Zinc Metallochaperones as Mutant p53 Reactivators: A New Paradigm in Cancer Therapeutics

{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. The Multi-Functional Role of p53
Over Three Decades the Spectrum of Cellular Functions of p53 Has Grown Substantially
1.2. p53 Mutations and Cancer: A Unique Tumor Suppressor
1.3. Classes of p53 Mutations
1.4. The Relationship between p53 Structure/Function and Zinc
1.5. Discovery of Thiosemicarbazones as Mutant p53 Reactivators
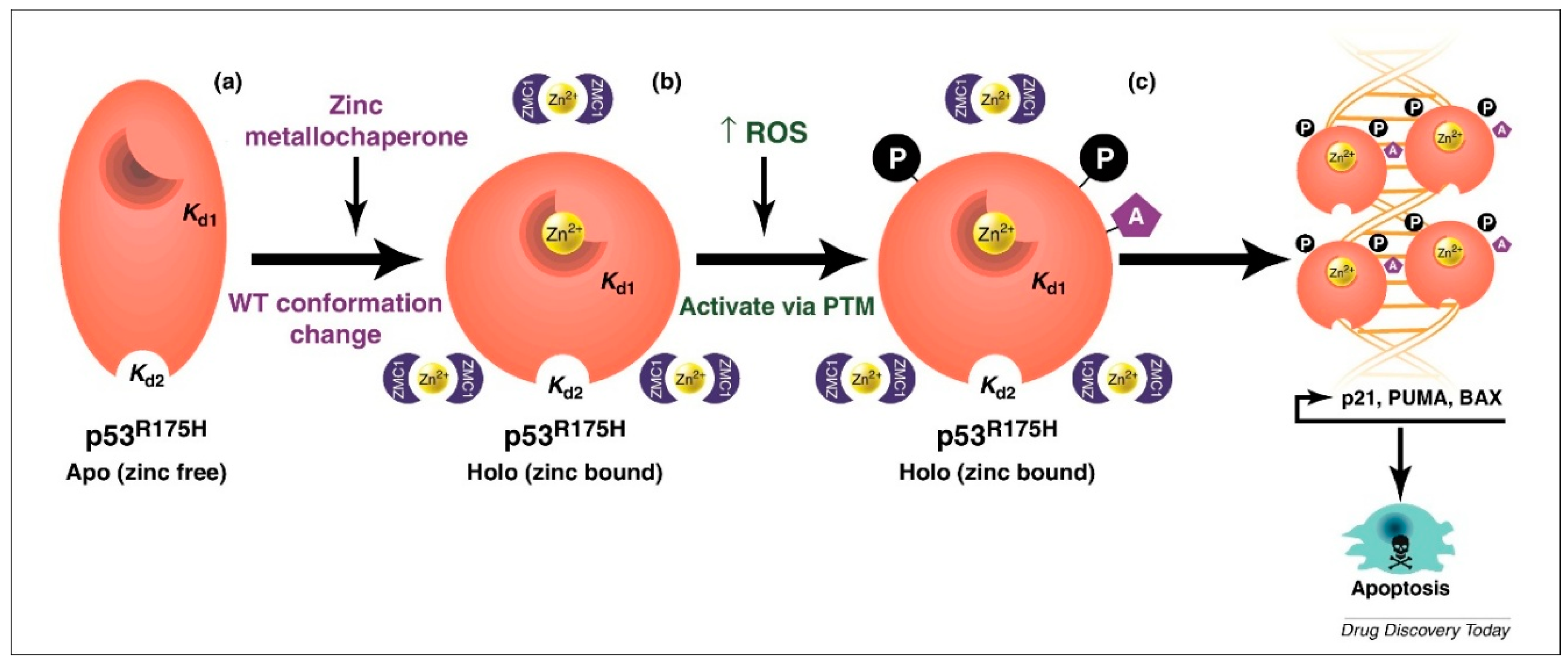
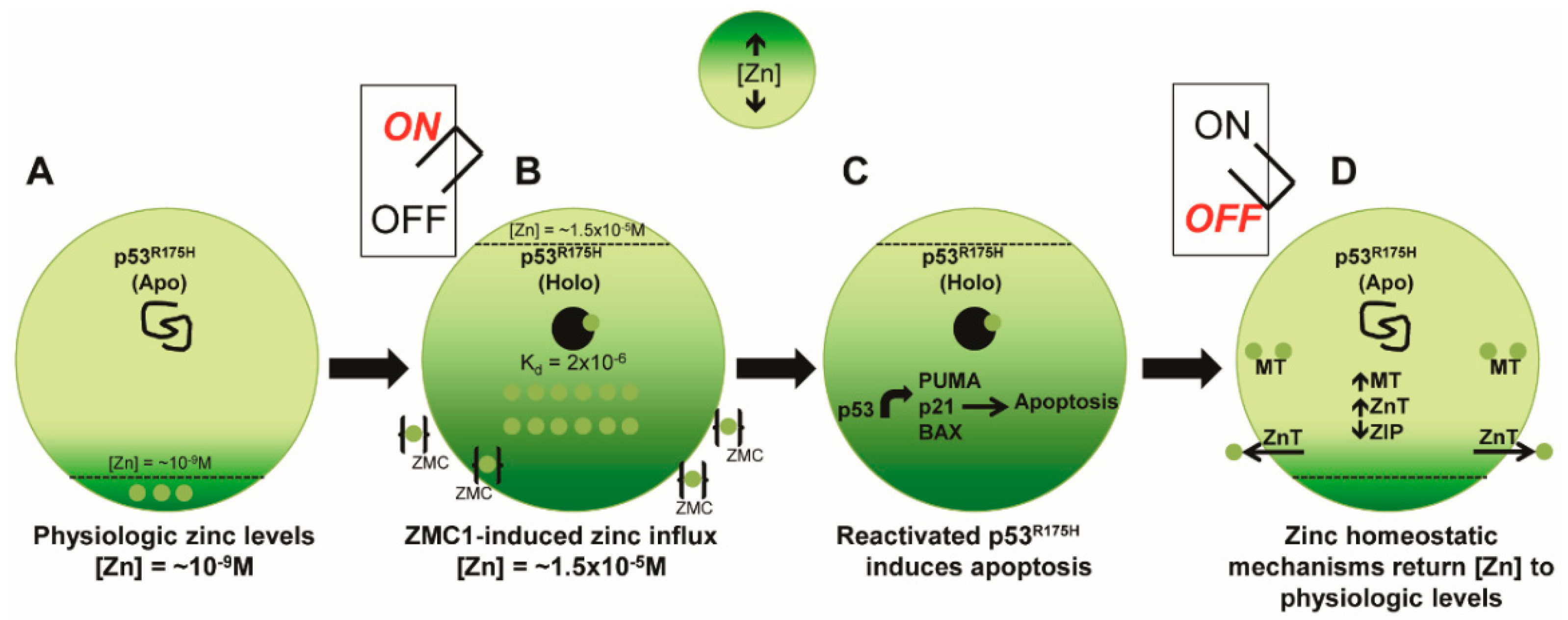
1.6. Elucidation of a Novel Mechanism of Action in Cancer Therapeutics
1.7. Pre-Clinical Translation of ZMCs
1.8. ZMCs Synthesized in Complex with Zinc: A Novel Drug Formulation
1.9. Pharmacokinetic Parameters That Distinguish ZMCs
1.10. Design Principles for Future Clinical Trials of a ZMC
1.10.1. Patient Selection
1.10.2. Proof-of-Concept Clinical Trial
1.10.3. Biomarkers of Sensitivity/Resistance
2. Conclusions
Funding
Conflicts of Interest
References
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Zhu, W.G. Surf the post-translational modification network of p53 regulation. Int. J. Biol. Sci. 2012, 8, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef] [PubMed]
- Leroy, B.; Anderson, M.; Soussi, T. TP53 mutations in human cancer: Database reassessment and prospects for the next decade. Hum. Mutat. 2014, 35, 672–688. [Google Scholar] [CrossRef] [PubMed]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Ang, H.C.; Joerger, A.C.; Mayer, S.; Fersht, A.R. Effects of common cancer mutations on stability and DNA binding of full-length p53 compared with isolated core domains. J. Biol. Chem. 2006, 281, 21934–21941. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Zerdoumi, Y.; Aury-Landas, J.; Bonaiti-Pellie, C.; Derambure, C.; Sesboue, R.; Renaux-Petel, M.; Frebourg, T.; Bougeard, G.; Flaman, J.M. Drastic effect of germline TP53 missense mutations in Li-Fraumeni patients. Hum. Mutat. 2013, 34, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-Awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015, 525, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F., Jr.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, R.C.; Sandrini, F.; Figueiredo, B.; Zambetti, G.P.; Michalkiewicz, E.; Lafferty, A.R.; DeLacerda, L.; Rabin, M.; Cadwell, C.; Sampaio, G. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc. Natl. Acad. Sci. USA 2001, 98, 9330–9335. [Google Scholar] [CrossRef] [PubMed]
