Toward the Discovery of a Novel Class of YAP–TEAD Interaction Inhibitors by Virtual Screening Approach Targeting YAP–TEAD Protein–Protein Interface


 , , and
, , and
Abstract
:1. Introduction
2. Results
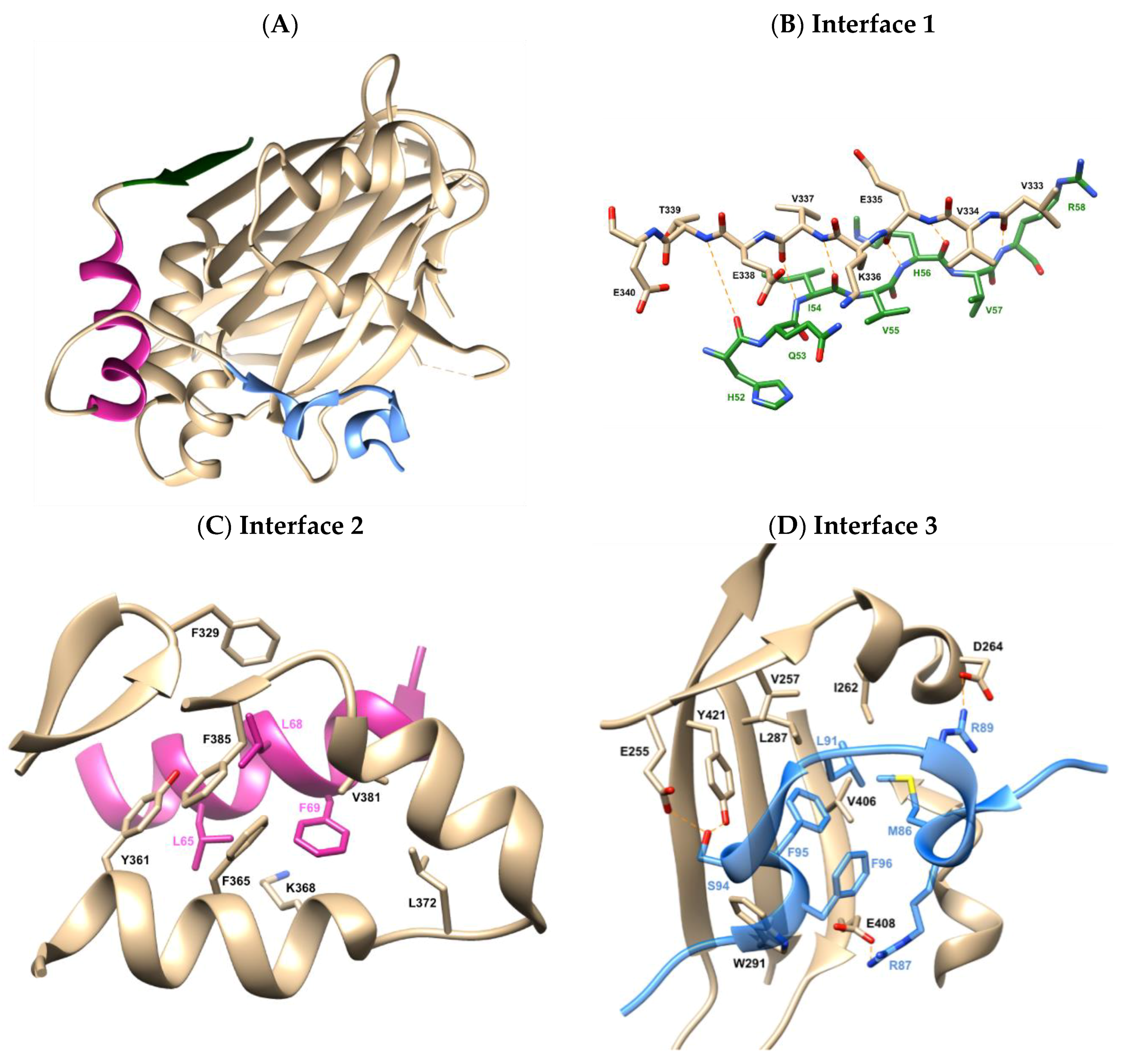
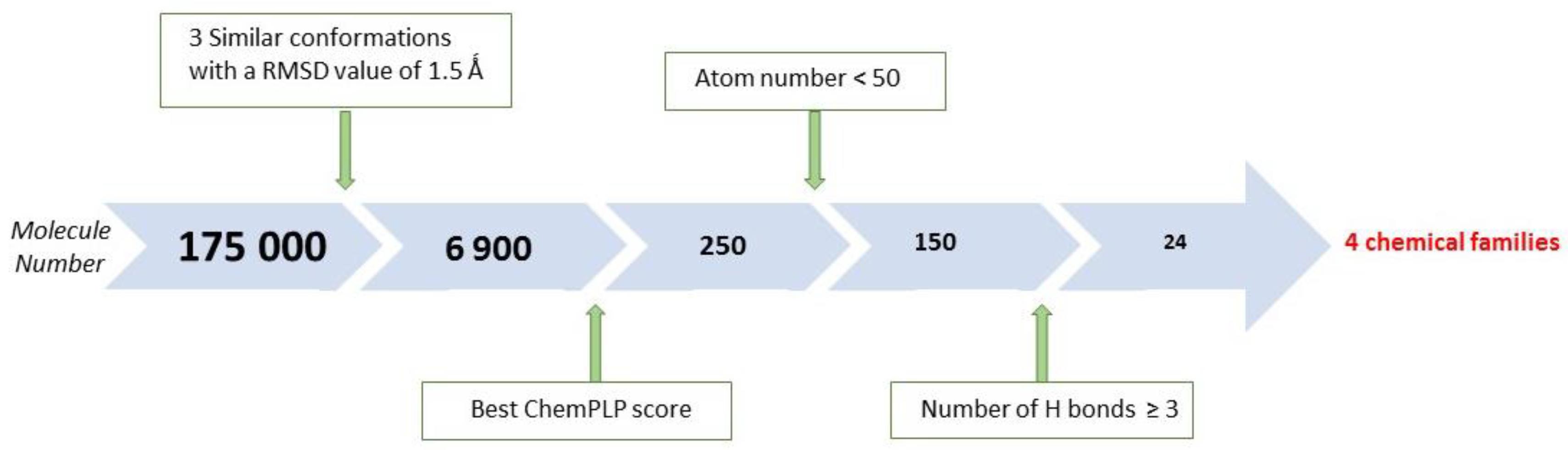
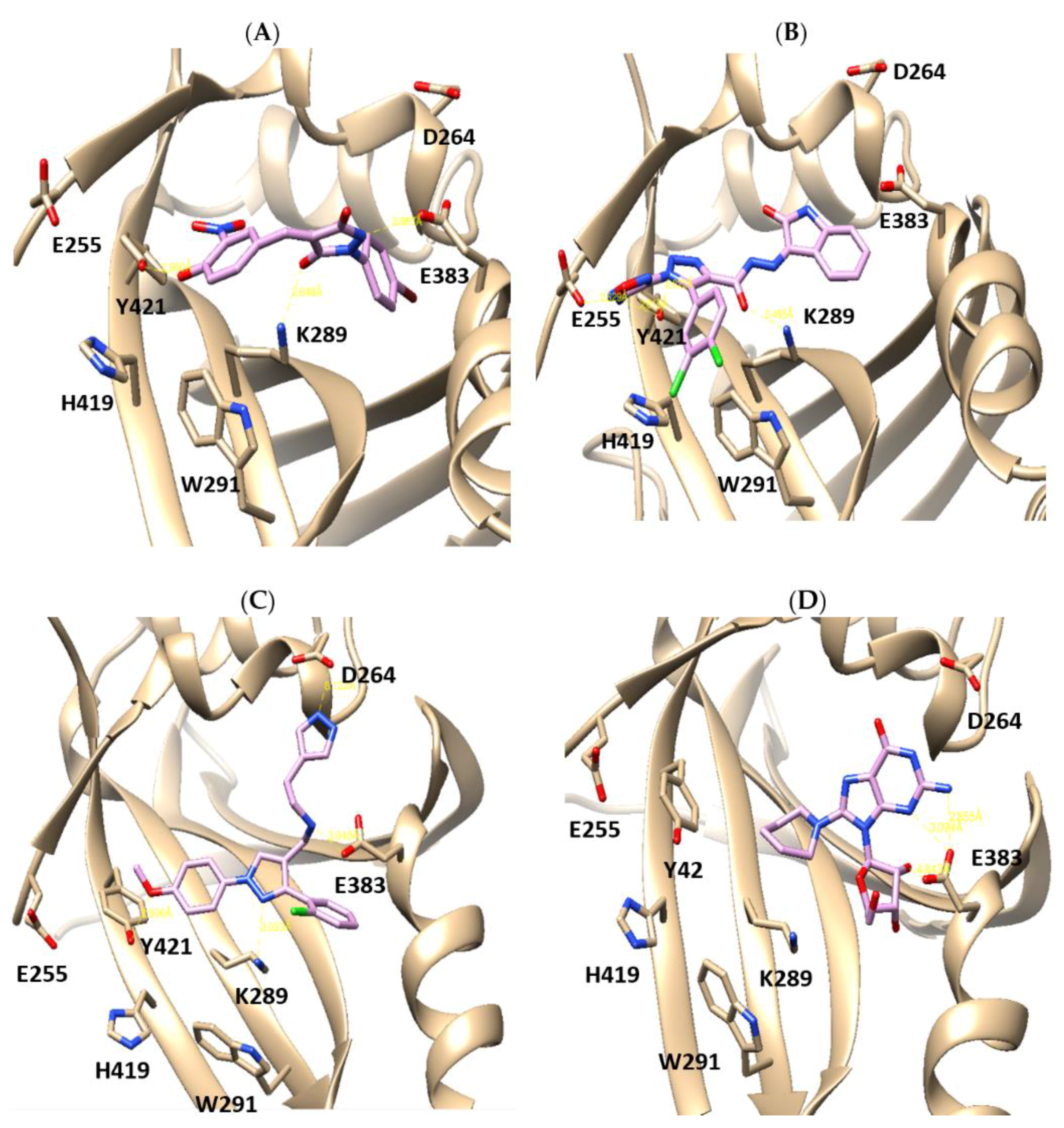
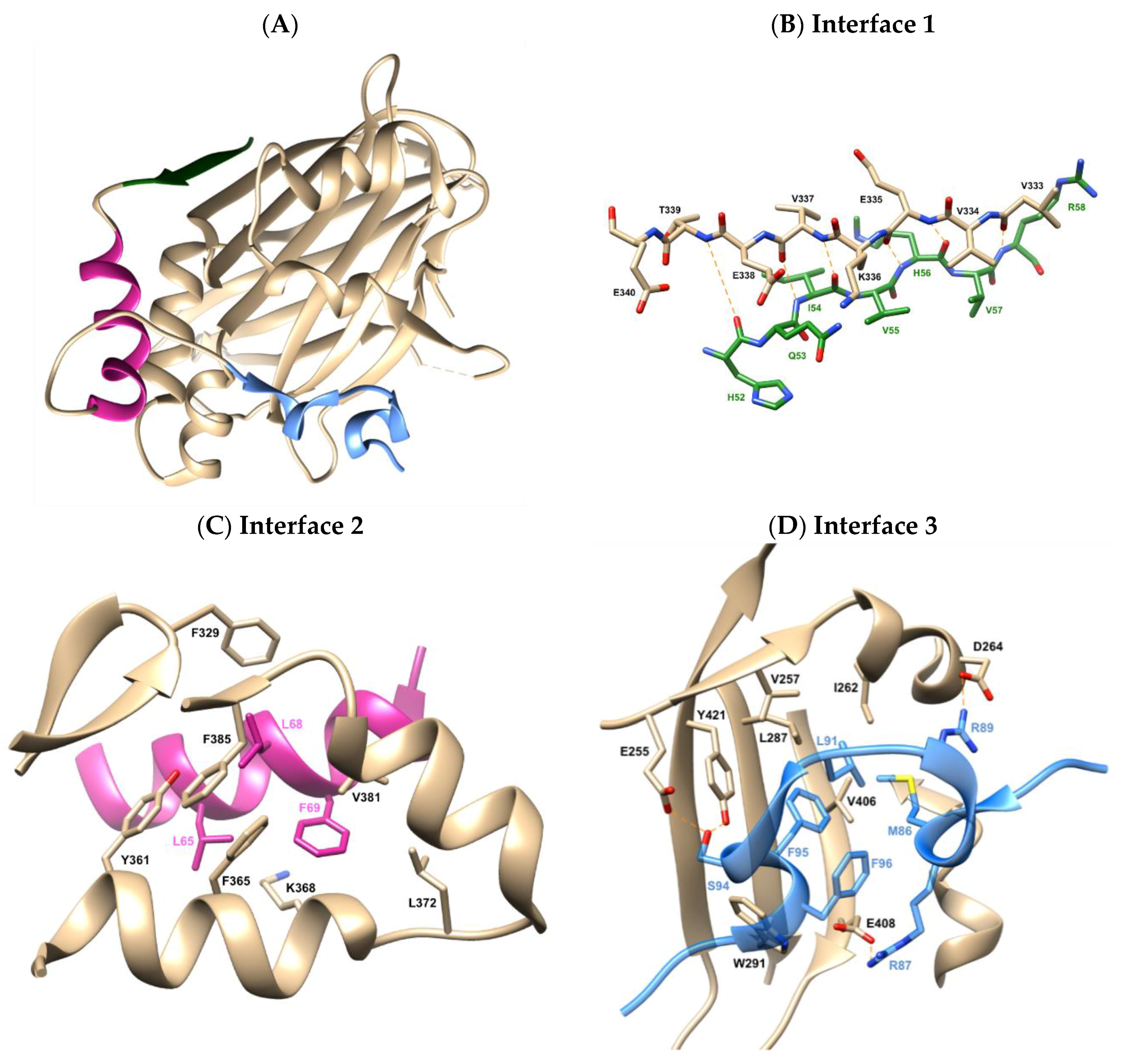
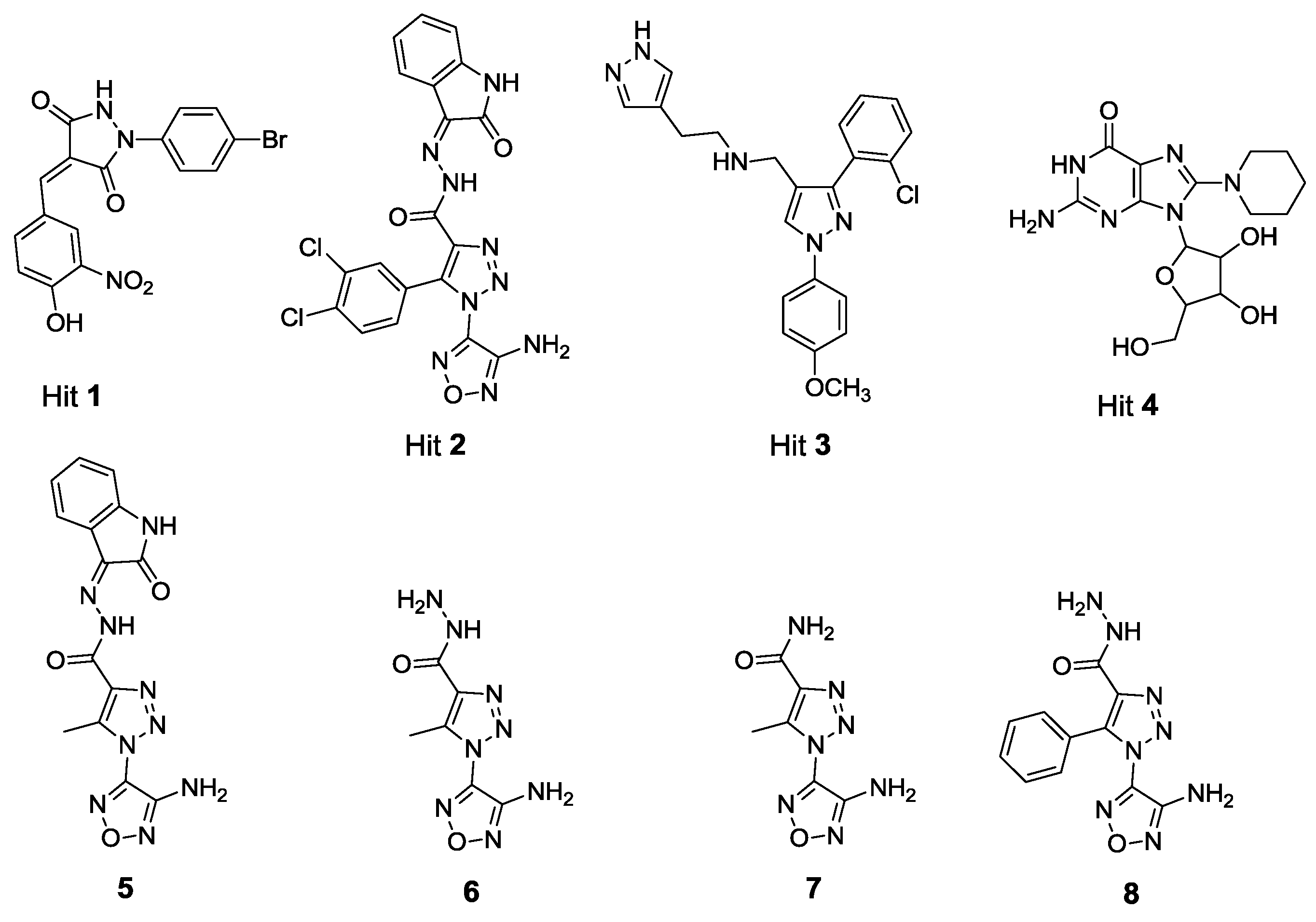
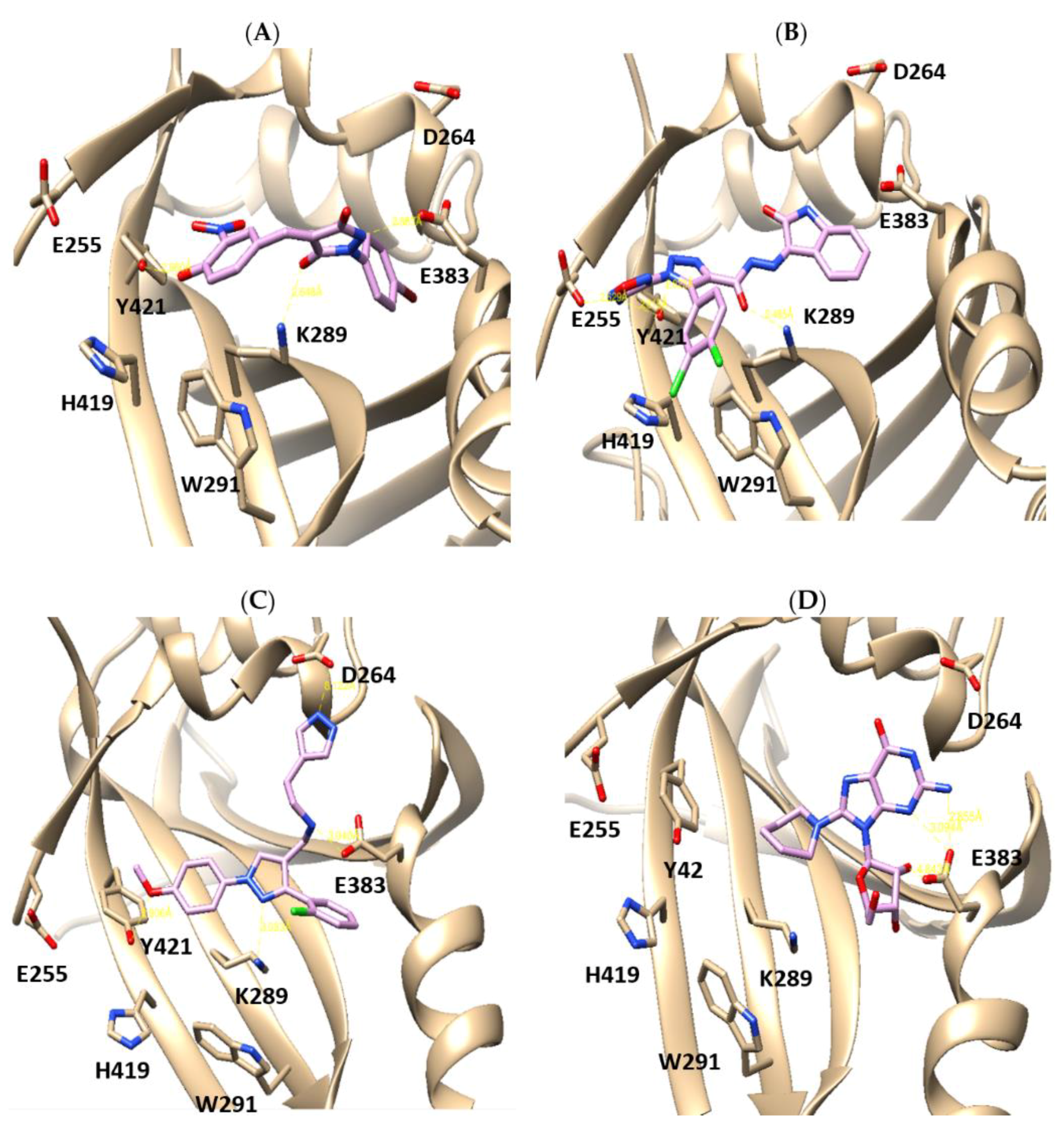
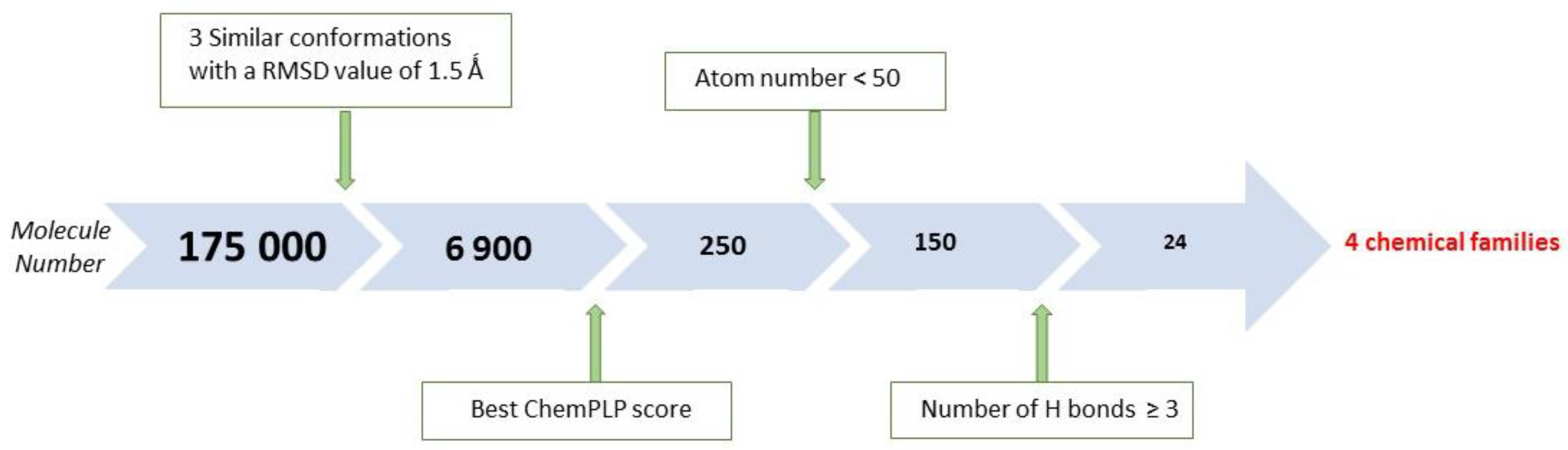
2.1. X-Ray Crystallographic Structure Analysis and Virtual Screening
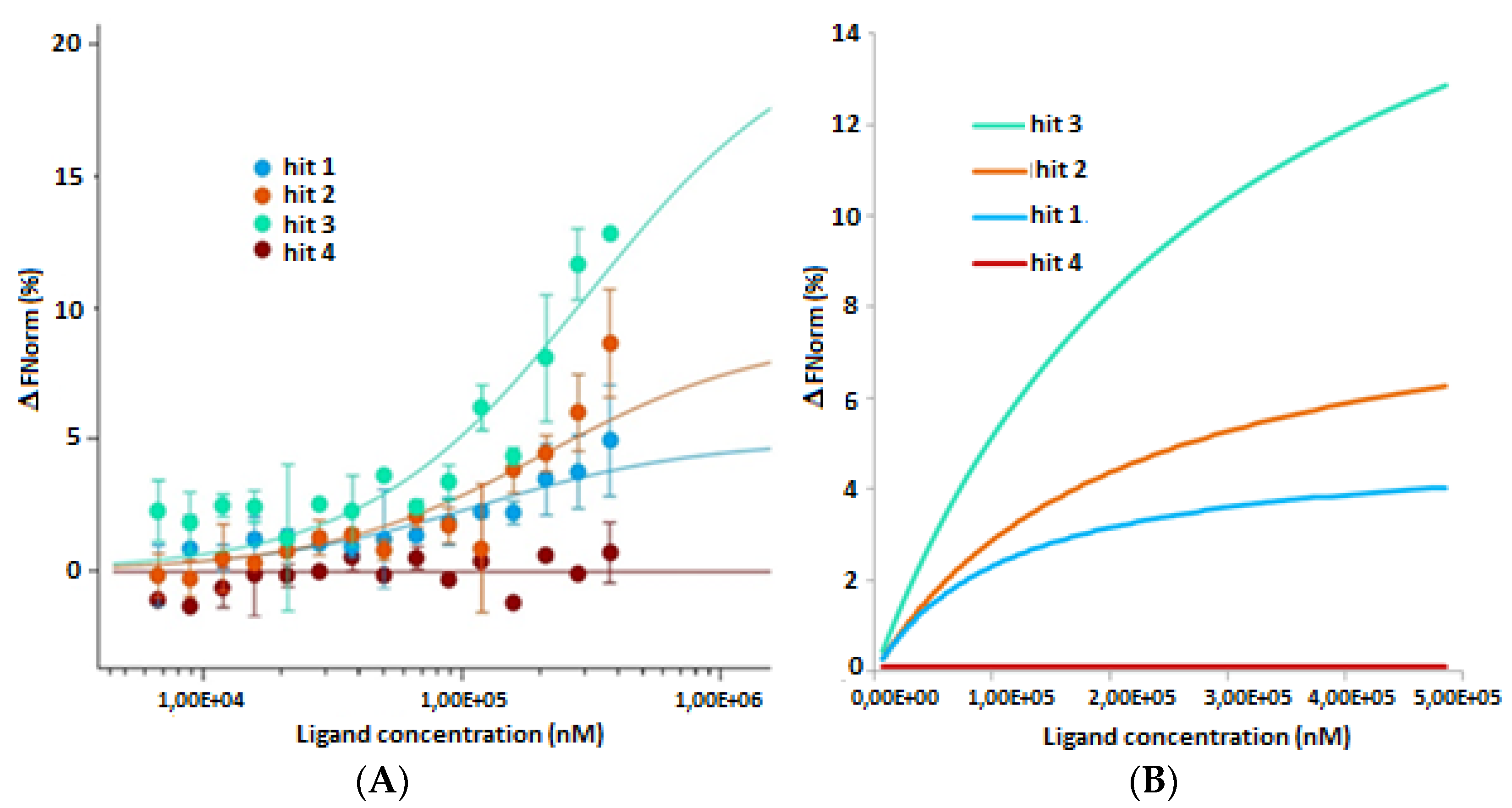
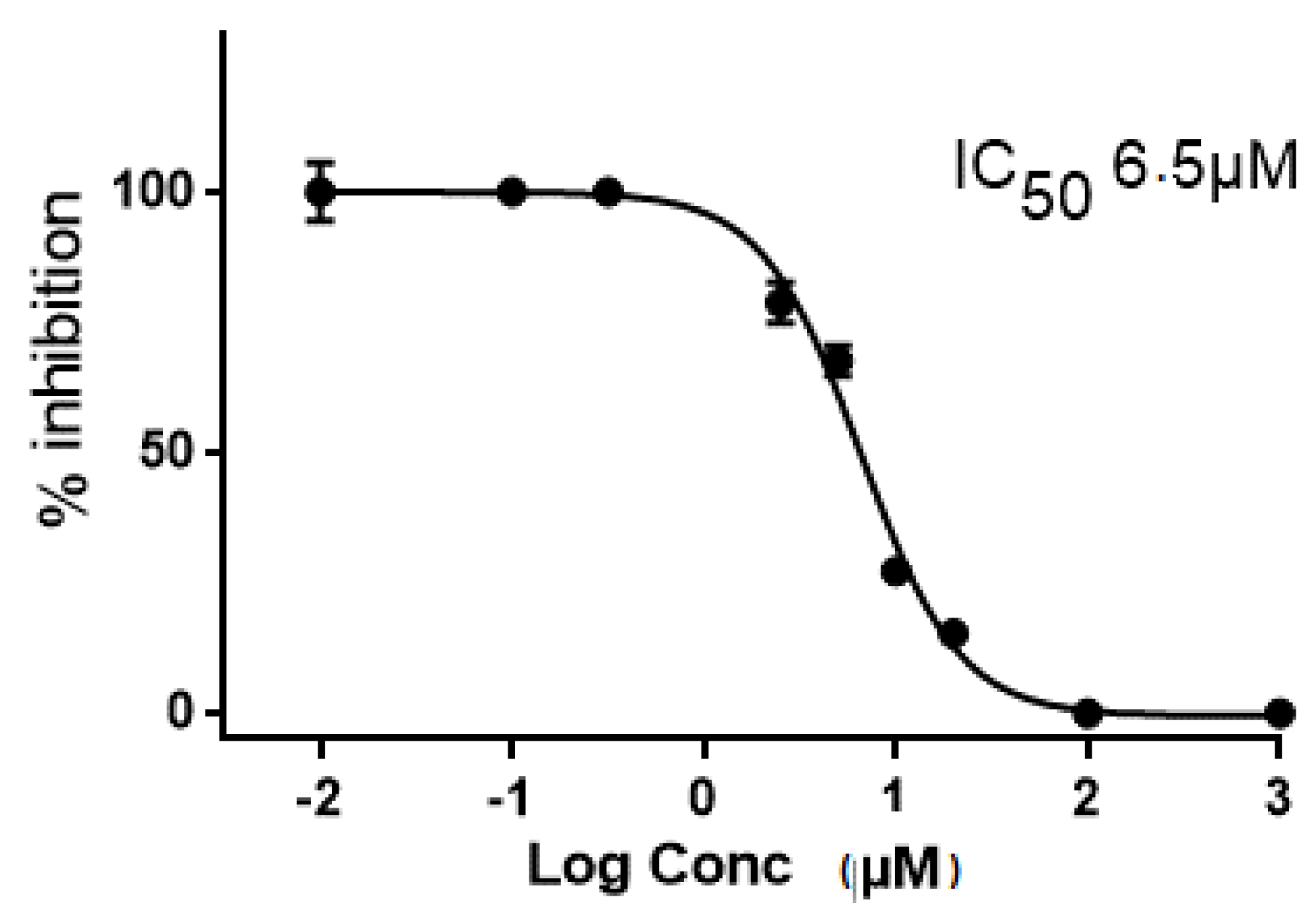
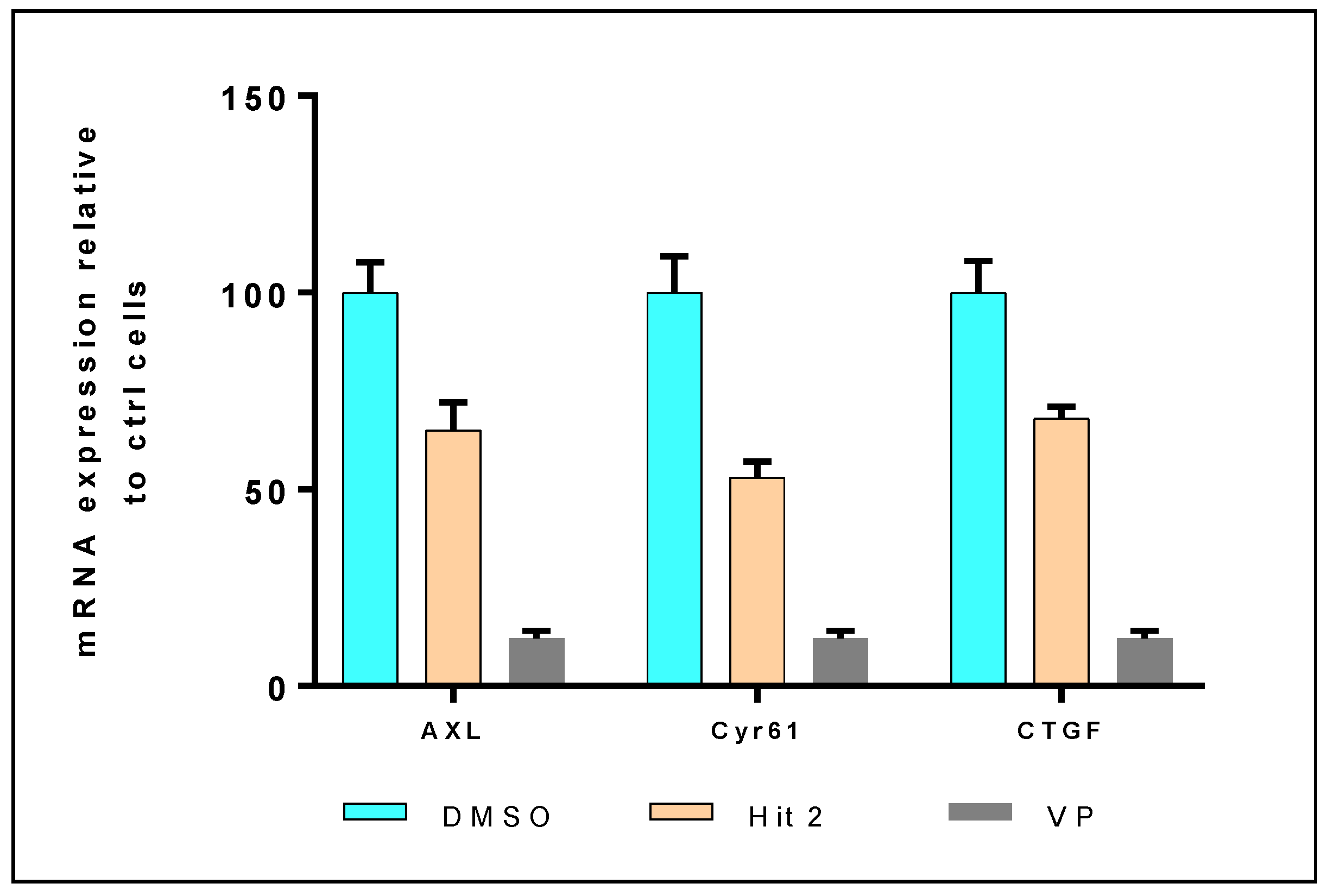
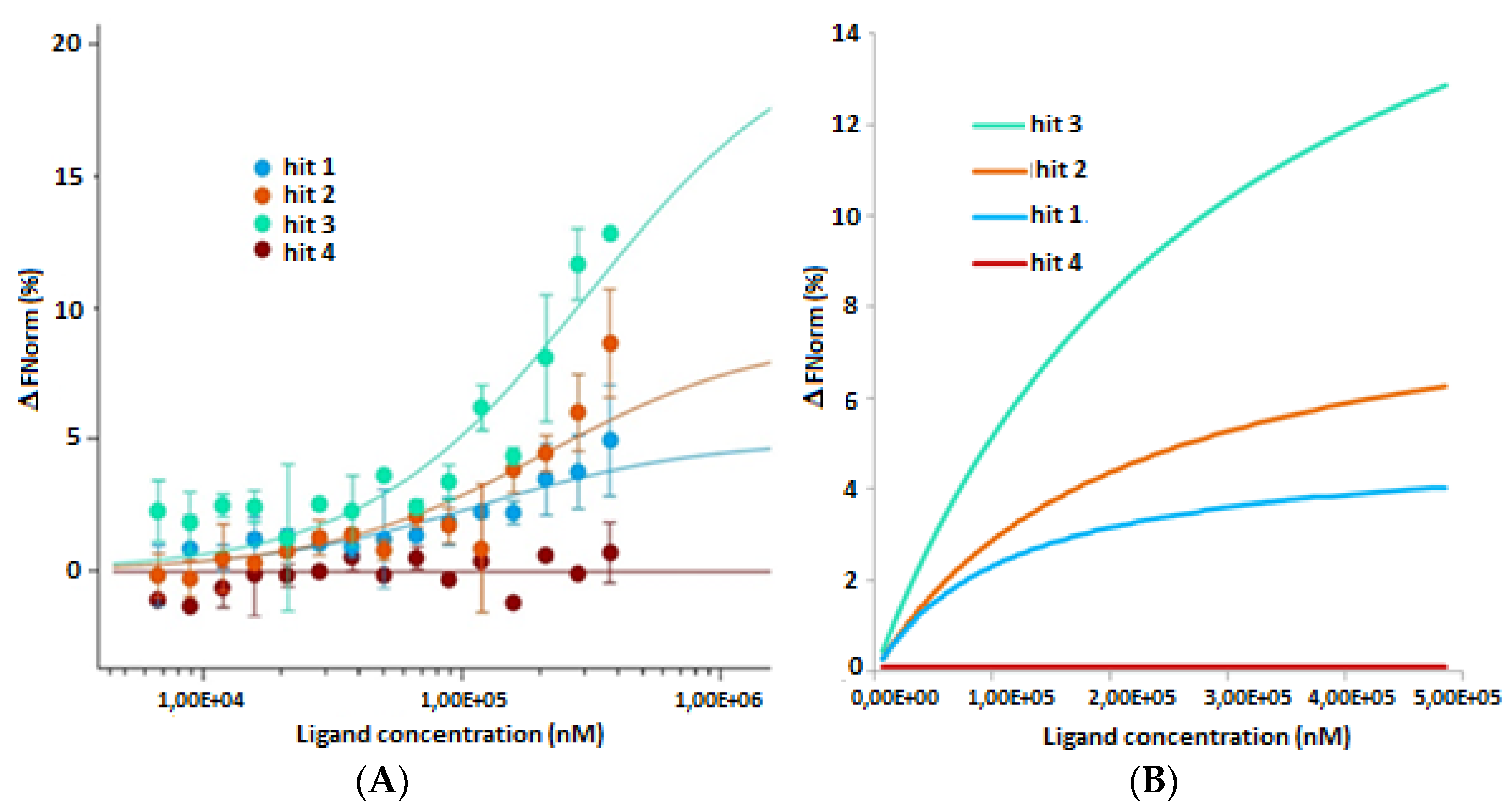
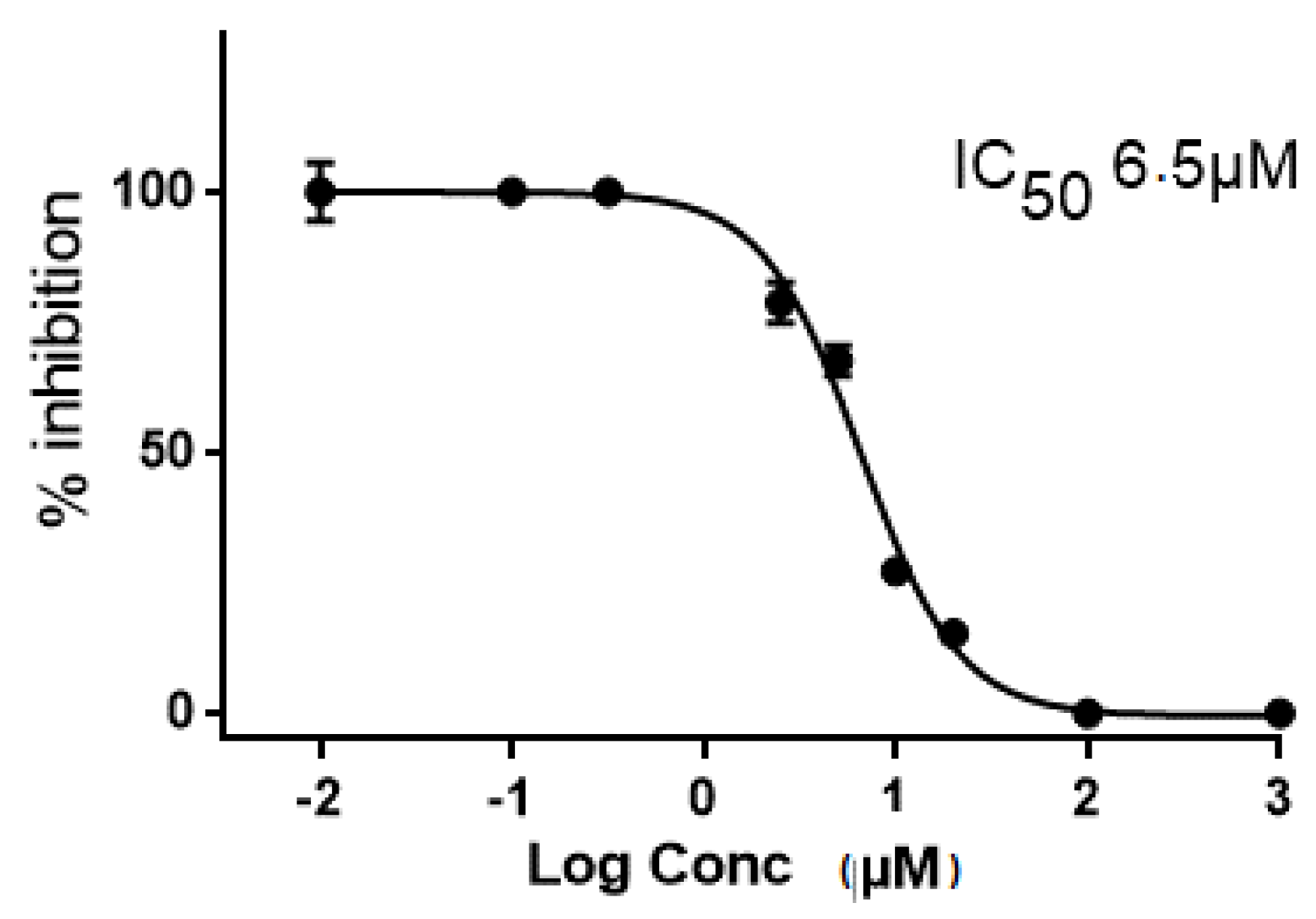
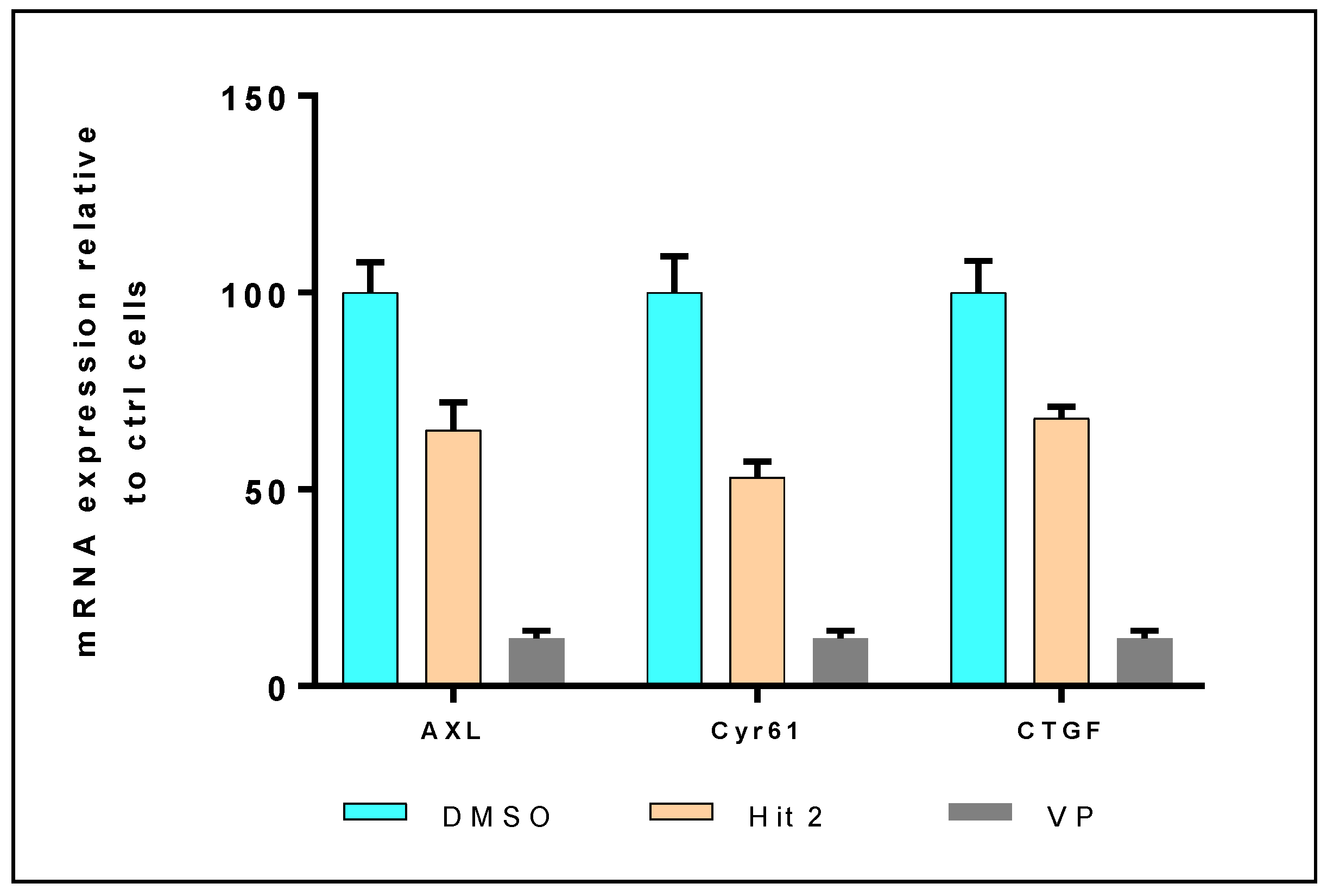
2.2. In Vitro and in Cellulo Binding Assays
3. Materials and Methods
3.1. Molecular Modelling
3.2. Chemicals
3.3. Binding Tests
3.3.1. Thermal Shift Assay
3.3.2. Microscale Thermophoresis
3.4. Biology
3.4.1. Cell Cultures
3.4.2. Luciferase Reporter Assay
3.4.3. mRNA Expression
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Johnson, R.; Halder, G. The two faces of Hippo: Targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Tumaneg, K.; Russel, R.C.; Guan, K.