The Role of Oncogenic Tyrosine Kinase NPM-ALK in Genomic Instability



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
The Oncogenic Tyrosine Kinase NPM-ALK
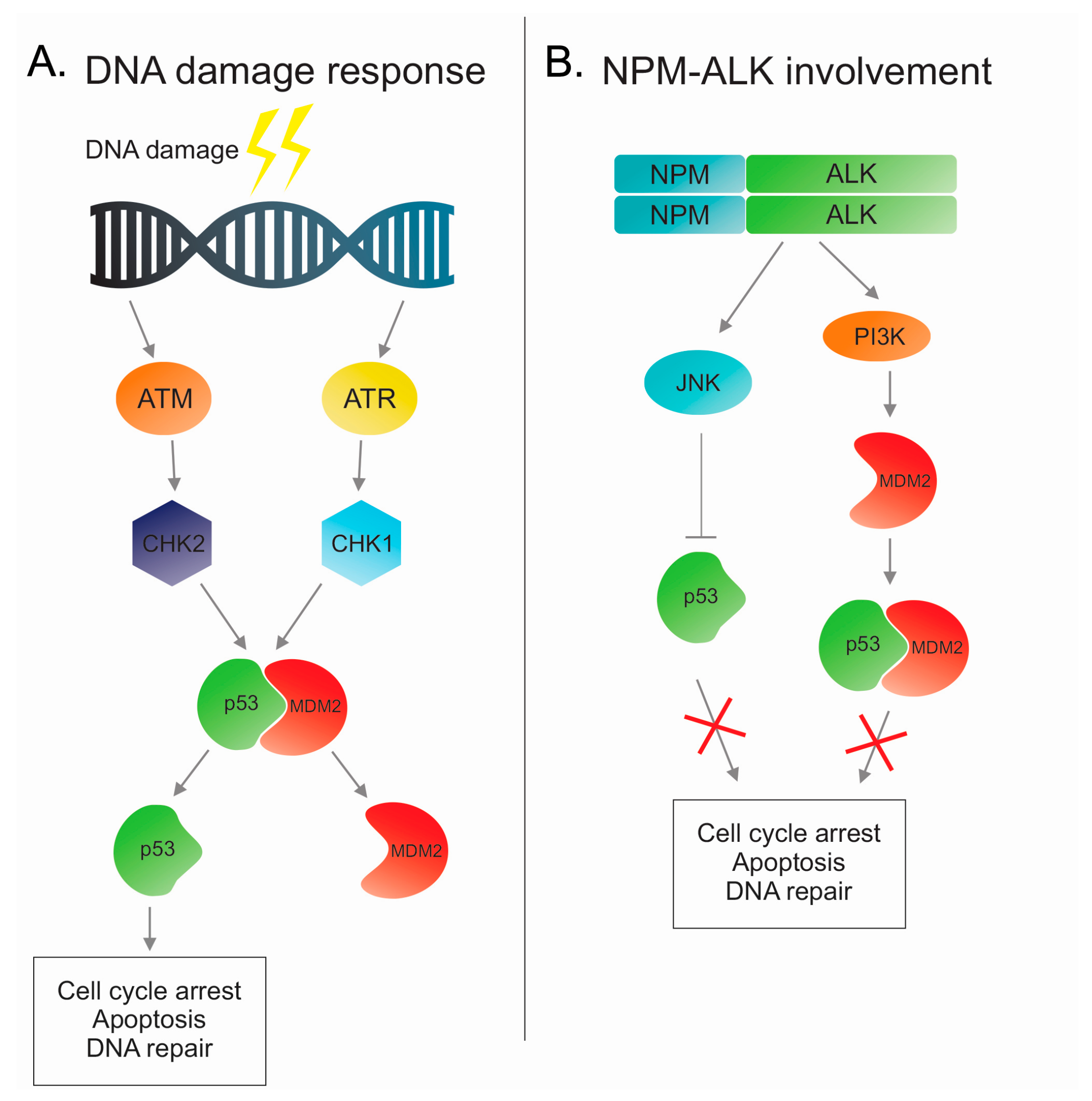
2. DNA Damage Response and Role of Tumor Suppressor p53
p53 in ALK-Positive Anaplastic Large Cell Lymphoma
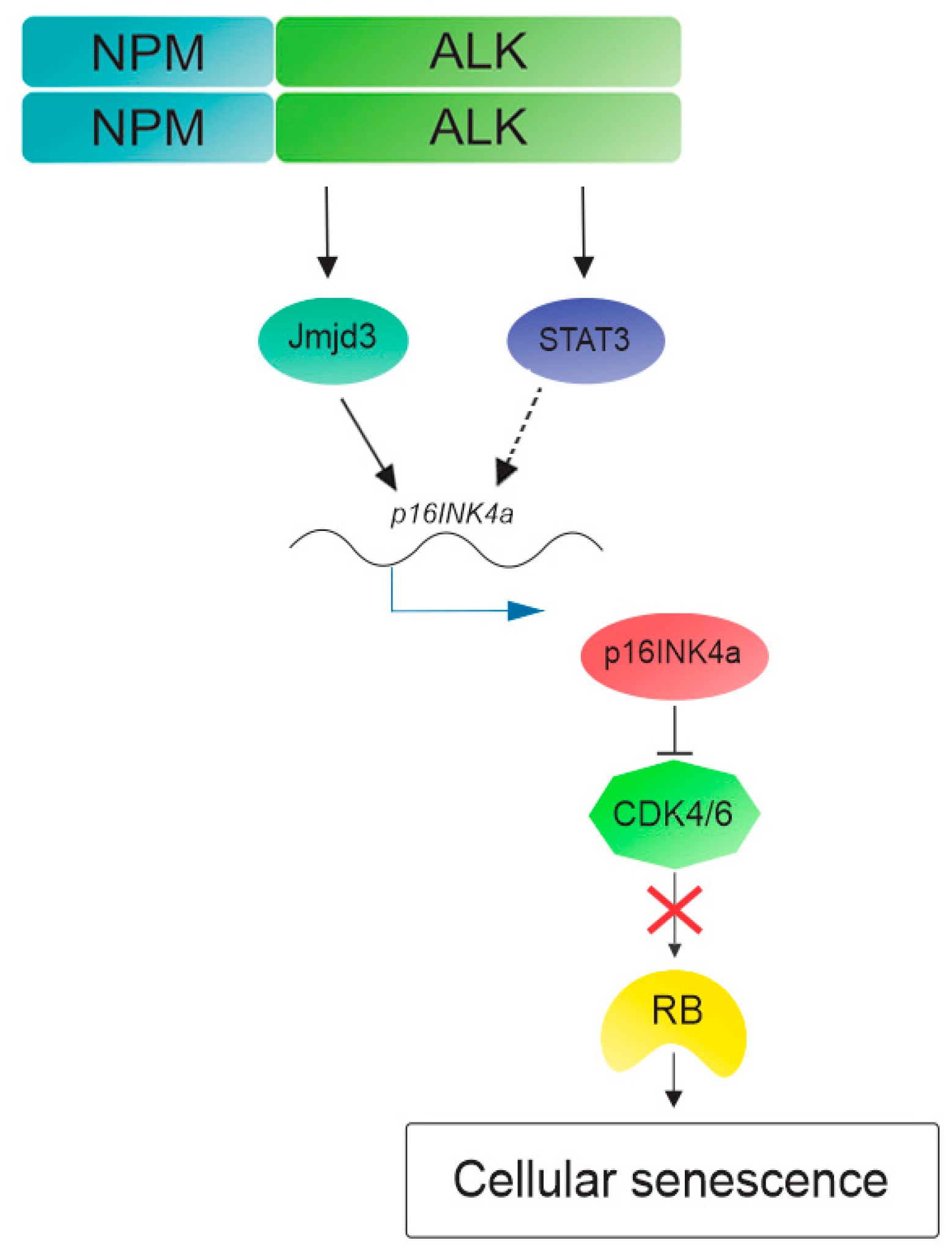
3. NPM-ALK Induces Cellular Senescence in Anaplastic Large Cell Lymphoma
4. NPM-ALK Overexpression Lead to DNA Damage Response activation
5. DNA Repair
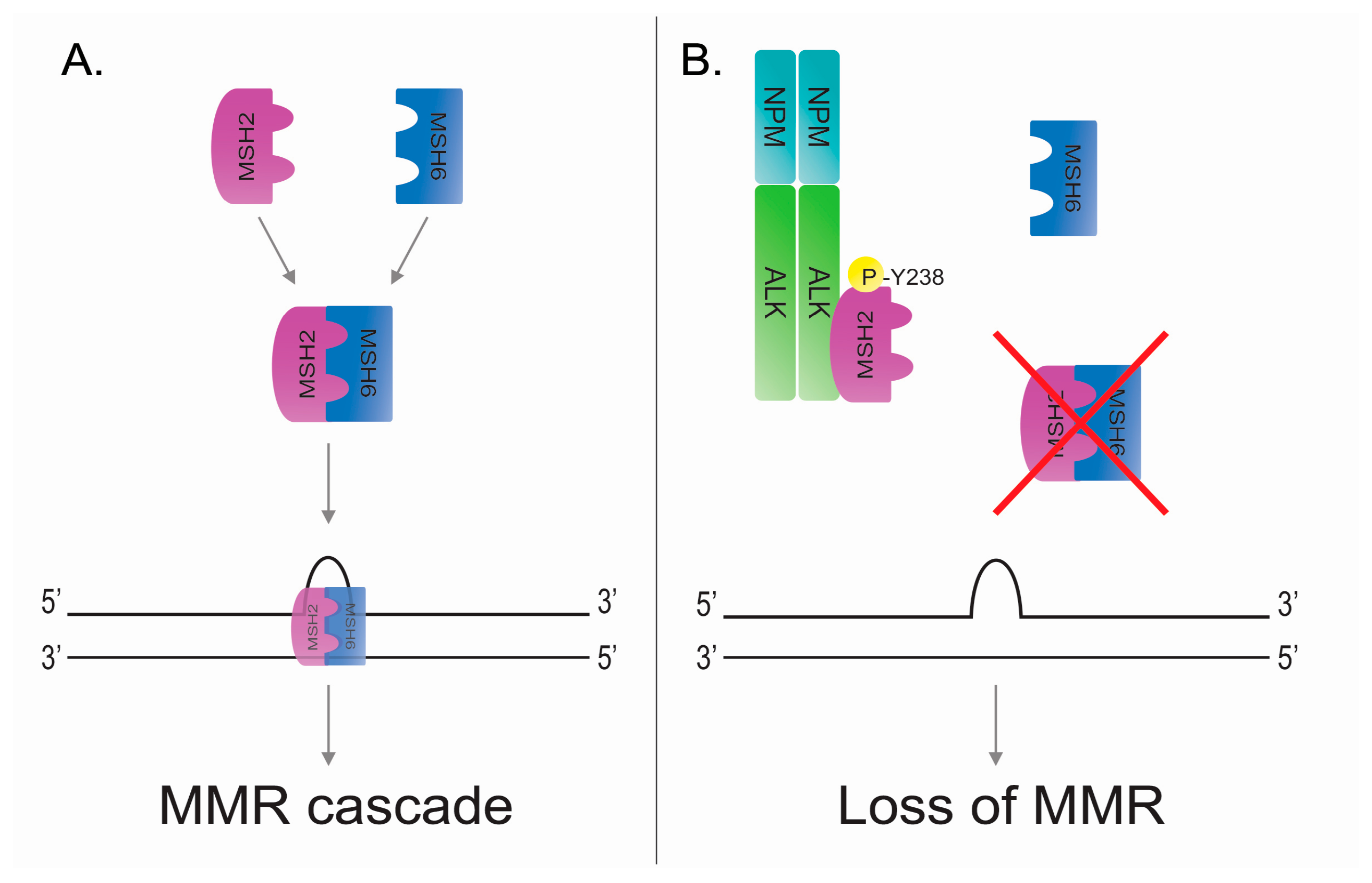
5.1. DNA Mismatch Repair
5.2. DNA Mismatch Repair in ALK-Positive Anaplastic Large Cell Lymphoma
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Niida, H.; Nakanishi, M. DNA damage checkpoints in mammals. Mutagenesis 2006, 21, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Skorski, T. Oncogenic tyrosine kinases and the DNA-damage response. Nat. Rev. Cancer 2002, 2, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.; Dugray, A.; AbdulKarim, B.; Marangoni, E.; Maggiorella, L.; Vaganay, S.; M’Kacher, R.; Rasy, S.D.; Eschwege, F.; Vainchenker, W.; et al. BCR-ABL down-regulates the DNA repair protein DNA-PKcs. Blood 2001, 97, 2084–2090. [Google Scholar] [CrossRef] [PubMed]
- Faderl, S.; Talpaz, M.; Estrov, Z.; O’Brien, S.; Kurzrock, R.; Kantarjian, H.M. The biology of chronic myeloid leukemia. N. Engl. J. Med. 1999, 341, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Benz-Lemoine, E.; Brizard, A.; Huret, J.L.; Babin, P.; Guilhot, F.; Couet, D.; Tanzer, J. Malignant histiocytosis: A specific t(2;5)(p23;q35) translocation? Review of the literature. Blood 1988, 72, 1045–1047. [Google Scholar] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Mano, H. Alkoma: A cancer subtype with a shared target. Cancer Discov. 2012, 2, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Pulford, K.; Pucciarini, A.; Carbone, A.; De Wolf-Peeters, C.; Cordell, J.; Fizzotti, M.; Santucci, A.; Pelicci, P.G.; Pileri, S.; et al. Lymphomas expressing ALK fusion protein(s) other than NPM-ALK. Blood 1999, 94, 3509–3515. [Google Scholar] [PubMed]
- Hernandez, L.; Bea, S.; Bellosillo, B.; Pinyol, M.; Falini, B.; Carbone, A.; Ott, G.; Rosenwald, A.; Fernandez, A.; Pulford, K.; et al. Diversity of genomic breakpoints in TFG-ALK translocations in anaplastic large cell lymphomas: Identification of a new TFG-ALK(XL) chimeric gene with transforming activity. Am. J. Pathol. 2002, 160, 1487–1494. [Google Scholar] [CrossRef]