- Latronico, A.C.; Pinto, E.M.; Domenice, S.; Fragoso, M.C.; Martin, R.M.; Zerbini, M.C.; Lucon, A.M.; Mendonca, B.B. An inherited mutation outside the highly conserved DNA-binding domain of the p53 tumor suppressor protein in children and adults with sporadic adrenocortical tumors. J. Clin. Endocrinol. Metab. 2001, 86, 4970–4973. [Google Scholar] [CrossRef] [PubMed]
- Di Giammarino, E.L.; Lee, A.S.; Cadwell, C.; Zhang, W.; Bothner, B.; Ribeiro, R.C.; Zambetti, G.; Kriwacki, R.W. A novel mechanism of tumorigenesis involving pH-dependent destabilization of a mutant p53 tetramer. Nat. Struct. Biol. 2002, 9, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. The common mechanisms of transformation by the small DNA tumor viruses: The inactivation of tumor suppressor gene products: P53. Virology 2009, 384, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Marine, J.C.; Francoz, S.; Maetens, M.; Wahl, G.; Toledo, F.; Lozano, G. Keeping p53 in check: Essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ. 2006, 13, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Kogan, S.; Carpizo, D. Pharmacological targeting of mutant p53. Transl. Cancer Res. 2016, 5, 698–706. [Google Scholar] [CrossRef]
- Yu, X.; Narayanan, S.; Vazquez, A.; Carpizo, D.R. Small molecule compounds targeting the p53 pathway: Are we finally making progress? Apoptosis 2014, 19, 1055–1068. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Ang, H.C.; Fersht, A.R. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl. Acad. Sci. USA 2006, 103, 15056–15061. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Henckel, J.; Fersht, A.R. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: Definition of mutant states for rescue in cancer therapy. Oncogene 2000, 19, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structure-function-rescue: The diverse nature of common p53 cancer mutants. Oncogene 2007, 26, 2226–2242. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.S.; Loh, S.N. Structure, function, and aggregation of the zinc-free form of the p53 DNA binding domain. Biochemistry 2003, 42, 2396–2403. [Google Scholar] [CrossRef] [PubMed]
