YAP and TAZ in Lung Cancer: Oncogenic Role and Clinical Targeting


Abstract
1. Introduction
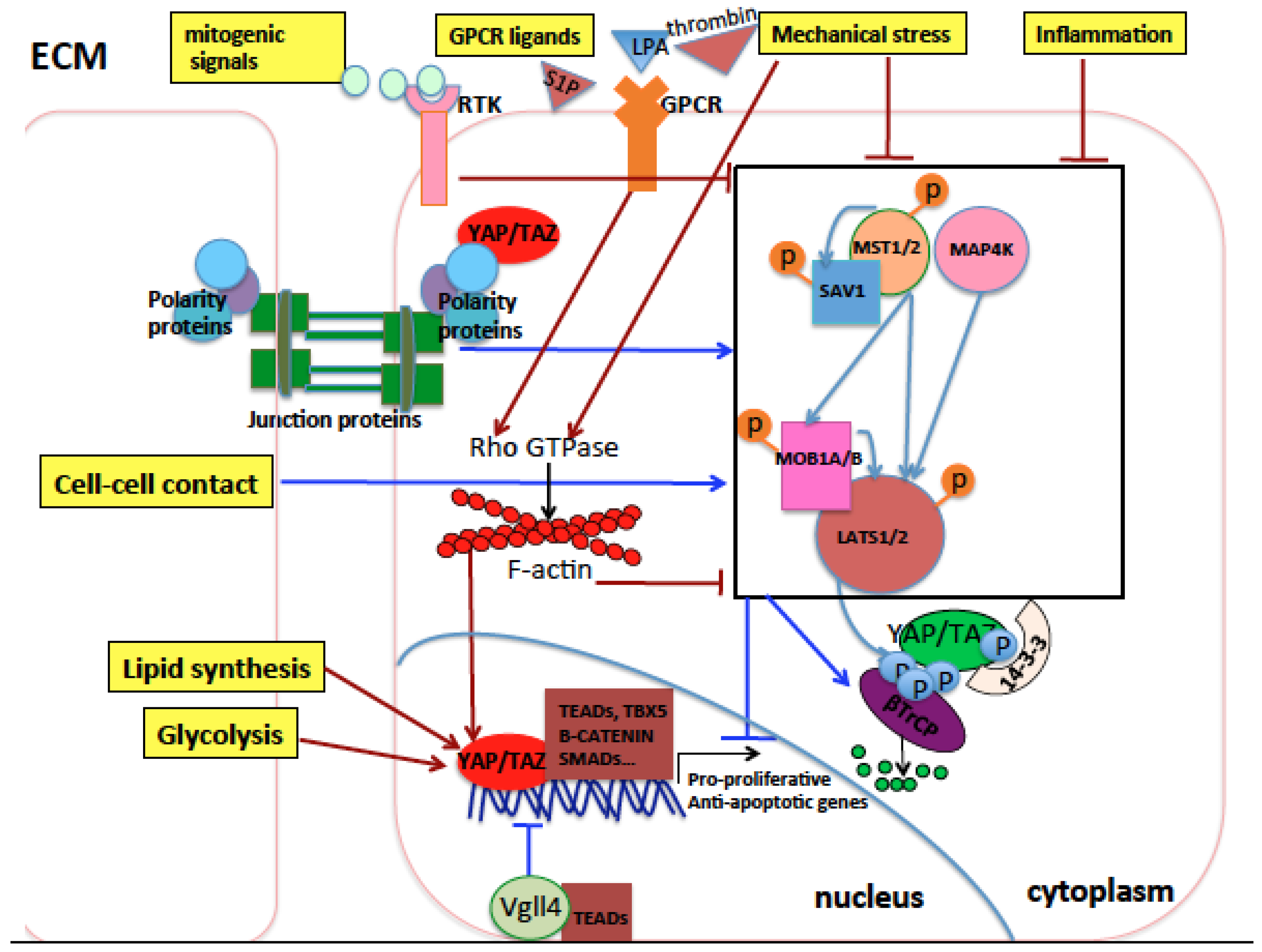
2. The Hippo Pathway
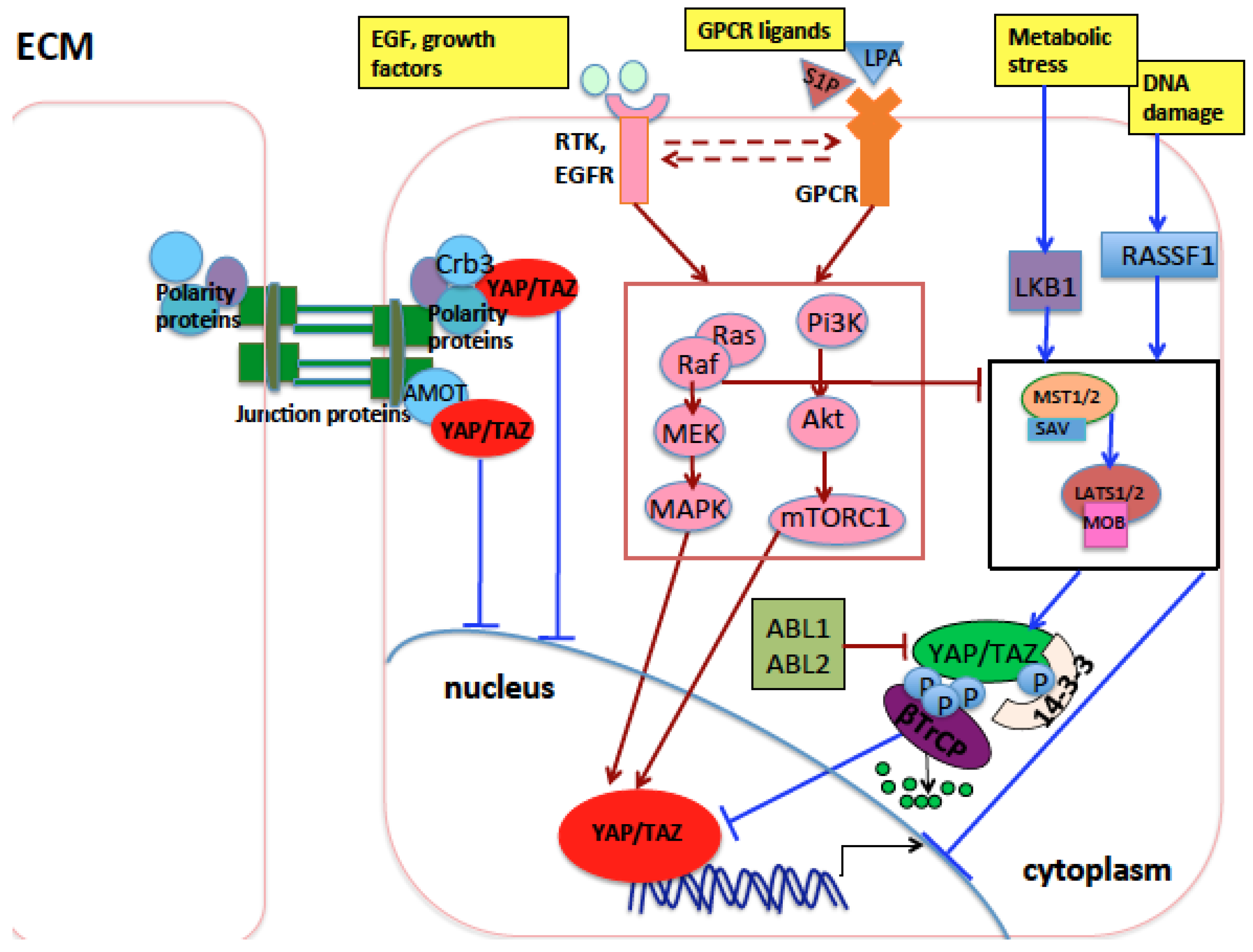
3. Regulation of the Hippo Pathway
4. YAP and TAZ are Pro-Oncogenic in Solid Tumors
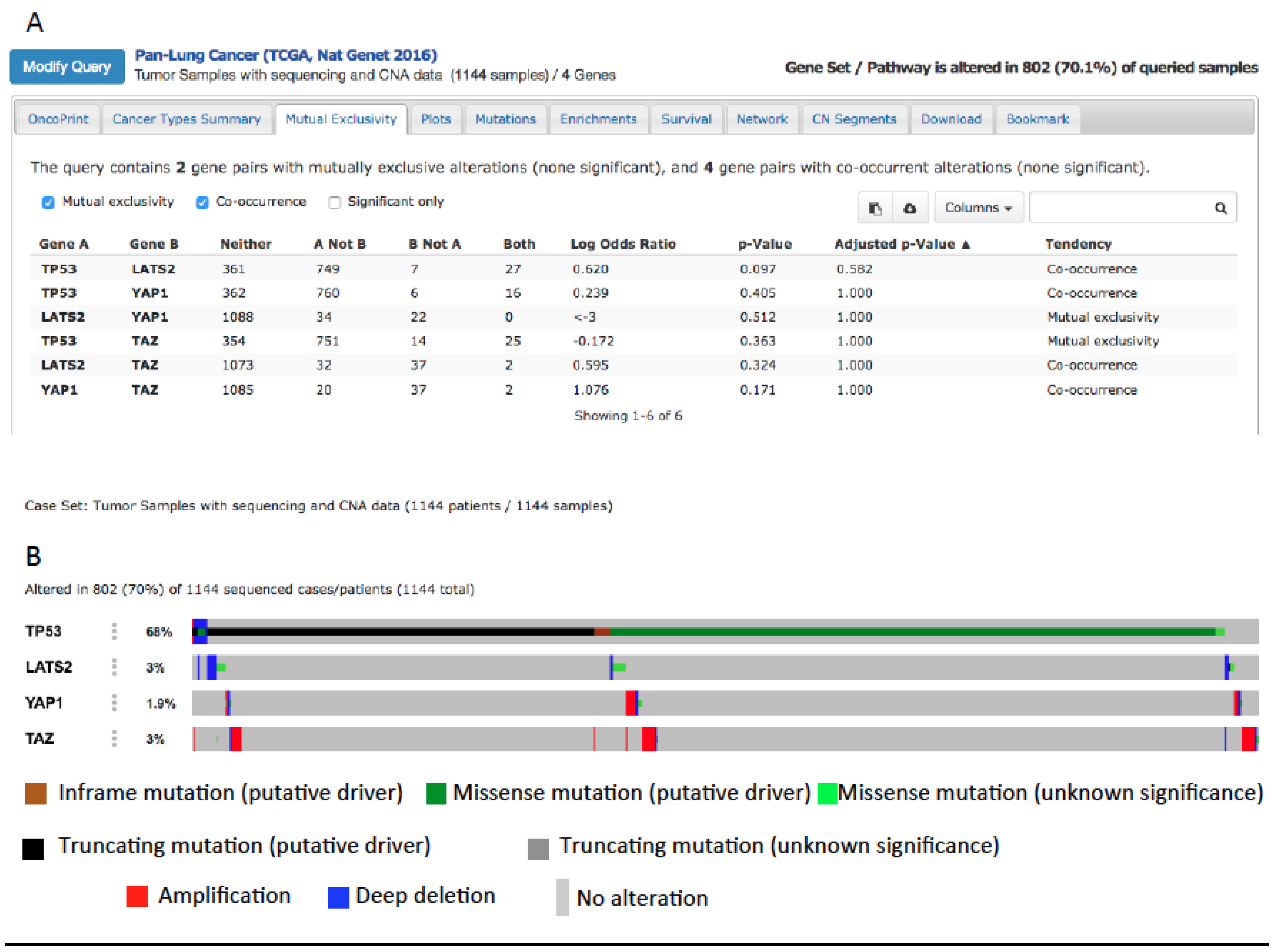
5. Hippo Pathway in Lung Cancer
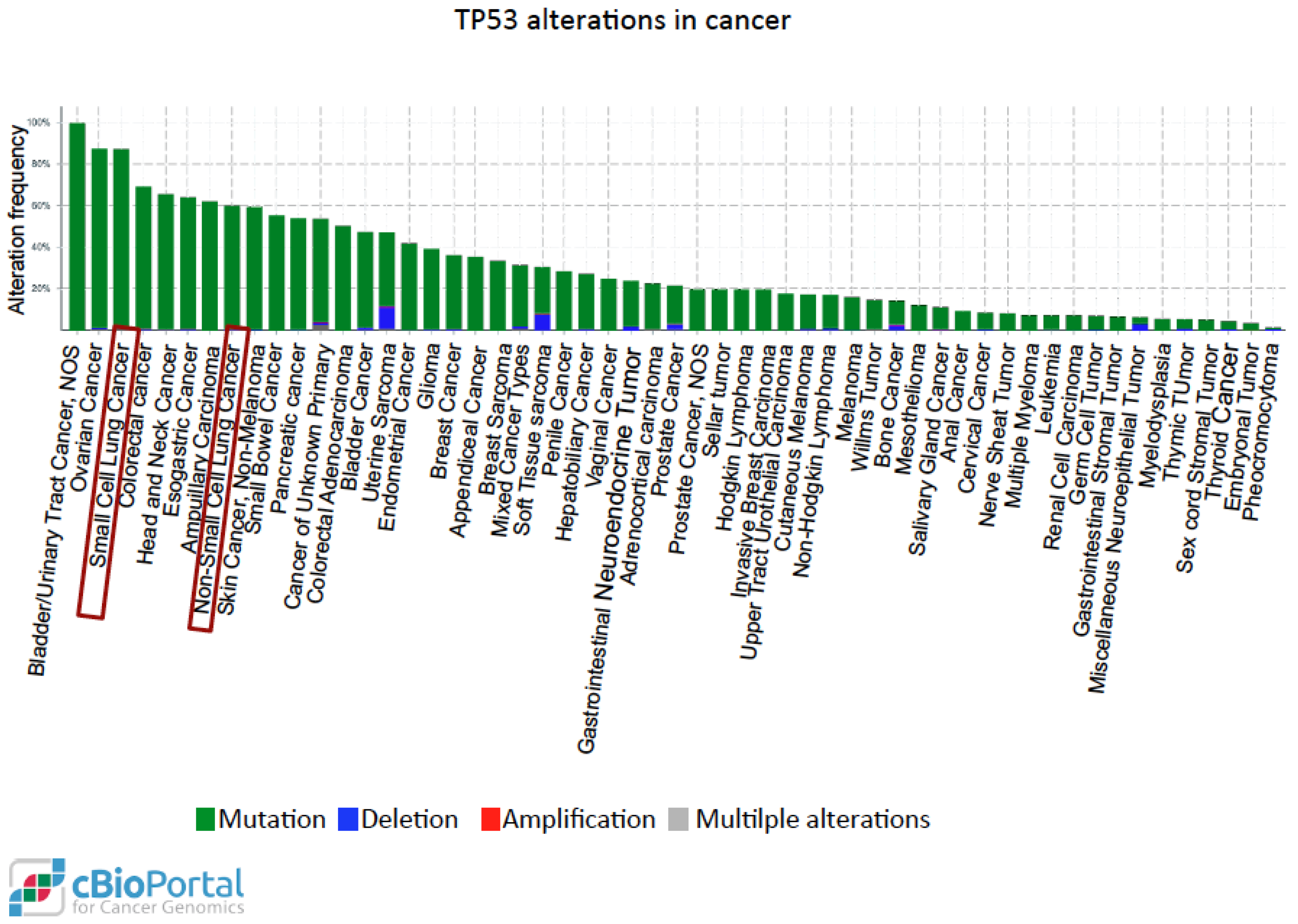
6. YAP and mutP53 Can Synergize in Lung Cancer
7. An Emerging Role of YAP/TAZ in microRNA Regulation in NSCLC
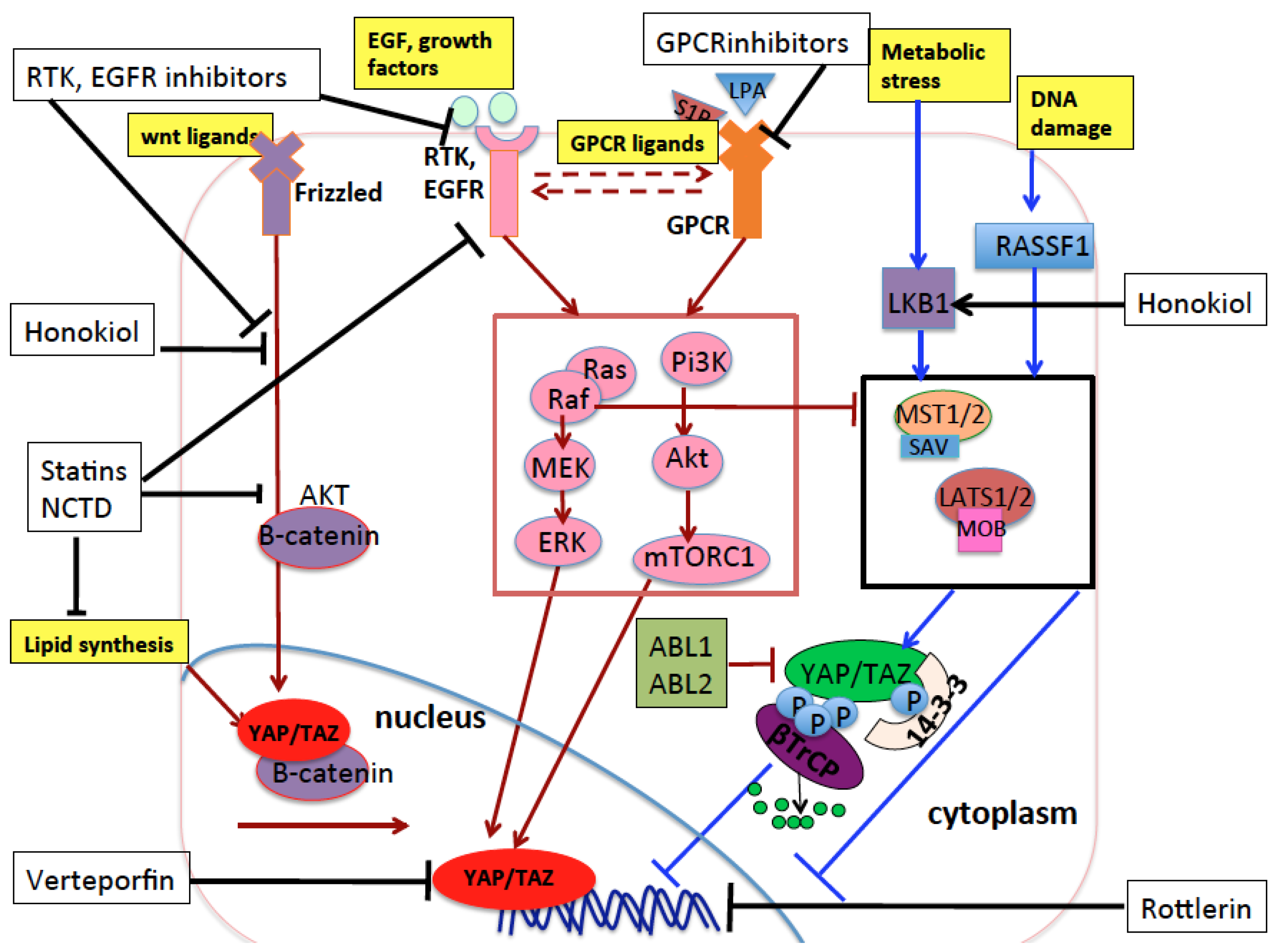
8. YAP/TAZ as Therapeutic Targets in NSCLC: State of the Art
9. Synergism between YAP/TAZ Inhibition and Current Therapies in NSCLC Experimental Models
10. Immune Evasion
11. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Yokota, J.; Kohno, T. Molecular footprints of human lung cancer progression. Cancer Sci. 2004, 95, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Eichler, A.F.; Chung, E.; Kodack, D.P.; Loeffler, J.S.; Fukumura, D.; Jain, R.K. The biology of brain metastases-translation to new therapies. Nat. Rev. Clin. Oncol. 2011, 8, 344–356. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Siegel, R.L.; Jemal, A. Lung cancer statistics. Adv. Exp. Med. Biol. 2016, 893, 1–19. [Google Scholar] [PubMed]
- Back, A.L.; Park, E.R.; Greer, J.A.; Jackson, V.A.; Jacobsen, J.C.; Gallagher, E.R.; Temel, J.S. Clinician roles in early integrated palliative care for patients with advanced cancer: A qualitative study. J. Palliat. Med. 2014, 17, 1244–1248. [Google Scholar] [CrossRef] [PubMed]
- Greer, J.A.; Jackson, V.A.; Meier, D.E.; Temel, J.S. Early integration of palliative care services with standard oncology care for patients with advanced cancer. CA Cancer J. Clin. 2013, 63, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Begnaud, A.; Hall, T.; Allen, T. Lung cancer screening with low-dose CT: Implementation amid changing public policy at one health care system. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, e468–e475. [Google Scholar] [CrossRef] [PubMed]
- Eberth, J.M.; Sercy, E. Implementation of lung cancer screening in the United States: Changing trends based on a survey of society of thoracic radiology members. J. Thorac. Imaging 2015, 30, W60–W62. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.H.; Ashraf, H. Implementation and organization of lung cancer screening. Ann. Transl. Med. 2016, 4, 152. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Moroishi, T.; Hansen, C.G.; Guan, K.L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the roots of cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell. Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Santinon, G.; Pocaterra, A.; Dupont, S. Control of YAP/TAZ activity by metabolic and nutrient-sensing pathways. Trends Cell Biol. 2016, 26, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, N.; Xie, R.; Wang, W.; Cai, J.; Choi, K.S.; David, K.K.; Huang, B.; Yabuta, N.; Nojima, H.; et al. Homeostatic control of Hippo signaling activity revealed by an endogenous activating mutation in YAP. Genes Dev. 2015, 29, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.Q.; Zhou, Z.C.; Tian, W. Research progresses on the molecular structure of the Hippo signaling pathway components. Yi Chuan 2017, 39, 659–674. [Google Scholar] [PubMed]
- Zheng, Y.; Wang, W.; Liu, B.; Deng, H.; Uster, E.; Pan, D. Identification of Happyhour/MAP4K as Alternative Hpo/Mst-like Kinases in the Hippo Kinase Cascade. Dev. Cell 2015, 34, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Mottier-Pavie, V.; Plouffe, S.W.; Hansen, C.G.; Hong, A.W.; Park, H.W.; Mo, J.S.; Lu, W.; Lu, S.; et al. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat. Commun. 2015, 6, 8357. [Google Scholar] [CrossRef] [PubMed]
- Maejima, Y.; Kyoi, S.; Zhai, P.; Liu, T.; Li, H.; Ivessa, A.; Sciarretta, S.; Del Re, D.P.; Zablocki, D.K.; Hsu, C.P.; et al. MST1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat. Med. 2013, 19, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Furth, N.; Aylon, Y. The LATS1 and LATS2 tumor suppressors: Beyond the Hippo pathway. Cell Death Differ. 2017, 24, 1488–1501. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.J.; Sahai, E. MST kinases in development and disease. J. Cell Biol. 2015, 210, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Oudhoff, M.J.; Freeman, S.A.; Couzens, A.L.; Antignano, F.; Kuznetsova, E.; Min, P.H.; Northrop, J.P.; Lehnertz, B.; Barsyte-Lovejoy, D.; Vedadi, M.; et al. Control of the Hippo pathway by Set7-dependent methylation of YAP. Dev. Cell 2013, 26, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Mao, B.; Hu, F.; Cheng, J.; Wang, P.; Xu, M.; Yuan, F.; Meng, S.; Wang, Y.; Yuan, Z.; Bi, W. SIRT1 regulates YAP2-mediated cell proliferation and chemoresistance in hepatocellular carcinoma. Oncogene 2014, 33, 1468–1474. [Google Scholar] [CrossRef] [PubMed]
- Hata, S.; Hirayama, J.; Kajiho, H.; Nakagawa, K.; Hata, Y.; Katada, T.; Furutani-Seiki, M.; Nishina, H. A novel acetylation cycle of transcription co-activator Yes-associated protein that is downstream of Hippo pathway is triggered in response to SN2 alkylating agents. J. Biol. Chem. 2012, 287, 22089–22098. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, V.; Gudmundsdottir, K.; Luong, P.; Leung, K.Y.; Knebel, A.; Basu, S. JNK phosphorylates Yes-associated protein (YAP) to regulate apoptosis. Cell Death Dis. 2010, 1, e29. [Google Scholar] [CrossRef] [PubMed]
- Keshet, R.; Adler, J.; Ricardo Lax, I.; Shanzer, M.; Porat, Z.; Reuven, N.; Shaul, Y. c-Abl antagonizes the YAP oncogenic function. Cell Death Differ. 2015, 22, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.J.; Rouse, C.; Xu, X.; Wang, J.; Onaitis, M.W.; Pendergast, A.M. Inactivation of ABL kinases suppresses non-small cell lung cancer metastasis. JCI Insight 2016, 1, e89647. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Basu, S.; Totty, N.F.; Irwin, M.S.; Sudol, M.; Downward, J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol. Cell 2003, 11, 11–23. [Google Scholar] [CrossRef]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. β-catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Kohli, P.; Bartram, M.P.; Habbig, S.; Pahmeyer, C.; Lamkemeyer, T.; Benzing, T.; Schermer, B.; Rinschen, M.M. Label-free quantitative proteomic analysis of the YAP/TAZ interactome. Am. J. Physiol. Cell Physiol. 2014, 306, C805–C818. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.R.; Camargo, F.D. The Hippo superhighway: Signaling crossroads converging on the Hippo/YAP pathway in stem cells and development. Curr. Opin. Cell Biol. 2013, 25, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.G.; Moroishi, T.; Guan, K.L. YAP and TAZ: A nexus for Hippo signaling and beyond. Trends Cell Biol. 2015, 25, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Finch-Edmondson, M.L.; Strauss, R.P.; Passman, A.M.; Sudol, M.; Yeoh, G.C.; Callus, B.A. TAZ protein accumulation is negatively regulated by YAP abundance in mammalian cells. J. Biol. Chem. 2015, 290, 27928–27938. [Google Scholar] [CrossRef] [PubMed]