- Lamant, L.; Dastugue, N.; Pulford, K.; Delsol, G.; Mariame, B. A new fusion gene tpm3-alk in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood 1999, 93, 3088–3095. [Google Scholar] [PubMed]
- Colleoni, G.W.; Bridge, J.A.; Garicochea, B.; Liu, J.; Filippa, D.A.; Ladanyi, M. ATIC-ALK: A novel variant ALK gene fusion in anaplastic large cell lymphoma resulting from the recurrent cryptic chromosomal inversion, inv(2)(p23q35). Am. J. Pathol. 2000, 156, 781–789. [Google Scholar] [CrossRef]
- Zamo, A.; Chiarle, R.; Piva, R.; Howes, J.; Fan, Y.; Chilosi, M.; Levy, D.E.; Inghirami, G. Anaplastic lymphoma kinase (ALK) activates STAT3 and protects hematopoietic cells from cell death. Oncogene 2002, 21, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Slupianek, A.; Nieborowska-Skorska, M.; Hoser, G.; Morrione, A.; Majewski, M.; Xue, L.; Morris, S.W.; Wasik, M.A.; Skorski, T. Role of phosphatidylinositol 3-kinase-AKT pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res. 2001, 61, 2194–2199. [Google Scholar] [PubMed]
- Bai, R.Y.; Dieter, P.; Peschel, C.; Morris, S.W.; Duyster, J. Nucleophosmin-anaplastic lymphoma kinase of large-cell anaplastic lymphoma is a constitutively active tyrosine kinase that utilizes phospholipase c-gamma to mediate its mitogenicity. Mol. Cell. Biol. 1998, 18, 6951–6961. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 2008, 8, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Rouse, J.; Jackson, S.P. Interfaces between the detection, signaling, and repair of DNA damage. Science 2002, 297, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.H.; Zou, L. Checkpoint and coordinated cellular responses to DNA damage. Results Probl. Cell Differ. 2006, 42, 65–92. [Google Scholar] [PubMed]
- D’Amours, D.; Jackson, S.P. The mre11 complex: At the crossroads of DNA repair and checkpoint signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. ATM activation by DNA double-strand breaks through the MRE11-RAD50-NBS1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Byun, T.S.; Pacek, M.; Yee, M.C.; Walter, J.C.; Cimprich, K.A. Functional uncoupling of mcm helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005, 19, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through atrip recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. CHK1 and CHK2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. Atm, atr, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Meulmeester, E.; Pereg, Y.; Shiloh, Y.; Jochemsen, A.G. ATM-mediated phosphorylations inhibit Mdmx/Mdm2 stabilization by HAUSP in favor of p53 activation. Cell Cycle 2005, 4, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Stommel, J.M.; Wahl, G.M. Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. EMBO J. 2004, 23, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W. Tumour suppression by p53: A role for the DNA damage response? Nat. Rev. Cancer 2009, 9, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Prives, C.; Hall, P.A. The p53 pathway. J. Pathol. 1999, 187, 112–126. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919. [Google Scholar] [CrossRef] [PubMed]
- Herrero, A.B.; Rojas, E.A.; Misiewicz-Krzeminska, I.; Krzeminski, P.; Gutierrez, N.C. Molecular mechanisms of p53 deregulation in cancer: An overview in multiple myeloma. Int. J. Mol. Sci. 2016, 17, 2003. [Google Scholar] [CrossRef] [PubMed]
- Bond, G.L.; Hu, W.; Bond, E.E.; Robins, H.; Lutzker, S.G.; Arva, N.C.; Bargonetti, J.; Bartel, F.; Taubert, H.; Wuerl, P.; et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 2004, 119, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Toledo, F.; Wahl, G.M. MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int. J. Biochem. Cell Biol. 2007, 39, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Feng, Z.; Ma, L.; Wagner, J.; Rice, J.J.; Stolovitzky, G.; Levine, A.J. A single nucleotide polymorphism in the MDM2 gene disrupts the oscillation of p53 and MDM2 levels in cells. Cancer Res. 2007, 67, 2757–2765. [Google Scholar] [CrossRef] [PubMed]
- Whibley, C.; Pharoah, P.D.; Hollstein, M. p53 polymorphisms: Cancer implications. Nat. Rev. Cancer 2009, 9, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.B.; Schumacher, B. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Takakuwa, T.; Fujita, S.; Ham, M.F.; Luo, W.J.; Daibata, M.; Aozasa, K. Alterations of DNA damage-response genes ATM and ATR in pyothorax-associated lymphoma. Lab. Investig. J. Tech. Methods Pathol. 2005, 85, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Schaffner, C.; Idler, I.; Stilgenbauer, S.; Dohner, H.; Lichter, P. Mantle cell lymphoma is characterized by inactivation of the ATM gene. Proc. Natl. Acad. Sci. USA 2000, 97, 2773–2778. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Rasi, S.; Spina, V.; Bruscaggin, A.; Monti, S.; Ciardullo, C.; Deambrogi, C.; Khiabanian, H.; Serra, R.; Bertoni, F.; et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood 2013, 121, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.; Martinez, P.; Wade, R.; Hockley, S.; Oscier, D.; Matutes, E.; Dearden, C.E.; Richards, S.M.; Catovsky, D.; Morgan, G.J. Mutational status of the Tp53 gene as a predictor of response and survival in patients with chronic lymphocytic leukemia: Results from the LRF CLL4 trial. J. Clin. Oncol. 2011, 29, 2223–2229. [Google Scholar] [CrossRef] [PubMed]
- Te Raa, G.D.; Malcikova, J.; Pospisilova, S.; Trbusek, M.; Mraz, M.; Garff-Tavernier, M.L.; Merle-Beral, H.; Lin, K.; Pettitt, A.R.; Merkel, O.; et al. Overview of available p53 function tests in relation to Tp53 and ATM gene alterations and chemoresistance in chronic lymphocytic leukemia. Leuk. Lymphoma 2013, 54, 1849–1853. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Inghirami, G.; Chadburn, A.; Knowles, D.M. High levels of p53 protein expression do not correlate with p53 gene mutations in anaplastic large cell lymphoma. Am. J. Pathol. 1993, 143, 845–856. [Google Scholar] [PubMed]
- Rassidakis, G.Z.; Thomaides, A.; Wang, S.; Jiang, Y.; Fourtouna, A.; Lai, R.; Medeiros, L.J. P53 gene mutations are uncommon but p53 is commonly expressed in anaplastic large-cell lymphoma. Leukemia 2005, 19, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Boi, M.; Rinaldi, A.; Kwee, I.; Bonetti, P.; Todaro, M.; Tabbo, F.; Piva, R.; Rancoita, P.M.; Matolcsy, A.; Timar, B.; et al. PRDM1/BLIMP1 is commonly inactivated in anaplastic large T-cell lymphoma. Blood 2013, 122, 2683–2693. [Google Scholar] [CrossRef] [PubMed]
- Leventaki, V.; Drakos, E.; Medeiros, L.J.; Lim, M.S.; Elenitoba-Johnson, K.S.; Claret, F.X.; Rassidakis, G.Z. NPM-ALK oncogenic kinase promotes cell-cycle progression through activation of JNK/CJUN signaling in anaplastic large-cell lymphoma. Blood 2007, 110, 1621–1630. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.X.; Kerby, A.; McDuff, F.K.; Ye, H.; Turner, S.D. NPM-ALK inhibits the p53 tumor suppressor pathway in an MDM2 and JNK-dependent manner. Blood 2009, 113, 5217–5227. [Google Scholar] [CrossRef] [PubMed]
- McDuff, F.K.; Turner, S.D. Aberrant anaplastic lymphoma kinase activity induces a p53 and RB-dependent senescence-like arrest in the absence of detectable p53 stabilization. PLoS ONE 2011, 6, e17854. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Drakos, E.; Atsaves, V.; Schlette, E.; Li, J.; Papanastasi, I.; Rassidakis, G.Z.; Medeiros, L.J. The therapeutic potential of p53 reactivation by nutlin-3a in ALK+ anaplastic large cell lymphoma with wild-type or mutated p53. Leukemia 2009, 23, 2290–2299. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular senescence in cancer and aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Serrano, M. The power and the promise of oncogene-induced senescence markers. Nat. Rev. Cancer 2006, 6, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; Edwards, C.D.; Kobzik, L.; Godleski, J.; Richards, W.; Sugarbaker, D.J.; Rollins, B.J. Reciprocal RB inactivation and p16ink4 expression in primary lung cancers and cell lines. Cancer Res. 1995, 55, 505–509. [Google Scholar] [PubMed]
- Martinelli, P.; Bonetti, P.; Sironi, C.; Pruneri, G.; Fumagalli, C.; Raviele, P.R.; Volorio, S.; Pileri, S.; Chiarle, R.; McDuff, F.K.; et al. The lymphoma-associated NPM-ALK oncogene elicits a P16INK4A/PRB-dependent tumor-suppressive pathway. Blood 2011, 117, 6617–6626. [Google Scholar] [CrossRef] [PubMed]
- Trotta, R.; Vignudelli, T.; Candini, O.; Intine, R.V.; Pecorari, L.; Guerzoni, C.; Santilli, G.; Byrom, M.W.; Goldoni, S.; Ford, L.P.; et al. BCR/ABL activates MDM2 mrna translation via the la antigen. Cancer Cell 2003, 3, 145–160. [Google Scholar] [CrossRef]
- Insinga, A.; Monestiroli, S.; Ronzoni, S.; Carbone, R.; Pearson, M.; Pruneri, G.; Viale, G.; Appella, E.; Pelicci, P.; Minucci, S. Impairment of p53 acetylation, stability and function by an oncogenic transcription factor. EMBO J. 2004, 23, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Ceccon, M.; Merlo, M.E.B.; Mologni, L.; Poggio, T.; Varesio, L.M.; Menotti, M.; Bombelli, S.; Rigolio, R.; Manazza, A.D.; Di Giacomo, F.; et al. Excess of NPM-ALK oncogenic signaling promotes cellular apoptosis and drug dependency. Oncogene 2016, 35, 3854–3865. [Google Scholar] [CrossRef] [PubMed]
- Ceccon, M.; Mologni, L.; Giudici, G.; Piazza, R.; Pirola, A.; Fontana, D.; Gambacorti-Passerini, C. Treatment efficacy and resistance mechanisms using the second-generation ALK inhibitor ap26113 in human NPM-ALK-positive anaplastic large cell lymphoma. Mol. Cancer Res. 2015, 13, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Zdzalik, D.; Dymek, B.; Grygielewicz, P.; Gunerka, P.; Bujak, A.; Lamparska-Przybysz, M.; Wieczorek, M.; Dzwonek, K. Activating mutations in ALK kinase domain confer resistance to structurally unrelated ALK inhibitors in NPM-ALK-positive anaplastic large-cell lymphoma. J. Cancer Res. Clin. Oncol. 