- Meplan, C.; Richard, M.J.; Hainaut, P. Metalloregulation of the tumor suppressor protein p53: Zinc mediates the renaturation of p53 after exposure to metal chelators in vitro and in intact cells. Oncogene 2000, 19, 5227–5236. [Google Scholar] [CrossRef] [PubMed]
- Puca, R.; Nardinocchi, L.; Givol, D.; D’Orazi, G. Regulation of p53 activity by HIPK2: Molecular mechanisms and therapeutical implications in human cancer cells. Oncogene 2010, 29, 4378–4387. [Google Scholar] [CrossRef] [PubMed]
- Puca, R.; Nardinocchi, L.; Gal, H.; Rechavi, G.; Amariglio, N.; Domany, E.; Notterman, D.A.; Scarsella, M.; Leonetti, C.; Sacchi, A.; et al. Reversible dysfunction of wild-type p53 following homeodomain-interacting protein kinase-2 knockdown. Cancer Res. 2008, 68, 3707–3714. [Google Scholar] [CrossRef] [PubMed]
- Puca, R.; Nardinocchi, L.; Bossi, G.; Sacchi, A.; Rechavi, G.; Givol, D.; D’orazi, G. Restoring wtp53 activity in HIPK2 depleted MCF7 cells by modulating metallothionein and zinc. Exp. Cell Res. 2009, 315, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Puca, R.; Nardinocchi, L.; Porru, M.; Simon, A.J.; Rechavi, G.; Leonetti, C.; Givol, D.; D’Orazi, G. Restoring p53 active conformation by zinc increases the response of mutant p53 tumor cells to anticancer drugs. Cell Cycle 2011, 10, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Vazquez, A.; Levine, A.J.; Carpizo, D.R. Allele-specific p53 mutant reactivation. Cancer Cell 2012, 21, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Kalinowski, D.S.; Kovacevic, Z.; Siafakas, A.R.; Jansson, P.J.; Stefani, C.; Lovejoy, D.B.; Sharpe, P.C.; Bernhardt, P.V.; Richardson, D.R. Thiosemicarbazones from the old to new: Iron chelators that are more than just ribonucleotide reductase inhibitors. J. Med. Chem. 2009, 52, 5271–5294. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, D.S.; Richardson, D.R. Future of toxicology—Iron chelators and differing modes of action and toxicity: The changing face of iron chelation therapy. Chem. Res. Toxicol. 2007, 20, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Blanden, A.R.; Tsang, A.; Zaman, S.; Liu, Y.; Bencivenga, A.F.; Kimball, S.D.; Loh, S.N.; Carpizo, D.R. Thiosemicarbazones Functioning as Zinc Metallochaperones to Reactivate Mutant p53. Mol. Pharmacol. 