- Kaan, H.Y.K.; Chan, S.W.; Tan, S.K.J.; Guo, F.; Lim, C.J.; Hong, W.; Song, H. Crystal structure of TAZ-TEAD complex reveals a distinct interaction mode from that of YAP-TEAD complex. Sci. Rep. 2017, 7, 2035. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Higashi, T.; Yokoyama, N.; Kaida, T.; Sakamoto, K.; Fukushima, Y.; Ishimoto, T.; Kuroki, H.; Nitta, H.; Hashimoto, D.; et al. An imbalance in TAZ and YAP expression in hepatocellular carcinoma confers cancer stem cell-like behaviors contributing to disease progression. Cancer Res. 2015, 75, 4985–4997. [Google Scholar] [CrossRef] [PubMed]
- Hau, J.C.; Erdmann, D.; Mesrouze, Y.; Furet, P.; Fontana, P.; Zimmermann, C.; Schmelzle, T.; Hofmann, F.; Chene, P. The TEAD4-YAP/TAZ protein-protein interaction: Expected similarities and unexpected differences. Chembiochem 2013, 14, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Elbediwy, A.; Vincent-Mistiaen, Z.I.; Spencer-Dene, B.; Stone, R.K.; Boeing, S.; Wculek, S.K.; Cordero, J.; Tan, E.H.; Ridgway, R.; Brunton, V.G.; et al. Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development 2016, 143, 1674–1687. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, A.; Yu, F.X. The Hippo pathway in tissue homeostasis and regeneration. Protein Cell 2017, 8, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Cottini, F.; Hideshima, T.; Xu, C.; Sattler, M.; Dori, M.; Agnelli, L.; ten Hacken, E.; Bertilaccio, M.T.; Antonini, E.; Neri, A.; et al. Rescue of Hippo coactivator YAP1 triggers DNA damage-induced apoptosis in hematological cancers. Nat. Med. 2014, 20, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Liu, Y.; Zheng, Y.; Dong, J.; Pan, D. The TEAD/TEF family protein scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev. Cell 2008, 14, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, A.; Kaneko, K.J.; Shu, H.; Zhao, Y.; DePamphilis, M.L. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev. 2001, 15, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Komuro, A.; Nagai, M.; Navin, N.E.; Sudol, M. WW domain-containing protein YAP associates with Erbb-4 and acts as a co-transcriptional activator for the carboxyl-terminal fragment of Erbb-4 that translocates to the nucleus. J. Biol. Chem. 2003, 278, 33334–33341. [Google Scholar] [CrossRef] [PubMed]
- Haskins, J.W.; Nguyen, D.X.; Stern, D.F. Neuregulin 1-activated Erbb4 interacts with YAP to induce Hippo pathway target genes and promote cell migration. Sci. Signal. 2014, 7, ra116. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, W.M., Jr.; Hong, J.H.; Yaffe, M.B.; Farrance, I.K. The transcriptional co-activator TAZ interacts differentially with transcriptional enhancer factor-1 (TEF-1) family members. Biochem. J. 2005, 388, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Nakagawa, M.; Olson, E.N.; Nakagawa, O. A WW domain protein TAZ is a critical coactivator for TBX5, a transcription factor implicated in Holt-Oram syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 18034–18039. [Google Scholar] [CrossRef] [PubMed]
- Di Agostino, S.; Sorrentino, G.; Ingallina, E.; Valenti, F.; Ferraiuolo, M.; Bicciato, S.; Piazza, S.; Strano, S.; Del Sal, G.; Blandino, G. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep. 2016, 17, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Tretiakova, M.S.; Silvis, M.R.; Lucas, J.; Klezovitch, O.; Coleman, I.; Bolouri, H.; Kutyavin, V.I.; Morrissey, C.; True, L.D.; et al. Erg activates the YAP1 transcriptional program and induces the development of age-related prostate tumors. Cancer Cell 2015, 27, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Alharbi, A.; Shan, H.; Hao, Y.; Snetsinger, B.; Rauh, M.J.; Yang, X. TAZ induces lung cancer stem cell properties and tumorigenesis by up-regulating ALDH1A1. Oncotarget 2017, 8, 38426–38443. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, M.; Kim, T.; Johnson, R.L.; Lim, D.S. Transcriptional co-repressor function of the Hippo pathway transducers YAP and TAZ. Cell Rep. 2015, 11, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Chen, Y.; Chen, L.; Cheng, H.; Mu, C.; Li, J.; Gao, R.; Zhou, C.; Cao, L.; Liu, J.; et al. Hypoxia regulates Hippo signalling through the SIAH2 ubiquitin E3 ligase. Nat. Cell Biol. 2015, 17, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.Y.; Zhuang, L.H.; Wang, D.D.; Zhou, T.Y.; Chang, L.L.; Gai, R.H.; Zhu, D.F.; Yang, B.; Zhu, H.; He, Q.J. Nuclear translocation and activation of YAP by hypoxia contributes to the chemoresistance of SN38 in hepatocellular carcinoma cells. Oncotarget 2016, 7, 6933–6947. [Google Scholar] [CrossRef] [PubMed]
- Low, B.C.; Pan, C.Q.; Shivashankar, G.V.; Bershadsky, A.; Sudol, M.; Sheetz, M. YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS Lett. 2014, 588, 2663–2670. [Google Scholar] [CrossRef] [PubMed]
- Pesic, M.; Greten, F.R. Inflammation and cancer: Tissue regeneration gone awry. Curr. Opin. Cell Biol. 2016, 43, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Enzo, E.; Santinon, G.; Pocaterra, A.; Aragona, M.; Bresolin, S.; Forcato, M.; Grifoni, D.; Pession, A.; Zanconato, F.; Guzzo, G.; et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J. 2015, 34, 1349–1370. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xiao, Z.D.; Li, X.; Aziz, K.E.; Gan, B.; Johnson, R.L.; Chen, J. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 2015, 17, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.S.; Meng, Z.; Kim, Y.C.; Park, H.W.; Hansen, C.G.; Kim, S.; Lim, D.S.; Guan, K.L. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat. Cell Biol. 2015, 17, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Cheng, C.; Tan, Z.; Li, N.; Tang, M.; Yang, L.; Cao, Y. Emerging roles of lipid metabolism in cancer metastasis. Mol. Cancer 2017, 16, 76. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Noto, A.; De Vitis, C.; Pisanu, M.E.; Roscilli, G.; Ricci, G.; Catizone, A.; Sorrentino, G.; Chianese, G.; Taglialatela-Scafati, O.; Trisciuoglio, D.; et al. Stearoyl-CoA-desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene 2017, 36, 4573–4584. [Google Scholar] [CrossRef] [PubMed]
- Mancini, R.; Noto, A.; Pisanu, M.E.; De Vitis, C.; Maugeri-Sacca, M.; Ciliberto, G. Metabolic features of cancer stem cells: The emerging role of lipid metabolism. Oncogene 2018. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.L.; Zhao, W.Y.; Zhang, W.J.; Song, H.; Ji, H.B.; Tang, N. Hippo signaling pathway in lung development, regeneration, and diseases. Yi Chuan 2017, 39, 597–606. [Google Scholar] [PubMed]
- Dai, Y.; Jablons, D.; You, L. Hippo pathway in lung development. J. Thorac. Dis. 2017, 9, 2246–2250. [Google Scholar] [CrossRef] [PubMed]
- Szymaniak, A.D.; Mahoney, J.E.; Cardoso, W.V.; Varelas, X. Crumbs3-mediated polarity directs airway epithelial cell fate through the Hippo pathway effector YAP. Dev. Cell 2015, 34, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Matsubara, D.; Tanaka, I.; Makiya, K.; Tanei, Z.I.; Kumagai, Y.; Shiu, S.J.; Nakaoka, H.J.; Ishikawa, S.; Isagawa, T.; et al. Loss of YAP1 defines neuroendocrine differentiation of lung tumors. Cancer Sci. 2016, 107, 1527–1538. [Google Scholar] [CrossRef] [PubMed]
- Horie, M.; Saito, A.; Ohshima, M.; Suzuki, H.I.; Nagase, T. YAP and TAZ modulate cell phenotype in a subset of small cell lung cancer. Cancer Sci. 2016, 107, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Oser, M.G.; Niederst, M.J.; Sequist, L.V.; Engelman, J.A. Transformation from non-small-cell lung cancer to small-cell lung cancer: Molecular drivers and cells of origin. Lancet Oncol. 2015, 16, e165–e172. [Google Scholar] [CrossRef]