2014, 140, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Altieri, F.; Grillo, C.; Maceroni, M.; Chichiarelli, S. DNA damage and repair: From molecular mechanisms to health implications. Antioxid. Redox Signaling 2008, 10, 891–937. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Barnes, D.E. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.M.; Lehmann, A.R. Conservation of eukaryotic DNA repair mechanisms. Intern. J. Radiat. Biol. 1998, 74, 277–286. [Google Scholar]
- Modrich, P.; Lahue, R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Ann. Rev. Biochem. 1996, 65, 101–133. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T.A.; Erie, D.A. DNA mismatch repair. Ann. Rev. Biochem. 2005, 74, 681–710. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Wilson, T.; Gradia, S.; Kane, M.F.; Guerrette, S.; Marsischky, G.T.; Kolodner, R.; Fishel, R. hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc. Natl. Acad. Sci. USA 1996, 93, 13629–13634. [Google Scholar] [CrossRef] [PubMed]
- Li, G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Modrich, P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 2006, 281, 30305–30309. [Google Scholar] [CrossRef] [PubMed]
- Prolla, T.A.; Christie, D.M.; Liskay, R.M. Dual requirement in yeast DNA mismatch repair for MLH1 and PMS1, two homologs of the bacterial mutL gene. Mol. Cell. Biol. 1994, 14, 407–415. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guo, S.; Presnell, S.R.; Yuan, F.; Zhang, Y.; Gu, L.; Li, G.M. Differential requirement for proliferating cell nuclear antigen in 5’ and 3’ nick-directed excision in human mismatch repair. J. Biol. Chem. 2004, 279, 16912–16917. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, F.C.; Jager, A.C.; Lutzen, A.; Bundgaard, J.R.; Rasmussen, L.J. Characterization of human exonuclease 1 in complex with mismatch repair proteins, subcellular localization and association with pcna. Oncogene 2004, 23, 1457–1468. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.T.; Erdeniz, N.; Symington, L.S.; Liskay, R.M. EXO1-a multi-tasking eukaryotic nuclease. DNA Repair 2004, 3, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Keijzers, G.; Rasmussen, L.J. DNA mismatch repair and its many roles in eukaryotic cells. Mutat. Res. 2017, 773, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Tiraby, J.G.; Fox, M.S. Marker discrimination in transformation and mutation of pneumococcus. Proc. Natl. Acad. Sci. USA 1973, 70, 3541–3545. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; de la Chapelle, A. Genetic susceptibility to non-polyposis colorectal cancer. J. Med. Genet. 1999, 36, 801–818. [Google Scholar] [PubMed]