2017, 91, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Kunos, C.A.; Radivoyevitch, T.; Waggoner, S.; Debernardo, R.; Zanotti, K.; Resnick, K.; Fusco, N.; Adams, R.; Redline, R.; Faulhaber, P.; et al. Radiochemotherapy plus 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, NSC #663249) in advanced-stage cervical and vaginal cancers. Gynecol. Oncol. 2013, 130, 75–80. [Google Scholar] [PubMed]
- Zeidner, J.F.; Karp, J.E.; Blackford, A.L.; Smith, B.D.; Gojo, I.; Gore, S.D.; Levis, M.J.; Carraway, H.E.; Greer, J.M.; Ivy, S.P.; et al. A phase II trial of sequential ribonucleotide reductase inhibition in aggressive myeloproliferative neoplasms. Haematologica 2014, 99, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Salim, K.Y.; Maleki Vareki, S.; Danter, W.R.; Koropatnick, J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget 2016, 7, 41363–41379. [Google Scholar] [CrossRef] [PubMed]
- Loh, S.N. The missing zinc: P53 misfolding and cancer. Metallomics 2010, 2, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Blanden, A.R.; Narayanan, S.; Jayakumar, L.; Lubin, D.; Augeri, D.; Kimball, S.D.; Loh, S.N.; Carpizo, D.R. Small molecule restoration of wildtype structure and function of mutant p53 using a novel zinc-metallochaperone based mechanism. Oncotarget 2014, 5, 8879–8892. [Google Scholar] [CrossRef] [PubMed]
- Blanden, A.R.; Yu, X.; Loh, S.N.; Levine, A.J.; Carpizo, D.R. Reactivating mutant p53 using small molecules as zinc metallochaperones: Awakening a sleeping giant in cancer. Drug Discov. Today 2015, 20, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Blanden, A.R.; Yu, X.; Wolfe, A.J.; Gilleran, J.A.; Augeri, D.J.; O’Dell, R.S.; Olson, E.C.; Kimball, S.D.; Emge, T.J.; Movileanu, L.; et al. Synthetic metallochaperone ZMC1 rescues mutant p53 conformation by transporting zinc into cells as an ionophore. Mol. Pharmacol. 2015, 87, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Kogan, S.; Chen, Y.; Tsang, A.T.; Withers, T.; Lin, H.; Buckley, B.; Moore, D.; Bertino, J.; Chan, C.; et al. Zinc Metallochaperones Reactivate Mutant p53 Using an ON/OFF Switch Mechanism: A New Paradigm in Cancer Therapeutics. Clin. Cancer Res. 2018, in press. [Google Scholar]