- Su, L.L.; Ma, W.X.; Yuan, J.F.; Shao, Y.; Xiao, W.; Jiang, S.J. Expression of Yes-associated protein in non-small cell lung cancer and its relationship with clinical pathological factors. Chin. Med. J. 2012, 125, 4003–4008. [Google Scholar] [PubMed]
- Wang, Y.; Dong, Q.; Zhang, Q.; Li, Z.; Wang, E.; Qiu, X. Overexpression of Yes-associated protein contributes to progression and poor prognosis of non-small-cell lung cancer. Cancer Sci. 2010, 101, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Lorenzetto, E.; Brenca, M.; Boeri, M.; Verri, C.; Piccinin, E.; Gasparini, P.; Facchinetti, F.; Rossi, S.; Salvatore, G.; Massimino, M.; et al. YAP1 acts as oncogenic target of 11q22 amplification in multiple cancer subtypes. Oncotarget 2014, 5, 2608–2621. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Kang, D.W.; Long, L.Z.; Huang, S.M.; Yeo, M.K.; Yi, E.S.; Kim, K.H. Differential expression of Yes-associated protein is correlated with expression of cell cycle markers and pathologic TNM staging in non-small-cell lung carcinoma. Hum. Pathol. 2011, 42, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wu, Y.; Yang, L.; Du, J.; Gong, K.; Chen, W.; Dai, J.; Li, X.; Xi, S. Repression of YAP by NCTD disrupts NSCLC progression. Oncotarget 2017, 8, 2307–2319. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.L.; Han, F.F.; Peng, X.H.; Chen, X.; Luan, C.Y.; Han, R.C.; Xu, W.G.; Guo, X.J. Yes-associated protein 1 promotes adenocarcinoma growth and metastasis through activation of the receptor tyrosine kinase AXL. Int. J. Immunopathol. Pharmacol. 2012, 25, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hao, Y.; Liu, N.; Raptis, L.; Tsao, M.S.; Yang, X. TAZ is a novel oncogene in non-small cell lung cancer. Oncogene 2011, 30, 2181–2186. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Zhang, L.; He, C.S.; Hou, J.H.; Lin, S.X.; Hu, Z.H.; Xu, F.; Zhao, H.Y. Prognostic significance of TAZ expression in resected non-small cell lung cancer. J. Thorac. Oncol. 2012, 7, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Saito, A.; Horie, M.; Mikami, Y.; Suzuki, H.I.; Morishita, Y.; Ohshima, M.; Abiko, Y.; Mattsson, J.S.; Konig, H.; et al. An integrative analysis of the tumorigenic role of TAZ in human non-small cell lung cancer. Clin. Cancer Res. 2014, 20, 4660–4672. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Yu, S.L.; Ho, B.C.; Su, K.Y.; Hsu, Y.C.; Chang, C.S.; Li, Y.C.; Yang, S.Y.; Hsu, P.Y.; Ho, H.; et al. R331W missense mutation of oncogene YAP1 is a germline risk allele for lung adenocarcinoma with medical actionability. J. Clin. Oncol. 2015, 33, 2303–2310. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.N.; Curtis, S.J.; Fillmore, C.M.; Rowbotham, S.P.; Mohseni, M.; Wagner, D.E.; Beede, A.M.; Montoro, D.T.; Sinkevicius, K.W.; Walton, Z.E.; et al. Tumor-propagating cells and YAP/TAZ activity contribute to lung tumor progression and metastasis. EMBO J. 2014, 33, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Gao, Y.; Li, F.; Tong, X.; Ren, Y.; Han, X.; Yao, S.; Long, F.; Yang, Z.; Fan, H.; et al. YAP promotes malignant progression of LKB1-deficient lung adenocarcinoma through downstream regulation of survivin. Cancer Res. 2015, 75, 4450–4457. [Google Scholar] [CrossRef] [PubMed]
- Mohseni, M.; Sun, J.; Lau, A.; Curtis, S.; Goldsmith, J.; Fox, V.L.; Wei, C.; Frazier, M.; Samson, O.; Wong, K.K.; et al. A genetic screen identifies an LKB1-mark signalling axis controlling the Hippo-YAP pathway. Nat. Cell Biol. 2014, 16, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Greuber, E.K.; Smith-Pearson, P.; Wang, J.; Pendergast, A.M. Role of ABL family kinases in cancer: From leukaemia to solid tumours. Nat. Rev. Cancer 2013, 13, 559–571. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar]
- Sos, M.L.; Michel, K.; Zander, T.; Weiss, J.; Frommolt, P.; Peifer, M.; Li, D.; Ullrich, R.; Koker, M.; Fischer, F.; et al. Predicting drug susceptibility of non-small cell lung cancers based on genetic lesions. J. Clin. Investig. 2009, 119, 1727–1740. [Google Scholar] [CrossRef] [PubMed]
- Testoni, E.; Stephenson, N.L.; Torres-Ayuso, P.; Marusiak, A.A.; Trotter, E.W.; Hudson, A.; Hodgkinson, C.L.; Morrow, C.J.; Dive, C.; Brognard, J. Somatically mutated ABL1 is an actionable and essential NSCLC survival gene. EMBO Mol. Med. 2016, 8, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Chou, H.C.; Fu, Y.N.; Yeh, C.L.; Cheng, H.W.; Chang, I.C.; Liu, K.J.; Chang, G.C.; Tsai, T.F.; Tsai, S.F.; et al. EGFR over-expression in non-small cell lung cancers harboring EGFR mutations is associated with marked down-regulation of CD82. Biochim. Biophys. Acta 2015, 1852, 1540–1549. [Google Scholar] [CrossRef] [PubMed]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar] [PubMed]
- Formisano, L.; D’Amato, V.; Servetto, A.; Brillante, S.; Raimondo, L.; Di Mauro, C.; Marciano, R.; Orsini, R.C.; Cosconati, S.; Randazzo, A.; et al. Src inhibitors act through different mechanisms in non-small cell lung cancer models depending on EGFR and RAS mutational status. Oncotarget 2015, 6, 26090–26103. [Google Scholar] [CrossRef] [PubMed]
- Rothschild, S.I.; Gautschi, O.; Haura, E.B.; Johnson, F.M. Src inhibitors in lung cancer: Current status and future directions. Clin. Lung Cancer 2010, 11, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G.; Zucali, P.A. Src as a potential therapeutic target in non-small-cell lung cancer. Ann. Oncol. 2008, 19, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Westcott, P.M.; To, M.D. The genetics and biology of KRAS in lung cancer. Chin. J. Cancer 2013, 32, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Rolfo, C.; Caparica, R. B-RAF mutation in non-small cell lung cancer: The sleeping beauty is waking up. Transl. Lung Cancer Res. 2016, 5, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Stinchcombe, T.E.; Johnson, G.L. MEK inhibition in non-small cell lung cancer. Lung Cancer 2014, 86, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Li, C. Convergence between WNT-β-catenin and EGFR signaling in cancer. Mol. Cancer 2010, 9, 236. [Google Scholar] [CrossRef] [PubMed]
- Moody, T.W.; Nuche-Berenguer, B.; Nakamura, T.; Jensen, R.T. EGFR transactivation by peptide g protein-coupled receptors in cancer. Curr. Drug Targets 2016, 17, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. Transactivation of epidermal growth factor receptor by G protein-coupled receptors: Recent progress, challenges and future research. Int. J. Mol. Sci. 2016, 17, 95. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.V.; Irvine, K.D. Regulation of Hippo signaling by EGFR-MAPK signaling through AJUBA family proteins. Dev. Cell 2013, 24, 459–471. [Google Scholar] [CrossRef] [PubMed]
- You, B.; Yang, Y.L.; Xu, Z.; Dai, Y.; Liu, S.; Mao, J.H.; Tetsu, O.; Li, H.; Jablons, D.M.; You, L. Inhibition of ERK1/2 down-regulates the Hippo/YAP signaling pathway in human NSCLC cells. Oncotarget 2015, 6, 4357–4368. [Google Scholar] [CrossRef] [PubMed]
