7, 1656–1663. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Trevino, A.; Castel, S.; Lopez-Iglesias, C.; Cortadellas, N.; Comas-Riu, J.; Mercade, E. Effects of adenovirus-mediated SV5 fusogenic glycoprotein expression on tumor cells. J. Gene Med. 2003, 5, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Opyrchal, M.; Dispenzieri, A.; Peng, K.W.; Federspiel, M.J.; Russell, S.J.; Galanis, E. Clinical trials with oncolytic measles virus: Current status and future prospects. Curr. Cancer Drug Targets 2018, 18, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Palese, P. Oncolytic newcastle disease virus for cancer therapy: Old challenges and new directions. Future Microbiol. 2012, 7, 347–367. [Google Scholar] [CrossRef] [PubMed]
- Csatary, L.K.; Eckhardt, S.; Bukosza, I.; Czegledi, F.; Fenyvesi, C.; Gergely, P.; Bodey, B.; Csatary, C.M. Attenuated veterinary virus vaccine for the treatment of cancer. Cancer Detect. Prev. 1993, 17, 619–627. [Google Scholar] [PubMed]
- Csatary, L.K.; Gosztonyi, G.; Szeberenyi, J.; Fabian, Z.; Liszka, V.; Bodey, B.; Csatary, C.M. MTH-68/H oncolytic viral treatment in human high-grade gliomas. J. Neurooncol. 2004, 67, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.I.; Zakay-Rones, Z.; Gomori, J.M.; Linetsky, E.; Rasooly, L.; Greenbaum, E.; Rozenman-Yair, S.; Panet, A.; Libson, E.; Irving, C.S.; et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol. Ther. 2006, 13, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Lorence, R.M.; Roberts, M.S.; O’Neil, J.D.; Groene, W.S.; Miller, J.A.; Mueller, S.N.; Bamat, M.K. Phase 1 clinical experience using intravenous administration of PV701, an oncolytic newcastle disease virus. Curr. Cancer Drug Targets 2007, 7, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Lorence, R.M.; Pecora, A.L.; Major, P.P.; Hotte, S.J.; Laurie, S.A.; Roberts, M.S.; Groene, W.S.; Bamat, M.K. Overview of phase I studies of intravenous administration of PV701, an oncolytic virus. Curr. Opin. Mol. Ther. 2003, 5, 618–624. [Google Scholar] [PubMed]
- Cassel, W.A.; Murray, D.R. A ten-year follow-up on stage II malignant melanoma patients treated postsurgically with newcastle disease virus oncolysate. Med. Oncol. Tumor Pharmacother. 1992, 9, 169–171. [Google Scholar] [PubMed]
- Batliwalla, F.M.; Bateman, B.A.; Serrano, D.; Murray, D.; Macphail, S.; Maino, V.C.; Ansel, J.C.; Gregersen, P.K.; Armstrong, C.A. A 15-year follow-up of AJCC stage III malignant melanoma patients treated postsurgically with newcastle disease virus (NDV) oncolysate and determination of alterations in the CD8 T cell repertoire. Mol. Med. 1998, 4, 783–794. [Google Scholar] [PubMed]
- Karcher, J.; Dyckhoff, G.; Beckhove, P.; Reisser, C.; Brysch, M.; Ziouta, Y.; Helmke, B.H.; Weidauer, H.; Schirrmacher, V.; Herold-Mende, C. Antitumor vaccination in patients with head and neck squamous cell carcinomas with autologous virus-modified tumor cells. Cancer Res. 2004, 64, 8057–8061. [Google Scholar] [CrossRef] [PubMed]
- Schulze, T.; Kemmner, W.; Weitz, J.; Wernecke, K.D.; Schirrmacher, V.; Schlag, P.M. Efficiency of adjuvant active specific immunization with newcastle disease virus modified tumor cells in colorectal cancer patients following resection of liver metastases: Results of a prospective randomized trial. Cancer Immunol. Immunother. 2009, 58, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Heinzerling, L.; Kunzi, V.; Oberholzer, P.A.; Kundig, T.; Naim, H.; Dummer, R. Oncolytic measles virus in cutaneous T-cell lymphomas mounts antitumor immune responses in vivo and targets interferon-resistant tumor cells. Blood 2005, 106, 2287–2294. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Hartmann, L.C.; Cliby, W.A.; Long, H.J.; Peethambaram, P.P.; Barrette, B.A.; Kaur, J.S.; Haluska, P.J., Jr.; Aderca, I.; Zollman, P.J.; et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res. 2010, 70, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Federspiel, M.J.; Peng, K.W.; Tong, C.; Dingli, D.; Morice, W.G.; Lowe, V.; O’Connor, M.K.; Kyle, R.A.; Leung, N.; et al. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin. Proc. 2014, 89, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Hasumi, K.; Okudaira, Y.; Yamanishi, K.; Takahashi, M. Immunotherapy of advanced gynecologic cancer patients utilizing mumps virus. Cancer Detect. Prev. 1988, 12, 487–495. [Google Scholar] [PubMed]
