Tumor Resistance against ALK Targeted Therapy-Where It Comes From and Where It Goes

 , ,
, , 
Abstract
1. Introduction
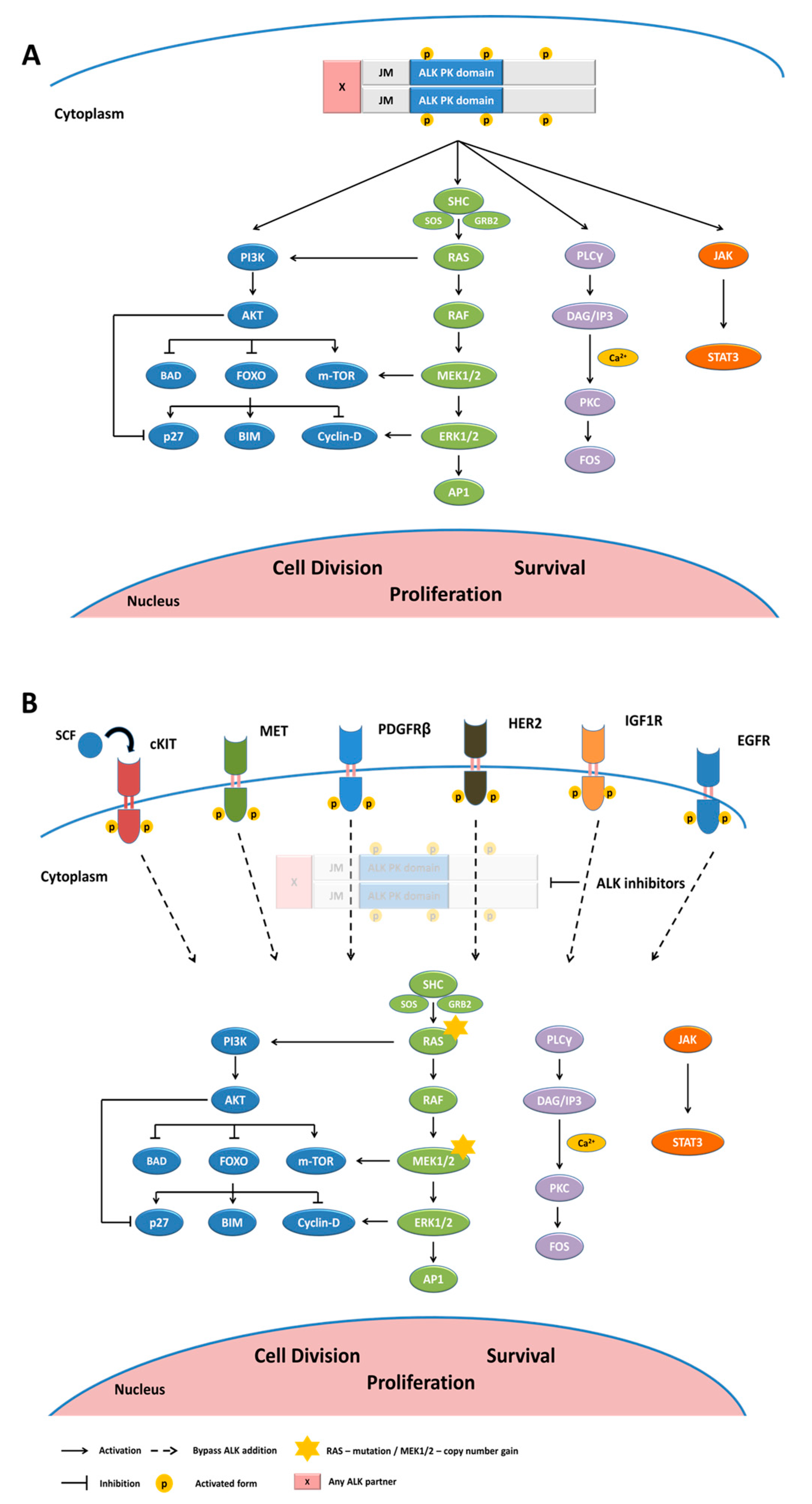
2. Anaplastic Lymphoma Kinase-Physiological Expression and Functional Role
3. ALK Gene Alterations in Cancers
4. ALK Inhibitors
4.1. Crizotinib: A First-Generation ALK Inhibitor
4.2. Second Generation ALK Inhibitors
4.2.1. Ceritinib (LDK378; Zykadia; Novartis)
4.2.2. Alectinib (CH5424802; Chugai-Roche)
4.2.3. Brigatinib (AP26113; Ariad)
4.3. Other ALK TKI Under Development
5. ALK TKI Resistance Mechanisms
5.1. ALK-Dependent Resistance Mechanisms
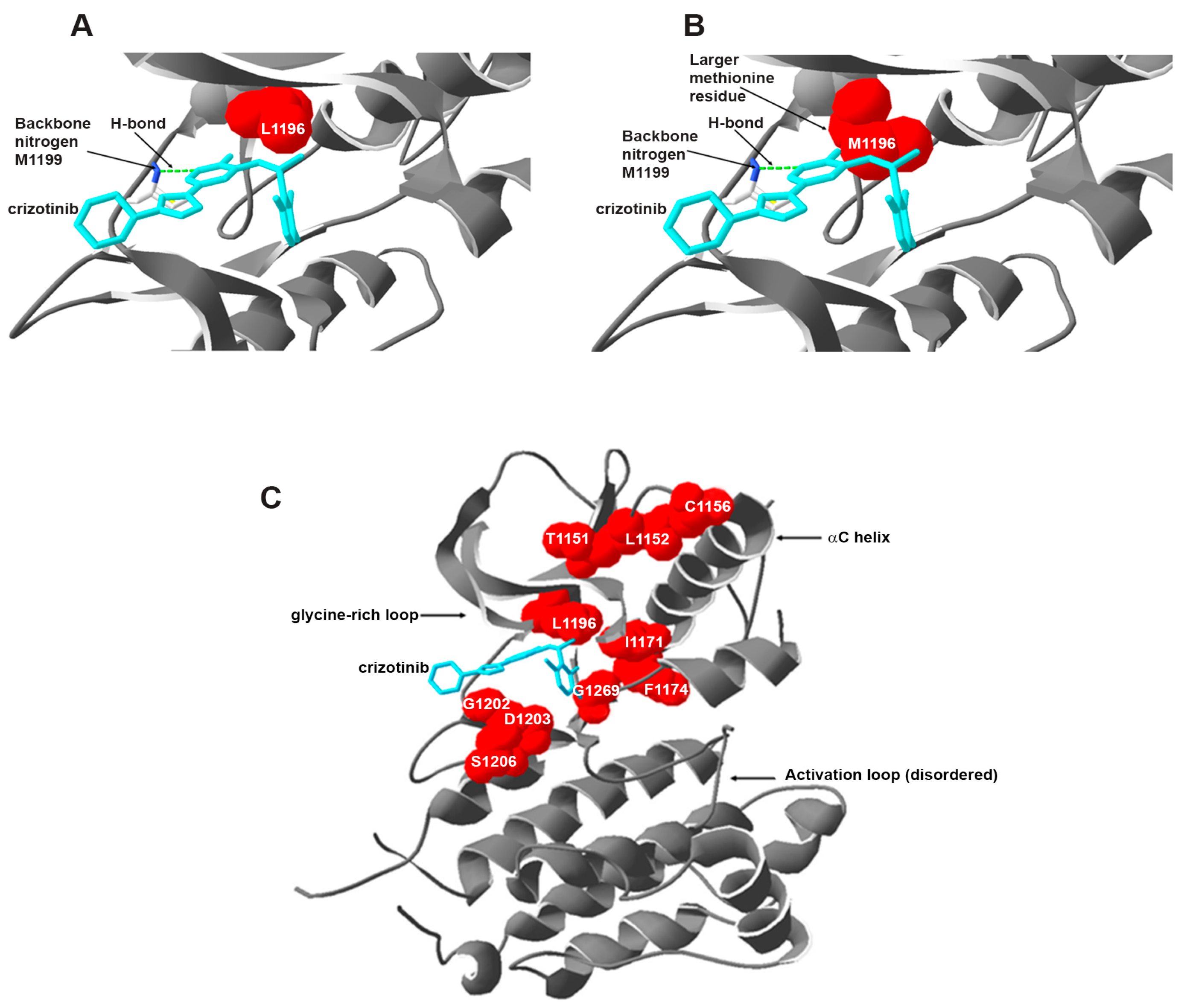
5.1.1. Secondary Mutations in the ALK Tyrosine Kinase Domain
Resistance against Crizotinib
Resistance to Second-Generation ALK TKIs
5.1.2. Amplification of ALK
5.2. ALK-Independent Resistance Mechanisms
5.2.1. Activation of Bypass Signaling Pathways
5.2.3. Other Mechanisms
6. Other Therapeutic Strategies to Overcome ALK-Related Resistance
6.1. ALK TKIs Combined with Other Inhibitors Targeting Different Kinases
6.2. ALK Inhibitors Combined with Immunotherapy
6.2.1. Immune Checkpoint Inhibitors
6.2.2. Vaccine Therapy
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Guilhot, F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.; Gattermann, N.; Deininger, M.W.; Silver, R.T.; Goldman, J.M.; Stone, R.M.; et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 2006, 355, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.B.; Lax, I.; Reshetnyak, A.; Ligon, G.F.; Lillquist, J.S.; Natoli, E.J., Jr.; Shi, X.; Folta-Stogniew, E.; Gunel, M.; Alvarado, D.; et al. Heparin is an activating ligand of the orphan receptor tyrosine kinase ALK. Sci. Signal. 2015, 8, ra6. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 2008, 8, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Mi, R.; Chen, W.; Hoke, A. Pleiotrophin is a neurotrophic factor for spinal motor neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 4664–4669. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Felip, E.; Bauer, T.M.; Besse, B.; Navarro, A.; Postel-Vinay, S.; Gainor, J.F.; Johnson, M.; Dietrich, J.; James, L.P.; et al. Lorlatinib in non-small-cell lung cancer with ALK or ros1 rearrangement: An international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 2017, 18, 1590–1599. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crino, L.; Ahn, M.J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.J.; Mok, T.; Kim, D.W.; Wu, Y.L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, D.W.; Mehra, R.; Tan, D.S.; Felip, E.; Chow, L.Q.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; De Pas, T.; et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 370, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Mehra, R.; Tan, D.S.; Felip, E.; Chow, L.Q.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; De Pas, T.; Riely, G.J.; et al. Activity and safety of ceritinib in patients with ALK-rearranged non-small-cell lung cancer (ascend-1): Updated results from the multicentre, open-label, phase 1 trial. Lancet Oncol. 2016, 17, 452–463. [Google Scholar] [CrossRef]
- Ou, S.H.; Ahn, J.S.; De Petris, L.; Govindan, R.; Yang, J.C.; Hughes, B.; Lena, H.; Moro-Sibilot, D.; Bearz, A.; Ramirez, S.V.; et al. Alectinib in crizotinib-refractory ALK-rearranged non-small-cell lung cancer: A phase ii global study. J. Clin. Oncol. 2016, 34, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Gandhi, L.; Gadgeel, S.; Riely, G.J.; Cetnar, J.; West, H.; Camidge, D.R.; Socinski, M.A.; Chiappori, A.; Mekhail, T.; et al. Alectinib in ALK-positive, crizotinib-resistant, non-small-cell lung cancer: A single-group, multicentre, phase 2 trial. Lancet Oncol. 2016, 17, 234–242. [Google Scholar] [CrossRef]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.W.; Ou, S.I.; Perol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Tiseo, M.; Ahn, M.J.; Reckamp, K.L.; Hansen, K.H.; Kim, S.W.; Huber, R.M.; West, H.L.; Groen, H.J.M.; Hochmair, M.J.; et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase-positive non-small-cell lung cancer: A randomized, multicenter phase II trial. J. Clin. Oncol. 2017, 35, 2490–2498. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti Passerini, C.; Farina, F.; Stasia, A.; Redaelli, S.; Ceccon, M.; Mologni, L.; Messa, C.; Guerra, L.; Giudici, G.; Sala, E.; et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. J. Natl. Cancer Inst. 2014, 106, djt378. [Google Scholar] [CrossRef] [PubMed]