- McGowan, M.; Kleinberg, L.; Halvorsen, A.R.; Helland, A.; Brustugun, O.T. NSCLC depend upon YAP expression and nuclear localization after acquiring resistance to EGFR inhibitors. Genes Cancer 2017, 8, 497–504. [Google Scholar] [PubMed]
- Strazisar, M.; Mlakar, V.; Glavac, D. LATS2 tumour specific mutations and down-regulation of the gene in non-small cell carcinoma. Lung Cancer 2009, 64, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Liu, H.; Li, Z.; Zhong, C.; Fang, W. Down-regulation of LATS2 in non-small cell lung cancer promoted the growth and motility of cancer cells. Tumour. Biol. 2015, 36, 2049–2057. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.A.; Khan, M.S.; Dar, M.; Hussain, M.U.; Shah, M.A.; Shafi, S.M.; Mudassar, S. Molecular alterations and expression dynamics of LATS1 and LATS2 genes in non-small-cell lung carcinoma. Pathol. Oncol. Res. 2018, 24, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Sun, M.; Liu, G.J.; Wei, C.C.; Zhang, E.B.; Kong, R.; Xu, T.P.; Huang, M.D.; Wang, Z.X. Long noncoding RNA PVT1 promotes non-small cell lung cancer cell proliferation through epigenetically regulating LATS2 expression. Mol. Cancer Ther. 2016, 15, 1082–1094. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sun, M.; Zang, C.; Ma, P.; He, J.; Zhang, M.; Huang, Z.; Ding, Y.; Shu, Y. Upregulated long non-coding RNA AGAP2-AS1 represses LATS2 and KLF2 expression through interacting with EZH2 and LSD1 in non-small-cell lung cancer cells. Cell Death Dis. 2016, 7, e2225. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.Y.; Sit, K.Y.; Sihoe, A.D.; Suen, W.S.; Au, W.K.; Tang, X.; Ma, E.S.; Chan, W.K.; Wistuba, I.I.; Minna, J.D.; et al. Aberrant large tumor suppressor 2 (LATS2) gene expression correlates with EGFR mutation and survival in lung adenocarcinomas. Lung Cancer 2014, 85, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.Y.; Zhang, X.P.; Wu, J.H.; Qiu, X.S.; Wang, E.H. Expression of LATS1 contributes to good prognosis and can negatively regulate YAP oncoprotein in non-small-cell lung cancer. Tumour Biol. 2014, 35, 6435–6443. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Bachman, J.; Lai, Z.C. Evidence for a tumor suppressor role for the large tumor suppressor genes LATS1 and LATS2 in human cancer. Genetics 2013, 195, 1193–1196. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hsu, Y.L.; Hung, J.Y.; Chou, S.H.; Huang, M.S.; Tsai, M.J.; Lin, Y.S.; Chiang, S.Y.; Ho, Y.W.; Wu, C.Y.; Kuo, P.L. Angiomotin decreases lung cancer progression by sequestering oncogenic YAP/TAZ and decreasing Cyr61 expression. Oncogene 2015, 34, 4056–4068. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.M.; Liu, W.W.; Liu, C.J.; Wen, C.; Lu, H.F.; Wan, F.S. MST1 overexpression inhibited the growth of human non-small cell lung cancer in vitro and in vivo. Cancer Gene Ther. 2013, 20, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Gao, Y.; Li, P.; Shi, Z.; Guo, T.; Li, F.; Han, X.; Feng, Y.; Zheng, C.; Wang, Z.; et al. VGLL4 functions as a new tumor suppressor in lung cancer by negatively regulating the YAP-TEAD transcriptional complex. Cell Res. 2014, 24, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Matallanas, D.; Romano, D.; Yee, K.; Meissl, K.; Kucerova, L.; Piazzolla, D.; Baccarini, M.; Vass, J.K.; Kolch, W.; O’Neill, E. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol. Cell 2007, 27, 962–975. [Google Scholar] [CrossRef] [PubMed]
- Scrace, S.F.; O’Neill, E. Rassf signalling and DNA damage: Monitoring the integrity of the genome? Mol. Biol. Int. 2012, 2012, 141732. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Zhang, X.; Pfeifer, G.P. The tumor suppressor RASSF1A prevents dephosphorylation of the mammalian STE20-like kinases MST1 and MST2. J. Biol. Chem. 2011, 286, 6253–6261. [Google Scholar] [CrossRef] [PubMed]
- Dammann, R.; Li, C.; Yoon, J.H.; Chin, P.L.; Bates, S.; Pfeifer, G.P. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat. Genet. 2000, 25, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Agathanggelou, A.; Honorio, S.; Macartney, D.P.; Martinez, A.; Dallol, A.; Rader, J.; Fullwood, P.; Chauhan, A.; Walker, R.; Shaw, J.A.; et al. Methylation associated inactivation of RASSF1A from region 3p21.3 in lung, breast and ovarian tumours. Oncogene 2001, 20, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Burbee, D.G.; Forgacs, E.; Zochbauer-Muller, S.; Shivakumar, L.; Fong, K.; Gao, B.; Randle, D.; Kondo, M.; Virmani, A.; Bader, S.; et al. Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J. Natl. Cancer Inst. 2001, 93, 691–699. [Google Scholar] [CrossRef] [PubMed]
- De Fraipont, F.; Levallet, G.; Creveuil, C.; Bergot, E.; Beau-Faller, M.; Mounawar, M.; Richard, N.; Antoine, M.; Rouquette, I.; Favrot, M.C.; et al. An apoptosis methylation prognostic signature for early lung cancer in the IFCT-0002 trial. Clin. Cancer Res. 2012, 18, 2976–2986. [Google Scholar] [CrossRef] [PubMed]
- Dubois, F.; Keller, M.; Calvayrac, O.; Soncin, F.; Hoa, L.; Hergovich, A.; Parrini, M.C.; Mazieres, J.; Vaisse-Lesteven, M.; Camonis, J.; et al. RASSF1A suppresses the invasion and metastatic potential of human non-small cell lung cancer cells by inhibiting YAP activation through the GEF-H1/RHOB pathway. Cancer Res. 2016, 76, 1627–1640. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.B.; Babcock, J.T.; Wells, C.D.; Quilliam, L.A. LKB1 tumor suppressor regulates AMP kinase/mTOR-independent cell growth and proliferation via the phosphorylation of YAP. Oncogene 2013, 32, 4100–4109. [Google Scholar] [CrossRef] [PubMed]
- Momcilovic, M.; Shackelford, D.B. Targeting LKB1 in cancer—Exposing and exploiting vulnerabilities. Br. J. Cancer 2015, 113, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Carretero, J.; Medina, P.P.; Pio, R.; Montuenga, L.M.; Sanchez-Cespedes, M. Novel and natural knockout lung cancer cell lines for the LKB1/STK11 tumor suppressor gene. Oncogene 2004, 23, 4037–4040. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Ramsey, M.R.; Hayes, D.N.; Fan, C.; McNamara, K.; Kozlowski, P.; Torrice, C.; Wu, M.C.; Shimamura, T.; Perera, S.A.; et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 2007, 448, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Cespedes, M.; Parrella, P.; Esteller, M.; Nomoto, S.; Trink, B.; Engles, J.M.; Westra, W.H.; Herman, J.G.; Sidransky, D. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002, 62, 3659–3662. [Google Scholar] [PubMed]
- Matsumoto, S.; Iwakawa, R.; Takahashi, K.; Kohno, T.; Nakanishi, Y.; Matsuno, Y.; Suzuki, K.; Nakamoto, M.; Shimizu, E.; Minna, J.D.; et al. Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene 2007, 26, 5911–5918. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zheng, F.; Zhang, S.; Lu, J. Loss of RhoA expression prevents proliferation and metastasis of SPCA1 lung cancer cells in vitro. Biomed. Pharmacother. 2015, 69, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Wu, X.; Zhang, J.; Zhang, J.; Li, X. Ski regulates Smads and TAZ signaling to suppress lung cancer progression. Mol. Carcinog. 2017, 56, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Bartucci, M.; Dattilo, R.; Moriconi, C.; Pagliuca, A.; Mottolese, M.; Federici, G.; Benedetto, A.D.; Todaro, M.; Stassi, G.; Sperati, F.; et al. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene 2015, 34, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Ajani, J.A.; Honjo, S.; Maru, D.M.; Chen, Q.; Scott, A.W.; Heallen, T.R.; Xiao, L.; Hofstetter, W.L.; Weston, B.; et al. Hippo coactivator YAP1 upregulates SOX9 and endows esophageal cancer cells with stem-like properties. Cancer Res. 2014, 74, 4170–4182. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Oren, M. The Hippo pathway, p53 and cholesterol. Cell Cycle 2016, 15, 2248–2255. [Google Scholar] [CrossRef] [PubMed]