- Tanemura, A.; Kiyohara, E.; Katayama, I.; Kaneda, Y. Recent advances and developments in the antitumor effect of the HVJ envelope vector on malignant melanoma: From the bench to clinical application. Cancer Gene Ther. 2013, 20, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Kyi, C.; Postow, M.A. Immune checkpoint inhibitor combinations in solid tumors: Opportunities and challenges. Immunotherapy 2016, 8, 821–837. [Google Scholar] [CrossRef] [PubMed]
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 2018, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Mrkic, B.; Pavlovic, J.; Rulicke, T.; Volpe, P.; Buchholz, C.J.; Hourcade, D.; Atkinson, J.P.; Aguzzi, A.; Cattaneo, R. Measles virus spread and pathogenesis in genetically modified mice. J. Virol. 1998, 72, 7420–7427. [Google Scholar] [PubMed]
- Cuadrado-Castano, S.; Ayllon, J.; Mansour, M.; de la Iglesia-Vicente, J.; Jordan, S.; Tripathi, S.; Garcia-Sastre, A.; Villar, E. Enhancement of the proapoptotic properties of newcastle disease virus promotes tumor remission in syngeneic murine cancer models. Mol. Cancer Ther. 2015, 14, 1247–1258. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Martinez-Sobrido, L.; Kelly, K.; Mansour, M.; Sheng, G.; Vigil, A.; Garcia-Sastre, A.; Palese, P.; Fong, Y. Enhancement of oncolytic properties of recombinant newcastle disease virus through antagonism of cellular innate immune responses. Mol. Ther. 2009, 17, 697–706. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Fusogenic Proteins | Viral Origin Family | Recipient Viral Backbone | Engineering and Effect | Literature |
|---|---|---|---|---|
| FAST protein | Reovirus Reoviridae | VSV | VSVΔM51 + p14 FAST protein
| Le Boeuf et al., 2017 [104] |
| GALV.fus | GALV Retroviridae | HSV | HSV-1 + truncated GALV.fus
| Fu et al., 2003 [105] |
HSV-1 (Synco-2D) + GALV.fus
| Nakamori et al., 2004 [106] | |||
HSV-1 + GALV + Fcy::Fur (OncoVEXGALV/CD)
| Simpson et al., 2006 [107] Price et al., 2010 [108] Wong et al., 2010 [109] Simpson et al., 2012 [110] | |||
| AdV | AdV + GALV attached to a blocking ligand via a MMP-cleavable linker (AdM40)
| Allen et al., 2004 [111] | ||
| AdV | AdV5 + GALV.fus (ICOVIR16) under major late promoter
| Guedan et al., 2008 [112] Guedan et al., 2012 [38] | ||
| Lentivirus | HIV-based self-inactivating vector with a transcriptionally disabled 3′ LTR + GALV
| Diaz et al., 2000 [113] | ||
| HIV envelope | HIV Retroviridae | AdV | AdV5 + HIVenv
| Li et al., 2001 [100] |
| MV-F | MV Paramyxo-viridae | VSV | VSVΔG + MV-F/HN
| Ayala-Breton et al., 2014 [101] |
| NDV-F | NDV Paramyxo-viridae | VSV | VSV + NDV-F (L289A)
| Ebert et al., 2004 [103] |
| SV5-F | SV5 Paramyxo-viridae | AdV | AdV5 + SV5-F
| Gómez-Treviño et al., 2003 [114] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krabbe, T.; Altomonte, J. Fusogenic Viruses in Oncolytic Immunotherapy. Cancers 2018, 10, 216. https://doi.org/10.3390/cancers10070216
Krabbe T, Altomonte J. Fusogenic Viruses in Oncolytic Immunotherapy. Cancers. 2018; 10(7):216. https://doi.org/10.3390/cancers10070216
Chicago/Turabian StyleKrabbe, Teresa, and Jennifer Altomonte. 2018. "Fusogenic Viruses in Oncolytic Immunotherapy" Cancers 10, no. 7: 216. https://doi.org/10.3390/cancers10070216
APA StyleKrabbe, T., & Altomonte, J. (2018). Fusogenic Viruses in Oncolytic Immunotherapy. Cancers, 10(7), 216. https://doi.org/10.3390/cancers10070216