- Hampel, H.; Frankel, W.L.; Martin, E.; Arnold, M.; Khanduja, K.; Kuebler, P.; Nakagawa, H.; Sotamaa, K.; Prior, T.W.; Westman, J.; et al. Screening for the lynch syndrome (hereditary nonpolyposis colorectal cancer). N. Engl. J. Med. 2005, 352, 1851–1860. [Google Scholar] [CrossRef] [PubMed]
- Barnetson, R.A.; Tenesa, A.; Farrington, S.M.; Nicholl, I.D.; Cetnarskyj, R.; Porteous, M.E.; Campbell, H.; Dunlop, M.G. Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N. Engl. J. Med. 2006, 354, 2751–2763. [Google Scholar] [CrossRef] [PubMed]
- Fink, D.; Nebel, S.; Norris, P.S.; Aebi, S.; Kim, H.K.; Haas, M.; Howell, S.B. The effect of different chemotherapeutic agents on the enrichment of DNA mismatch repair-deficient tumour cells. Br. J. Cancer 1998, 77, 703–708. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Edelmann, W.; Yang, K.; Umar, A.; Heyer, J.; Lau, K.; Fan, K.; Liedtke, W.; Cohen, P.E.; Kane, M.F.; Lipford, J.R.; et al. Mutation in the mismatch repair gene msh6 causes cancer susceptibility. Cell 1997, 91, 467–477. [Google Scholar] [CrossRef]
- Reitmair, A.H.; Schmits, R.; Ewel, A.; Bapat, B.; Redston, M.; Mitri, A.; Waterhouse, P.; Mittrucker, H.W.; Wakeham, A.; Liu, B.; et al. MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nat. Genet. 1995, 11, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.M.; Plug, A.W.; Prolla, T.A.; Bronner, C.E.; Harris, A.C.; Yao, X.; Christie, D.M.; Monell, C.; Arnheim, N.; Bradley, A.; et al. Involvement of mouse MLH1 in DNA mismatch repair and meiotic crossing over. Nat. Genet. 1996, 13, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Prolla, T.A.; Baker, S.M.; Harris, A.C.; Tsao, J.L.; Yao, X.; Bronner, C.E.; Zheng, B.; Gordon, M.; Reneker, J.; Arnheim, N.; et al. Tumour susceptibility and spontaneous mutation in mice deficient in MLH1, PMS1 and PMS2 DNA mismatch repair. Nat. Genet. 1998, 18, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.; Clark, A.B.; Wong, E.; Kane, M.F.; Mazur, D.J.; Parris, T.; Kolas, N.K.; Russell, R.; Hou, H., Jr.; Kneitz, B.; et al. Inactivation of exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes Dev. 2003, 17, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Wang, P.; Young, L.C.; Lai, R.; Li, L. Proteome-wide identification of novel binding partners to the oncogenic fusion gene protein, NPM-ALK, using tandem affinity purification and mass spectrometry. Am. J. Pathol. 