- Lo Sardo, F.; Forcato, M.; Sacconi, A.; Capaci, V.; Zanconato, F.; di Agostino, S.; Del Sal, G.; Pandolfi, P.P.; Strano, S.; Bicciato, S.; et al. MCM7 and its hosted miR-25, 93 and 106b cluster elicit YAP/TAZ oncogenic activity in lung cancer. Carcinogenesis 2017, 38, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Zhang, Z.; Rodriguez-Barrueco, R.; Borczuk, A.; Liu, H.; Yu, J.; Silva, J.M.; Cheng, S.K.; Perez-Soler, R.; Halmos, B. Functional genomics screen identifies YAP1 as a key determinant to enhance treatment sensitivity in lung cancer cells. Oncotarget 2016, 7, 28976–28988. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Park, H.S.; Lee, D.; Yoo, G.; Kim, T.; Jeon, H.; Yeo, M.K.; Lee, C.S.; Moon, J.Y.; Jung, S.S.; et al. Hippo pathway effector YAP inhibition restores the sensitivity of EGFR-TKI in lung adenocarcinoma having primary or acquired EGFR-TKI resistance. Biochem. Biophys. Res. Commun. 2016, 474, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. P53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T. P53 antibodies in the sera of patients with various types of cancer: A review. Cancer Res. 2000, 60, 1777–1788. [Google Scholar] [PubMed]
- Lubin, R.; Zalcman, G.; Bouchet, L.; Tredanel, J.; Legros, Y.; Cazals, D.; Hirsch, A.; Soussi, T. Serum p53 antibodies as early markers of lung cancer. Nat. Med. 1995, 1, 701–702. [Google Scholar] [CrossRef] [PubMed]
- Mitsudomi, T.; Hamajima, N.; Ogawa, M.; Takahashi, T. Prognostic significance of p53 alterations in patients with non-small cell lung cancer: A meta-analysis. Clin. Cancer Res. 2000, 6, 4055–4063. [Google Scholar] [PubMed]
- Steels, E.; Paesmans, M.; Berghmans, T.; Branle, F.; Lemaitre, F.; Mascaux, C.; Meert, A.P.; Vallot, F.; Lafitte, J.J.; Sculier, J.P. Role of p53 as a prognostic factor for survival in lung cancer: A systematic review of the literature with a meta-analysis. Eur. Respir. J. 2001, 18, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, Z.; Yu, L.; Chen, M.; Li, K. Assessment of the potential diagnostic value of serum p53 antibody for cancer: A meta-analysis. PLoS ONE 2014, 9, e99255. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Kim, Y.; Lee, J.H.; Lee, E.Y.; Kim, H.S. Usefulness of serum anti-p53 antibody assay for lung cancer diagnosis. Arch. Pathol. Lab. Med. 2011, 135, 1570–1575. [Google Scholar] [CrossRef] [PubMed]
- Mattioni, M.; Soddu, S.; Prodosmo, A.; Visca, P.; Conti, S.; Alessandrini, G.; Facciolo, F.; Strigari, L. Prognostic role of serum p53 antibodies in lung cancer. BMC Cancer 2015, 15, 148. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Narendran, A.; Ganjavi, H.; Morson, N.; Connor, A.; Barlow, J.W.; Keystone, E.; Malkin, D.; Freedman, M.H. Mutant p53 in bone marrow stromal cells increases VEGF expression and supports leukemia cell growth. Exp. Hematol. 2003, 31, 693–701. [Google Scholar] [CrossRef]
- Madar, S.; Harel, E.; Goldstein, I.; Stein, Y.; Kogan-Sakin, I.; Kamer, I.; Solomon, H.; Dekel, E.; Tal, P.; Goldfinger, N.; et al. Mutant p53 attenuates the anti-tumorigenic activity of fibroblasts-secreted interferon beta. PLoS ONE 2013, 8, e61353. [Google Scholar] [CrossRef] [PubMed]
- Patocs, A.; Zhang, L.; Xu, Y.; Weber, F.; Caldes, T.; Mutter, G.L.; Platzer, P.; Eng, C. Breast-cancer stromal cells with TP53 mutations and nodal metastases. N. Engl. J. Med. 2007, 357, 2543–2551. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, Y.; Wang, H.; Zhang, Y.; Mei, L.; Fang, X.; Zhang, X.; Zhang, F.; Chen, H.; Liu, Y.; et al. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc. Natl. Acad. Sci. USA 2014, 111, E89–E98. [Google Scholar] [CrossRef] [PubMed]
- Taccioli, C.; Sorrentino, G.; Zannini, A.; Caroli, J.; Beneventano, D.; Anderlucci, L.; Lolli, M.; Bicciato, S.; Del Sal, G. MDP, a database linking drug response data to genomic information, identifies dasatinib and statins as a combinatorial strategy to inhibit YAP/TAZ in cancer cells. Oncotarget 2015, 6, 38854–38865. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Merino Salvador, M.; Gomez de Cedron, M.; Merino Rubio, J.; Falagan Martinez, S.; Sanchez Martinez, R.; Casado, E.; Ramirez de Molina, A.; Sereno, M. Lipid metabolism and lung cancer. Crit. Rev. Oncol. Hematol. 2017, 112, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Beyer, T.A.; Weiss, A.; Khomchuk, Y.; Huang, K.; Ogunjimi, A.A.; Varelas, X.; Wrana, J.L. Switch enhancers interpret TGF-β and Hippo signaling to control cell fate in human embryonic stem cells. Cell Rep. 2013, 5, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Murakami, H.; Fujii, M.; Ishiguro, F.; Tanaka, I.; Kondo, Y.; Akatsuka, S.; Toyokuni, S.; Yokoi, K.; Osada, H.; et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene 2012, 31, 5117–5122. [Google Scholar] [CrossRef] [PubMed]
- Lian, I.; Kim, J.; Okazawa, H.; Zhao, J.; Zhao, B.; Yu, J.; Chinnaiyan, A.; Israel, M.A.; Goldstein, L.S.; Abujarour, R.; et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 2010, 24, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Stanger, B.Z. YAP regulates s-phase entry in endothelial cells. PLoS ONE 2015, 10, e0117522. [Google Scholar] [CrossRef] [PubMed]
- Tumaneng, K.; Schlegelmilch, K.; Russell, R.C.; Yimlamai, D.; Basnet, H.; Mahadevan, N.; Fitamant, J.; Bardeesy, N.; Camargo, F.D.; Guan, K.L. YAP mediates crosstalk between the Hippo and PI(3)K-TOR pathways by suppressing PTEN via miR-29. Nat. Cell Biol. 2012, 14, 1322–1329. [Google Scholar] [CrossRef] [PubMed]
- Bertero, T.; Oldham, W.M.; Cottrill, K.A.; Pisano, S.; Vanderpool, R.R.; Yu, Q.; Zhao, J.; Tai, Y.; Tang, Y.; Zhang, Y.Y.; et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Investig. 2016, 126, 3313–3335. [Google Scholar] [CrossRef] [PubMed]
- Chaulk, S.G.; Lattanzi, V.J.; Hiemer, S.E.; Fahlman, R.P.; Varelas, X. The Hippo pathway effectors TAZ/YAP regulate dicer expression and microRNA biogenesis through Let-7. J. Biol. Chem. 2014, 289, 1886–1891. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Triboulet, R.; Mohseni, M.; Schlegelmilch, K.; Shrestha, K.; Camargo, F.D.; Gregory, R.I. Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell 2014, 156, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Ivanovska, I.; Ball, A.S.; Diaz, R.L.; Magnus, J.F.; Kibukawa, M.; Schelter, J.M.; Kobayashi, S.V.; Lim, L.; Burchard, J.; Jackson, A.L.; et al. Micrornas in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Mol. Cell Biol. 2008, 28, 2167–2174. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Inoue, I. Conditional expression of MCM7 increases tumor growth without altering DNA replication activity. FEBS Lett. 2003, 553, 213–217. [Google Scholar] [CrossRef]