-L. Organ size control by Hippo and TOR pathways. Curr. Biol. 2012, 22, R368–R379. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Guan, K.-L. The Hippo pathway: Regulators and regulations. Genes Dev. 2013, 27, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Guan, K.-L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Guan, K.-L. The YAP and TAZ transcription co-activators: Key downstream effectors of the mammalian Hippo pathway. Semin. Cell Dev. Biol. 2012, 23, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Ota, M.; Sasaki, H. Mammalian TEAD proteins regulate cell proliferation and contact inhibition as transcriptional mediators of Hippo signaling. Development 2008, 135, 4059–4069. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.-Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.-L. The Hippo-YAP pathway in organ size control and tumorigenesis: An updated version. Genes Dev. 2010, 24, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Santucci, M.; Vignudelli, T.; Ferrari, S.; Mor, M.; Scalvini, L.; Bolognesi, M.L.; Uliassi, E.; Costi, M.P. The Hippo pathway and YAP/TAZ–TEAD protein–protein interaction as targets for regenerative medicine and cancer treatment: Miniperspective. J. Med. Chem. 2015, 58, 4857–4873. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Z.; Chan, S.W.; Liu, A.M.; Wong, K.F.; Fan, S.T.; Chen, J.; Poon, R.T.; Zender, L.; Lowe, S.W.; Hong, W.; et al. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene 2011, 30, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Pobbati, A.V.; Hong, W. Emerging roles of TEAD transcription factors and its coactivators in cancers. Cancer Biol. Ther. 2013, 14, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Moroishi, T.; Hansen, C.G.; Guan, K.-L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the roots of cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD–YAP complex suppresses the oncogenic Activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Gibault, F.; Bailly, F.; Corvaisier, M.; Coevoet, M.; Huet, G.; Melnyk, P.; Cotelle, P. Molecular features of the YAP inhibitor verteporfin: Synthesis of hexasubstituted dipyrrins as potential inhibitors of YAP/TAZ, the downstream effectors of the Hippo pathway. Chem. Med. Chem. 2017, 12, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Basu, D.; Lettan, R.; Damodaran, K.; Strellec, S.; Reyes-Mugica, M.; Rebbaa, A. Identification, mechanism of action, and antitumor activity of a small molecule inhibitor of Hippo, TGF-β, and Wnt signaling pathways. Mol. Cancer Ther. 2014, 13, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.-X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.-X.; Zhang, Y.; Park, H.W.; Jewell, J.L.; Chen, Q.; Deng, Y.; Pan, D.; Taylor, S.S.; Lai, Z.-C.; Guan, K.-L. Protein kinase A activates the Hippo pathway to modulate cell proliferation and differentiation. Genes Dev. 2013, 27, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Oku, Y.; Nishiya, N.; Shito, T.; Yamamoto, R.; Yamamoto, Y.; Oyama, C.; Uehara, Y. Small molecules inhibiting the nuclear localization of YAP/TAZ for chemotherapeutics and chemosensitizers against breast cancers. FEBS Open Bio. 2015, 5, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Gibault, F.; Sturbaut, M.; Bailly, F.; Melnyk, P.; Cotelle, P. Targeting transcriptional enhanced associate domains (TEADs). J. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hu, T.; Xu, Z.; Lin, Z.; Zhang, Z.; Feng, T.; Zhu, L.; Rong, Y.; Shen, H.; Luk, J.M.; et al. Targeting Hippo pathway by specific interruption of YAP-TEAD interaction using cyclic YAP-like peptides. FASEB J. 2015, 29, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Wang, H.; Shi, Z.; Dong, A.; Zhang, W.; Song, X.; He, F.; Wang, Y.; Zhang, Z.; Wang, W.; et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 2014, 25, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Crook, Z.R.; Sevilla, G.P.; Friend, D.; Brusniak, M.Y.; Bandaranayake, A.D.; Clarke, M.; Gewe, M.; Mhyre, A.J.; Baker, D.; Strong, R.K.; et