- Jeong, J.; Eide, D.J. The SLC39 family of zinc transporters. Mol. Aspects Med. 2013, 34, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Tepaamorndech, S. The SLC30 family of zinc transporters—A review of current understanding of their biological and pathophysiological roles. Mol. Aspects Med. 2013, 34, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Babula, P.; Masarik, M.; Adam, V.; Eckschlager, T.; Stiborova, M.; Trnkova, L.; Skutkova, H.; Provaznik, I.; Hubalek, J.; Kizek, R. Mammalian metallothioneins: Properties and functions. Metallomics 2012, 4, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kambe, T. The Functions of Metallothionein and ZIP and ZnT Transporters: An Overview and Perspective. Int. J. Mol. Sci. 2016, 17, 336. [Google Scholar] [CrossRef] [PubMed]
- Samuel, K. (Rutgers University, Piscataway, NJ, USA); Darren, R.C. (Rutgers University, Piscataway, NJ, USA). Personal communication, 2018.
- Taylor, K.M.; Morgan, H.E.; Smart, K.; Zahari, N.M.; Pumford, S.; Ellis, I.O.; Robertson, J.F.; Nicholson, R.I. The emerging role of the LIV-1 subfamily of zinc transporters in breast cancer. Mol. Med. 2007, 13, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes of BioMedical Research; Saxena, R.; Voight, B.F.; Lyssenko, V.; Burtt, N.P.; de Bakker, P.I.; Chen, H.; Roix, J.J.; Kathiresan, S.; Hirschhorn, J.N.; et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007, 316, 1331–1336. [Google Scholar] [PubMed]
- Huang, L.; Kirschke, C.P.; Zhang, Y. Decreased intracellular zinc in human tumorigenic prostate epithelial cells: A possible role in prostate cancer progression. Cancer Cell Int. 2006, 6, 10. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kogan, S.; Carpizo, D.R. Zinc Metallochaperones as Mutant p53 Reactivators: A New Paradigm in Cancer Therapeutics. Cancers 2018, 10, 166. https://doi.org/10.3390/cancers10060166
Kogan S, Carpizo DR. Zinc Metallochaperones as Mutant p53 Reactivators: A New Paradigm in Cancer Therapeutics. Cancers. 2018; 10(6):166. https://doi.org/10.3390/cancers10060166
Chicago/Turabian StyleKogan, Samuel, and Darren R. Carpizo. 2018. "Zinc Metallochaperones as Mutant p53 Reactivators: A New Paradigm in Cancer Therapeutics" Cancers 10, no. 6: 166. https://doi.org/10.3390/cancers10060166
APA StyleKogan, S., & Carpizo, D. R. (2018). Zinc Metallochaperones as Mutant p53 Reactivators: A New Paradigm in Cancer Therapeutics. Cancers, 10(6), 166. https://doi.org/10.3390/cancers10060166