- Ren, B.; Yu, G.; Tseng, G.C.; Cieply, K.; Gavel, T.; Nelson, J.; Michalopoulos, G.; Yu, Y.P.; Luo, J.H. MCM7 amplification and overexpression are associated with prostate cancer progression. Oncogene 2006, 25, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Erkan, E.P.; Strobel, T.; Lewandrowski, G.; Tannous, B.; Madlener, S.; Czech, T.; Saydam, N.; Saydam, O. Depletion of minichromosome maintenance protein 7 inhibits glioblastoma multiforme tumor growth in vivo. Oncogene 2014, 33, 4778–4785. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, S.; Shomori, K.; Nishihara, K.; Yamaga, K.; Nosaka, K.; Araki, K.; Haruki, T.; Taniguchi, Y.; Nakamura, H.; Ito, H. Expression of minichromosome maintenance 7 (MCM7) in small lung adenocarcinomas (pT1): Prognostic implication. Lung Cancer 2009, 65, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Toyokawa, G.; Masuda, K.; Daigo, Y.; Cho, H.S.; Yoshimatsu, M.; Takawa, M.; Hayami, S.; Maejima, K.; Chino, M.; Field, H.I.; et al. Minichromosome maintenance protein 7 is a potential therapeutic target in human cancer and a novel prognostic marker of non-small cell lung cancer. Mol. Cancer 2011, 10, 65. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Z.; Jiang, Y.Y.; Hao, J.J.; Lu, S.S.; Zhang, T.T.; Shang, L.; Cao, J.; Song, X.; Wang, B.S.; Cai, Y.; et al. Prognostic significance of MCM7 expression in the bronchial brushings of patients with non-small cell lung cancer (NSCLC). Lung Cancer 2012, 77, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, L.; Salmena, L.; Riccardi, L.; Fornari, A.; Song, M.S.; Hobbs, R.M.; Sportoletti, P.; Varmeh, S.; Egia, A.; Fedele, G.; et al. Identification of the miR-106b~25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci. Signal. 2010, 3, ra29. [Google Scholar] [CrossRef] [PubMed]
- Vitolo, M.I.; Anglin, I.E.; Mahoney, W.M., Jr.; Renoud, K.J.; Gartenhaus, R.B.; Bachman, K.E.; Passaniti, A. The RUNX2 transcription factor cooperates with the Yes-associated protein, YAP65, to promote cell transformation. Cancer Biol. Ther. 2007, 6, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, T.; Imoto, I.; Matsui, T.; Kozaki, K.; Haruki, S.; Sudol, M.; Shimada, Y.; Tsuda, H.; Kawano, T.; Inazawa, J. YAP is a candidate oncogene for esophageal squamous cell carcinoma. Carcinogenesis 2011, 32, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Yang, J.J.; Zhou, X.; Deng, Z.Y.; Shi, K.H.; Li, J. Emerging role of long noncoding RNAs in lung cancer: Current status and future prospects. Respir. Med. 2016, 110, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Kunz, M.; Wolf, B.; Schulze, H.; Atlan, D.; Walles, T.; Walles, H.; Dandekar, T. Non-coding RNAs in lung cancer: Contribution of bioinformatics analysis to the development of non-invasive diagnostic tools. Genes 2016, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Mayekar, M.K.; Bivona, T.G. Current landscape of targeted therapy in lung cancer. Clin. Pharmacol. Ther. 2017, 102, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Tomasini, P.; Walia, P.; Labbe, C.; Jao, K.; Leighl, N.B. Targeting the KRAS pathway in non-small cell lung cancer. Oncologist 2016, 21, 1450–1460. [Google Scholar] [CrossRef] [PubMed]
- Tiefenbacher, A.; Pirker, R. EGFR tyrosine kinase inhibitors as first-line therapy in advanced EGFR mutation-positive non-small cell lung cancer: Strategies to improve clinical outcome. J. Thorac. Dis. 2017, 9, 4208–4211. [Google Scholar] [CrossRef] [PubMed]
- Heist, R.S.; Engelman, J.A. Snapshot: Non-small cell lung cancer. Cancer Cell 2012, 21, 448.e442. [Google Scholar] [CrossRef] [PubMed]
- Komiya, T.; Perez, R.P.; Erickson, K.D.; Huang, C.H. Systematic analysis of design and stratification for phase III trials in first-line advanced non-small cell lung cancer. Thorac. Cancer 2016, 7, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Maione, P.; Sacco, P.C.; Sgambato, A.; Casaluce, F.; Rossi, A.; Gridelli, C. Overcoming resistance to targeted therapies in NSCLC: Current approaches and clinical application. Ther. Adv. Med. Oncol. 2015, 7, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Shaw, A.T. Resisting resistance: Targeted therapies in lung cancer. Trends Cancer 2016, 2, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.F.; Chen, Y.J.; Chou, C.H.; Wang, F.W.; Kuo, C.D. Norcantharidin induces cell cycle arrest and inhibits progression of human leukemic Jurkat T cells through mitogen-activated protein kinase-mediated regulation of interleukin-2 production. Toxicol. In Vitro 2011, 25, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.Z.; Zhao, Z.M.; Fu, J.Y.; Chen, C.Q.; Sun, W. Norcantharidin inhibits growth of human gallbladder carcinoma xenografted tumors in nude mice by inducing apoptosis and blocking the cell cycle in vivo. Hepatobiliary Pancreat. Dis. Int. 2010, 9, 414–422. [Google Scholar] [PubMed]
- Chen, Y.J.; Chang, W.M.; Liu, Y.W.; Lee, C.Y.; Jang, Y.H.; Kuo, C.D.; Liao, H.F. A small-molecule metastasis inhibitor, norcantharidin, downregulates matrix metalloproteinase-9 expression by inhibiting Sp1 transcriptional activity in colorectal cancer cells. Chem. Biol. Interact. 2009, 181, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Li, Y.; Du, H.; Fang, J.; Song, X.; Zhang, J. The synthetic compound norcantharidin induced apoptosis in mantle cell lymphoma in vivo and in vitro through the PI3K-Akt-NF-κB signaling pathway. Evid. Based Complement. Alternat. Med. 2013, 2013, 461487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, G.; Ma, X.; Wang, Y.; Liu, G.; Feng, L.; Zhao, Y.; Zhang, G.; Wu, Y.; Ye, X.; et al. Norcantharidin enhances ABT-737-induced apoptosis in hepatocellular carcinoma cells by transcriptional repression of Mcl-1. Cell. Signal. 2012, 24, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.H.; Yang, Y.Y.; Huang, Y.F.; Chow, K.C.; Chen, M.F. Induction of apoptosis in human Hep3B hepatoma cells by norcantharidin through a p53 independent pathway via TRAIL/DR5 signal transduction. Chin. J. Integr. Med. 2012, 18, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Tsai, Y.M.; Kuo, C.D.; Ku, K.L.; Shie, H.S.; Liao, H.F. Norcantharidin is a small-molecule synthetic compound with anti-angiogenesis effect. Life Sci. 2009, 85, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, W.; Zhang, W.Z.; Ge, C.Y.; Zhang, J.T.; Liu, Z.Y.; Fan, Y.Z. Inhibition of tumor vasculogenic mimicry and prolongation of host survival in highly aggressive gallbladder cancers by norcantharidin via blocking the ephrin type a receptor 2/focal adhesion kinase/paxillin signaling pathway. PLoS ONE 2014, 9, e96982. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.T.; Fan, Y.Z.; Chen, C.Q.; Zhao, Z.M.; Sun, W. Norcantharidin: A potential antiangiogenic agent for gallbladder cancers in vitro and in vivo. Int. J. Oncol. 2012, 40, 1501–1514. [Google Scholar] [PubMed]
- Zhao, Z.; Zheng, N.; Wang, L.; Hou, Y.; Zhou, X.; Wang, Z. Rottlerin exhibits antitumor activity via down-regulation of TAZ in non-small cell lung cancer. Oncotarget 2017, 8, 7827–7838. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Nagalingam, A.; Muniraj, N.; Bonner, M.Y.; Mistriotis, P.; Afthinos, A.; Kuppusamy, P.; Lanoue, D.; Cho, S.; Korangath, P.; et al. Activation of tumor suppressor LKB1 by honokiol abrogates cancer stem-like phenotype in breast cancer via inhibition of oncogenic Stat3. Oncogene 2017, 36, 5709–5721. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.Q.; Qiao, X.R.; Su, L.; Chen, S.Z. Honokiol inhibits EMT-mediated motility and migration of human non-small cell lung cancer cells in vitro by targeting c-FLIP. Acta Pharmacol. Sin. 2016, 37, 1574–1586. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.; Katiyar, S.K. Honokiol inhibits non-small cell lung cancer cell migration by targeting PGE(2)-mediated activation of β-catenin signaling. PLoS ONE 2013, 8, e60749. [Google Scholar]