al. Mammalian display screening of diverse cystine-dense peptides for difficult to drug targets. Nat. Commun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Barth, M.; Contal, S.; Montalbetti, C.; Spitzer, L. Preparation of New 4-[(E)-[(1,1-dioxo-1,2-benzothiazol-3-yl)hydrazono]methyl]-2-methoxyphenols as Inhibitors of the YAP/TAZ-TEAD Interaction and Their Use in the Treatment of Malignant Mesothelioma. PCT Int. Appl. WO 2017064277 A1, 20 April 2017. [Google Scholar]
- Pobbati, A.V.; Han, X.; Hung, A.W.; Weiguang, S.; Huda, N.; Chen, G.-Y.; Kang, C.; Chia, C.S.B.; Luo, X.; Hong, W.; et al. Targeting the central pocket in human transcription factor TEAD as a potential cancer therapeutic strategy. Structure 2015, 23, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- Xu, W. TEAD Transcription Factor Autopalmitoylation Inhibitors. PCT Int. Appl. WO 2017053706 A1, 30 March 2017. [Google Scholar]
- Bakail, M.; Ochsenbein, F. Targeting protein-protein interactions, a wide open field for drug design. C. R. Chim. 2016, 19, 19–27. [Google Scholar] [CrossRef]
- Song, S.; Xie, M.; Scott, A.W.; Jin, J.; Ma, L.; Dong, X.; Skinner, H.D.; Johnson, R.L.; Ding, S.; Ajani, J.A. A novel YAP1 inhibitor targets CSCs-enriched radiation cells and exerts strong antitumor activity in esophageal adenocarcinoma. Mol. Cancer Ther. 2018, 17, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhao, B.; Wang, P.; Chen, F.; Dong, Z.; Yang, H.; Guan, K.-L.; Xu, Y. Structural insights into the YAP and TEAD complex. Genes Dev. 2010, 24, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chan, S.W.; Zhang, X.; Walsh, M.; Lim, C.J.; Hong, W.; Song, H. Structural basis of YAP recognition by TEAD4 in the Hippo pathway. Genes Dev. 2010, 24, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Hau, J.C.; Erdmann, D.; Mesrouze, Y.; Furet, P.; Fontana, P.; Zimmermann, C.; Schmelzle, T.; Hofmann, F.; Chène, P. The TEAD4-YAP/TAZ protein-protein interaction: Expected similarities and unexpected differences. ChemBioChem 2013, 14, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Yu, J.; Tomchick, D.R.; Pan, D.; Luo, X. Structural and functional analysis of the YAP-binding domain of human TEAD2. Proc. Natl. Acad. Sci. USA 2010, 107, 7293–7298. [Google Scholar] [CrossRef] [PubMed]
- Labbé, C.M.; Laconde, G.; Kuenemann, M.A.; Villoutreix, B.O.; Sperandio, O. IPPI-DB: A manually curated and interactive database of small non-peptide inhibitors of protein–protein interactions. Drug Discov. Today 2013, 18, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, O.; Petitjean, M.; Tuffery, P. wwLigCSre: A 3D ligand-based served for hit identification and optimization. Nucleic Acids Res. 2009, 37, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Mesrouze, Y.; Meyerhofer, M.; Bokhovchuk, F.; Fontana, P.; Zimmermann, C.; Martin, T.; Delaunay, C.; Izaac, A.; Kallen, J.; Schmelzle, T.; et al. Effect of the acylation of TEAD4 on its interaction with co-activators YAP and TAZ. Protein Sci. 2017, 12, 2399–2409. [Google Scholar] [CrossRef] [PubMed]
- Magnez, R.; Thiroux, B.; Taront, S.; Segaoula, Z.; Quesnel, B.; Thuru, X. PD-1/PD-L1 binding studies using microscale thermophoresis. Sci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ΔTm (°C) a | Kd (μM) b | % Luciferase Activity c |
|---|---|---|---|
| 1 | 4.2 | 392 | 103 ± 5 |
| 2 | 3.4 | 650 | 36 ± 2 |
| 3 | 4.1 | 363 | 80 ± 5 |
| 4 | 0.6 | n. d. | 100 ± 5 |
| Niflumic acid | 3.5 | n. d. | 86 ± 3 |
| 5 | 1.5 | n. d. | 86 ± 5 |
| 6 | 0.1 | n. d. | 103 ± 9 |
| 7 | 0.1 | n. d. | 94 ± 6 |
| 8 | 0.3 | n. d. | 103 ± 4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gibault, F.; Coevoet, M.; Sturbaut, M.; Farce, A.; Renault, N.; Allemand, F.; Guichou, J.-F.; Drucbert, A.-S.; Foulon, C.; Magnez, R.; et al. Toward the Discovery of a Novel Class of YAP–TEAD Interaction Inhibitors by Virtual Screening Approach Targeting YAP–TEAD Protein–Protein Interface. Cancers 2018, 10, 140. https://doi.org/10.3390/cancers10050140