2009, 174, 361–370. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Young, L.C.; Bone, K.M.; Wang, P.; Wu, F.; Adam, B.A.; Hegazy, S.; Gelebart, P.; Holovati, J.; Li, L.; Andrew, S.E.; et al. Fusion tyrosine kinase NPM-ALK deregulates MSH2 and suppresses DNA mismatch repair function novel insights into a potent oncoprotein. Am. J. Pathol. 2011, 179, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Bone, K.M.; Wang, P.; Wu, F.; Wu, C.; Li, L.; Bacani, J.T.; Andrew, S.E.; Lai, R. Npm-alk mediates phosphorylation of msh2 at tyrosine 238, creating a functional deficiency in MSH2 and the loss of mismatch repair. Blood Cancer J. 2015, 5, e311. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Savooji, J.; Liu, D. Second- and third-generation ALK inhibitors for non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Andreeff, M.; Kelly, K.R.; Yee, K.; Assouline, S.; Strair, R.; Popplewell, L.; Bowen, D.; Martinelli, G.; Drummond, M.W.; Vyas, P.; et al. Results of the phase I trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin. Cancer Res. 2016, 22, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Zhang, Z.; Liu, J.J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- Herting, F.; Herter, S.; Friess, T.; Muth, G.; Bacac, M.; Sulcova, J.; Umana, P.; Dangl, M.; Klein, C. Antitumour activity of the glycoengineered type II anti-CD20 antibody obinutuzumab (GA101) in combination with the MDM2-selective antagonist idasanutlin (RG7388). Eur. J. Haematol. 2016, 97, 461–470. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lobello, C.; Bikos, V.; Janikova, A.; Pospisilova, S. The Role of Oncogenic Tyrosine Kinase NPM-ALK in Genomic Instability. Cancers 2018, 10, 64. https://doi.org/10.3390/cancers10030064
Lobello C, Bikos V, Janikova A, Pospisilova S. The Role of Oncogenic Tyrosine Kinase NPM-ALK in Genomic Instability. Cancers. 2018; 10(3):64. https://doi.org/10.3390/cancers10030064
Chicago/Turabian StyleLobello, Cosimo, Vasilis Bikos, Andrea Janikova, and Sarka Pospisilova. 2018. "The Role of Oncogenic Tyrosine Kinase NPM-ALK in Genomic Instability" Cancers 10, no. 3: 64. https://doi.org/10.3390/cancers10030064
APA StyleLobello, C., Bikos, V., Janikova, A., & Pospisilova, S. (2018). The Role of Oncogenic Tyrosine Kinase NPM-ALK in Genomic Instability. Cancers, 10(3), 64. https://doi.org/10.3390/cancers10030064