- Pan, J.; Lee, Y.; Zhang, Q.; Xiong, D.; Wan, T.C.; Wang, Y.; You, M. Honokiol decreases lung cancer metastasis through inhibition of the Stat3 signaling pathway. Cancer Prev. Res. 2017, 10, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Gugger, M.; White, R.; Song, S.; Waser, B.; Cescato, R.; Riviere, P.; Reubi, J.C. GPR87 is an overexpressed G-protein coupled receptor in squamous cell carcinoma of the lung. Dis. Markers 2008, 24, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R., Jr. Estrogen-induced activation of ERK-1 and ERK-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.C.; You, B.; Yang, Y.L.; Zhang, W.Q.; Wang, Y.C.; Xu, Z.; Dai, Y.; Liu, S.; Yang, C.T.; Li, H.; et al. YAP promotes erlotinib resistance in human non-small cell lung cancer cells. Oncotarget 2016, 7, 51922–51933. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. YAP1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol. Cell 2008, 29, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Dobbelstein, M.; Strano, S.; Roth, J.; Blandino, G. P73-induced apoptosis: A question of compartments and cooperation. Biochem. Biophys. Res. Commun. 2005, 331, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.G.; Hwang, K.L.; Brown, K.K.; Evason, K.; Beltz, S.; Tsomides, A.; O’Connor, K.; Galli, G.G.; Yimlamai, D.; Chhangawala, S.; et al. YAP reprograms glutamine metabolism to increase nucleotide biosynthesis and enable liver growth. Nat. Cell Biol. 2016, 18, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Willers, H.; Azzoli, C.G.; Santivasi, W.L.; Xia, F. Basic mechanisms of therapeutic resistance to radiation and chemotherapy in lung cancer. Cancer J. 2013, 19, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Rosano, L.; Bagnato, A. Disrupting the endothelin and WNT relationship to overcome chemoresistance. Mol. Cell Oncol. 2015, 2, e995025. [Google Scholar] [CrossRef] [PubMed]
- Cianfrocca, R.; Rosano, L.; Tocci, P.; Sestito, R.; Caprara, V.; Di Castro, V.; De Maria, R.; Bagnato, A. Blocking endothelin-1-receptor/β-catenin circuit sensitizes to chemotherapy in colorectal cancer. Cell Death Differ. 2017, 24, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Dubrovska, A.; Linge, A.; Baumann, M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv. Drug Deliv. Rev. 2017, 109, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the WNT response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pan, P.; Wang, Z.; Zhang, Y.; Xie, P.; Geng, D.; Jiang, Y.; Yu, R.; Zhou, X. β-catenin-mediated YAP signaling promotes human glioma growth. J. Exp. Clin. Cancer Res. 2017, 36, 136. [Google Scholar] [CrossRef] [PubMed]
- Deng, F.; Peng, L.; Li, Z.; Tan, G.; Liang, E.; Chen, S.; Zhao, X.; Zhi, F. YAP triggers the WNT/β-catenin signalling pathway and promotes enterocyte self-renewal, regeneration and tumorigenesis after DSS-induced injury. Cell Death Dis. 2018, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Ghiso, E.; Migliore, C.; Ciciriello, V.; Morando, E.; Petrelli, A.; Corso, S.; De Luca, E.; Gatti, G.; Volante, M.; Giordano, S. YAP-dependent AXL overexpression mediates resistance to EGFR inhibitors in NSCLC. Neoplasia 2017, 19, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Tricker, E.M.; Xu, C.; Uddin, S.; Capelletti, M.; Ercan, D.; Ogino, A.; Pratilas, C.A.; Rosen, N.; Gray, N.S.; Wong, K.K.; et al. Combined EGFR/MEK inhibition prevents the emergence of resistance in EGFR-mutant lung cancer. Cancer Discov. 2015, 5, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. Met amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Zhang, Z.; Miao, L.; Yang, Z.; Yang, J.; Wang, Y.; Qian, D.; Cai, H.; Wang, Y. Anexelekto (AXL) increases resistance to EGFR-TKI and activation of AKT and ERK1/2 in non-small cell lung cancer cells. Oncol. Res. 2016, 24, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Wakelee, H.A.; Aragaki, A.K.; Tang, J.Y.; Kurian, A.W.; Manson, J.E.; Stefanick, M.L. Protective effects of statins in cancer: Should they be prescribed for high-risk patients? Curr. Atheroscler. Rep. 2016, 18, 72. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fu, J.; Yuan, X.; Hu, C. Simvastatin inhibits the proliferation of A549 lung cancer cells through oxidative stress and up-regulation of SOD2. Pharmazie 2014, 69, 610–614. [Google Scholar] [PubMed]
- Yu, X.; Pan, Y.; Ma, H.; Li, W. Simvastatin inhibits proliferation and induces apoptosis in human lung cancer cells. Oncol. Res. 2013, 20, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, Z.; Li, Y.; Li, W.; Chen, Y. Simvastatin prevents proliferation and bone metastases of lung adenocarcinoma in vitro and in vivo. Neoplasma 2013, 60, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Cardwell, C.R.; Mc Menamin, U.; Hughes, C.M.; Murray, L.J. Statin use and survival from lung cancer: A population-based cohort study. Cancer Epidemiol. Biomarkers Prev. 2015, 24, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Ezer, N.; Sigel, K.; Mhango, G.; Wisnivesky, J.P. The effect of statins on survival in patients with stage IV lung cancer. Lung Cancer 2016, 99, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.S.; Chen, I.C.; Lee, C.P.; Huang, R.J.; Chen, P.C.; Tsai, Y.H.; Yang, Y.H. Statin improves survival in patients with EGFR-TKI lung cancer: A nationwide population-based study. PLoS ONE 2017, 12, e0171137. [Google Scholar] [CrossRef] [PubMed]
- Fiala, O.; Pesek, M.; Finek, J.; Minarik, M.; Benesova, L.; Bortlicek, Z.; Topolcan, O. Statins augment efficacy of EGFR-TKIs in patients with advanced-stage non-small cell lung cancer harbouring KRAS mutation. Tumour Biol. 2015, 36, 5801–5805. [Google Scholar] [CrossRef] [PubMed]
- Mantha, A.J.; McFee, K.E.; Niknejad, N.; Goss, G.; Lorimer, I.A.; Dimitroulakos, J. Epidermal growth factor receptor-targeted therapy potentiates lovastatin-induced apoptosis in head and neck squamous cell carcinoma cells. J. Cancer Res. Clin. Oncol. 2003, 129, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Hwang, K.E.; Kwon, S.J.; Kim, Y.S.; Park, D.S.; Kim, B.R.; Yoon, K.H.; Jeong, E.T.; Kim, H.R. Effect of simvastatin on the resistance to EGFR tyrosine kinase inhibitors in a non-small cell lung cancer with the T790M mutation of EGFR. Exp. Cell Res. 2014, 323, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Giroux Leprieur, E.; Dumenil, C.; Julie, C.; Giraud, V.; Dumoulin, J.; Labrune, S.; Chinet, T. Immunotherapy revolutionises non-small-cell lung cancer therapy: Results, perspectives and new challenges. Eur. J. Cancer 2017, 78, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-dependent MDSC infiltration impairs tumor progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef] [PubMed]