Gibault F, Coevoet M, Sturbaut M, Farce A, Renault N, Allemand F, Guichou J-F, Drucbert A-S, Foulon C, Magnez R, et al. Toward the Discovery of a Novel Class of YAP–TEAD Interaction Inhibitors by Virtual Screening Approach Targeting YAP–TEAD Protein–Protein Interface. Cancers. 2018; 10(5):140. https://doi.org/10.3390/cancers10050140
Chicago/Turabian StyleGibault, Floriane, Mathilde Coevoet, Manon Sturbaut, Amaury Farce, Nicolas Renault, Frédéric Allemand, Jean-François Guichou, Anne-Sophie Drucbert, Catherine Foulon, Romain Magnez, and et al. 2018. "Toward the Discovery of a Novel Class of YAP–TEAD Interaction Inhibitors by Virtual Screening Approach Targeting YAP–TEAD Protein–Protein Interface" Cancers 10, no. 5: 140. https://doi.org/10.3390/cancers10050140
APA StyleGibault, F., Coevoet, M., Sturbaut, M., Farce, A., Renault, N., Allemand, F., Guichou, J.-F., Drucbert, A.-S., Foulon, C., Magnez, R., Thuru, X., Corvaisier, M., Huet, G., Chavatte, P., Melnyk, P., Bailly, F., & Cotelle, P. (2018). Toward the Discovery of a Novel Class of YAP–TEAD Interaction Inhibitors by Virtual Screening Approach Targeting YAP–TEAD Protein–Protein Interface. Cancers, 10(5), 140. https://doi.org/10.3390/cancers10050140