- Solito, S.; Marigo, I.; Pinton, L.; Damuzzo, V.; Mandruzzato, S.; Bronte, V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann. N. Y. Acad. Sci. 2014, 1319, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, M.L.; Lu, L.; Ramachandran, I.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the development of lung cancer. Cancer Immunol. Res. 2014, 2, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Huang, H.; Zhu, Y.; Yu, G.; Gao, X.; Xu, Y.; Liu, C.; Hou, J.; Zhang, X. A novel subset of B7-H3+CD14+HLA-DR−/low myeloid-derived suppressor cells are associated with progression of human NSCLC. Oncoimmunology 2015, 4, e977164. [Google Scholar] [CrossRef] [PubMed]
- Adah, D.; Hussain, M.; Qin, L.; Qin, L.; Zhang, J.; Chen, X. Implications of MDSCs-targeting in lung cancer chemo-immunotherapeutics. Pharmacol. Res. 2016, 110, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Hsu, P.C.; Yang, Y.L.; Xu, Z.; Dai, Y.; Wang, Y.; Chan, G.; Huang, Z.; Hu, B.; Li, H.; et al. YAP regulates PD-L1 expression in human NSCLC cells. Oncotarget 2017, 8, 114576–114587. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Park, D.I.; Lee, D.H.; Lee, J.E.; Yeo, M.K.; Park, Y.H.; Lim, D.S.; Choi, W.; Lee, D.H.; Yoo, G.; et al. Hippo effector YAP directly regulates the expression of PD-L1 transcripts in EGFR-TKI-resistant lung adenocarcinoma. Biochem. Biophys. Res. Commun. 2017, 491, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv324. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Fang, W.; Yu, J.; Chen, N.; Zhan, J.; Ma, Y.; Yang, Y.; Huang, Y.; Zhao, H.; Zhang, L. Expression of programmed death ligand-1 on tumor cells varies pre and post chemotherapy in non-small cell lung cancer. Sci. Rep. 2016, 6, 20090. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, G.; Wang, Y.; Wang, Y.; Zhao, S.; Haihong, P.; Zhao, H.; Wang, Y. Pd-L1 expression in lung cancer and its correlation with driver mutations: A meta-analysis. Sci. Rep. 2017, 7, 10255. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hippo Pathway Component/ Regulator | Increase/Decrease/Prognosis | Reference | N |
|---|---|---|---|
| YAP | Protein level increases in 70% NSCLC tissues. Bad prognosis. Correlation with late T stage, TNM stage and lymph node metastasis. | Su, L.L., et al.,2012 [72] | 40 |
| Protein level increased in 66.3% NSCLC tissues and predominantly nuclear. Bad prognosis. Correlation with TNM stage, lymph node metastasis, shorter O.S. | Wang, Y., et al.,2010 [73] | 92 | |
| YAP gene amplified in NSCLC cell lines and in 23% NSCLC tissues. | Lorenzetto E., et al, 2014 [74] | 77 | |
| Protein Level and Nuclear localization Increased in LAC. Bad prognosis. correlation with TNM stage, cyclinA overexpression, increased EGFR copy number. | Kim, J.M., et al., 2011 [75] | 168 | |
| Increased Nuclear localization. Increased nuclear and reduced cytoplasmic expression in NSCLC cell lines and tissues. | Guo, J., et al., 2017 [76] | 4 | |
| Protein level increased in 87.8% LAC. Bad prognosis. | Cui, Z.L., et al., 2012 [77] | 49 | |
| YAP1 R331W Missense germline mutation in 1.1% patients with LAC with respect to 0.18% in healthy control. Predictive of LAC predisposition. | Chen, H.Y., et al.,2015 [81] | 1312 LAC 1135 healthy controls | |
| Bad prognosis. Shorter survival when overexpressed with TAZ, SCD1 and β-catenin. | Noto, A., et al., 2017 [64] | 10 | |
| TAZ | Bad prognosis. Shorter survival when overexpressed with YAP, SCD1 and β-catenin. | Noto, A., et al., 2017 [64] | 10 |
| Overexpressed in NSCLC cells. | Zhou, Z., et al., 2011 [78] | cells | |
| Overexpressed in 66.8% NSCLC. Bad prognosis. Correlated with TNM stage and lymph node metastasis, invasion, shorter O.S and D.F.S. | Xie, M., et al., 2012 [79] | 181 | |
| Bad prognosis. Shorter survival. High TAZ expression at the genomic, mRNA, and protein levels in Squamous cell Carcinoma patients with respect to controls. | Noguchi, S., et al., 2014 [80] | 345 NSCLC patients 18 healthy controls | |
| LATS2 | Transcript Downregulated in NSCLC. | Strazisar, M., et al., 2009 [103] | 129 |
| Promoter hypermethylated in 71% NSCLC. | Malik, S.A., et al., 2017 [105] | 79 | |
| Lower protein and trasnscript expression in LAC. Good prognosis. Increased O.S and D.F.S. | Luo, S., et al., 2014 [108] | 79 | |
| LATS1 | Promoter hypermethylated in 66.66% NSCLC. | Malik, S.A., et al., 2017 [105] | 79 |
| Good prognosis. correlated with TNM stage, lymph node metastasis. | Lin, X.Y., et al., 2014 [109] | 136 | |
| AMOT | Downregulated in 45.8% NSCLC patients | Hsu, Y.L., et al., 2014 [111] | 24 |
| ABL1, ABL2 | ABL1 and 2 are somatically mutated in 1.5% and 4% NSCLC patients, ABL2 is amplified in 8% NSCLC. | Testoni, E., et al., 2016 [88] | N.A |
| LKB1 | Gene mutated in 18% LAC patients. | Ding, L., et al., 2008 [124] | 188 |
| Gene inactivated in 54% of LAC cell lines. | Carretero, J., et al., 2004 [125] | 11 | |
| Gene inactivated in 33% NSCLC patients. | Sanchez-Cespedes, M., et al., 2002 [127] | 20 | |
| Gene mutated in 8% NSCLC patients (7/91) and in 39% (20/51) NSCLC cell lines. Good prognosis. | Matsumoto, S., et al., 2007 [128] | 91 patients 51 cell lines | |
| RASSF1A | Promoter hypermethylated in 40% of primary lung tumors, missense mutation in 10% of primary lung tumors. | Dammann, R., et al., 2000 [117] | 60 |
| Promoter hypermethylated in 72% of SCLC, 34% of NSCLC. Loss of heterozigosity (LOH) in 86% SCLC (31/36), 91% (10/11) Squamous cell carcinoma, 71% (15/21) LAC. | Agathanggelou, A., et al., 2001 [118] | 29 SCLC, 41 NSCLC (hypermet study) 36 SCLC, 48 NSCLC (LOH study) | |
| Transcript Not expressed in 100% SCLC, 64% NSCLC. Promoter hypermethylated in 100% SCLC. Good prognosis. Hypermethylation was associated with impaired patient survival. | Burbee, D.G., et al., 2001 [119] | 47 SCLC, 107 NSCLC | |
| Promoter hypermethylated in 21.8% NSCLC patients. Good prognosis in patients expressing higher levels of RASSF1A. | de Fraipont, F., et al., 2012 [120] | 202 | |
| VGLL4 | Transcript downregulated in LAC tumor respect to non tumoral tissues (29/30) Lower levels of nuclear protein in LAC compared to normal lungs: 92.6% of patients (25/27) exhibited high nuclear VGLL4 expression in their normal lungs, whereas only 22.1% of patients (17 out of 77) had high nuclear expression of VGLL4 in their lung ADCs | Zhang, W., et al., 2014 [113] | 30 |
| Ski | Promoter hypermethylated in 56% of NSCLC tumor samples and in NSCLC cell lines Good prognosis. | Xie, M., et al., 2017 [130] | 168 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lo Sardo, F.; Strano, S.; Blandino, G. YAP and TAZ in Lung Cancer: Oncogenic Role and Clinical Targeting. Cancers 2018, 10, 137. https://doi.org/10.3390/cancers10050137
Lo Sardo F, Strano S, Blandino G. YAP and TAZ in Lung Cancer: Oncogenic Role and Clinical Targeting. Cancers. 2018; 10(5):137. https://doi.org/10.3390/cancers10050137
Chicago/Turabian StyleLo Sardo, Federica, Sabrina Strano, and Giovanni Blandino. 2018. "YAP and TAZ in Lung Cancer: Oncogenic Role and Clinical Targeting" Cancers 10, no. 5: 137. https://doi.org/10.3390/cancers10050137
APA StyleLo Sardo, F., Strano, S., & Blandino, G. (2018). YAP and TAZ in Lung Cancer: Oncogenic Role and Clinical Targeting. Cancers, 10(5), 137. https://doi.org/10.3390/cancers10050137