- Butrynski, J.E.; D’Adamo, D.R.; Hornick, J.L.; Dal Cin, P.; Antonescu, C.R.; Jhanwar, S.C.; Ladanyi, M.; Capelletti, M.; Rodig, S.J.; Ramaiya, N.; et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N. Engl. J. Med. 2010, 363, 1727–1733. [Google Scholar] [CrossRef] [PubMed]
- Richly, H.; Kim, T.M.; Schuler, M.; Kim, D.W.; Harrison, S.J.; Shaw, A.T.; Boral, A.L.; Yovine, A.; Solomon, B. Ceritinib in patients with advanced anaplastic lymphoma kinase-rearranged anaplastic large-cell lymphoma. Blood 2015, 126, 1257–1258. [Google Scholar] [CrossRef] [PubMed]
- Iwahara, T.; Fujimoto, J.; Wen, D.; Cupples, R.; Bucay, N.; Arakawa, T.; Mori, S.; Ratzkin, B.; Yamamoto, T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997, 14, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.H.; Vernersson, E.; Grabbe, C.; Hallberg, B. Anaplastic lymphoma kinase: Signalling in development and disease. Biochem. J. 2009, 420, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Naeve, C.; Mathew, P.; James, P.L.; Kirstein, M.N.; Cui, X.; Witte, D.P. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene 1997, 14, 2175–2188. [Google Scholar] [CrossRef] [PubMed]
- Loren, C.E.; Englund, C.; Grabbe, C.; Hallberg, B.; Hunter, T.; Palmer, R.H. A crucial role for the anaplastic lymphoma kinase receptor tyrosine kinase in gut development in drosophila melanogaster. EMBO Rep. 2003, 4, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Bazigou, E.; Apitz, H.; Johansson, J.; Loren, C.E.; Hirst, E.M.; Chen, P.L.; Palmer, R.H.; Salecker, I. Anterograde jelly belly and ALK receptor tyrosine kinase signaling mediates retinal axon targeting in drosophila. Cell 2007, 128, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Rohrbough, J.; Broadie, K. Anterograde jelly belly ligand to ALK receptor signaling at developing synapses is regulated by mind the gap. Development 2010, 137, 3523–3533. [Google Scholar] [CrossRef] [PubMed]
- Gouzi, J.Y.; Moressis, A.; Walker, J.A.; Apostolopoulou, A.A.; Palmer, R.H.; Bernards, A.; Skoulakis, E.M. The receptor tyrosine kinase ALK controls neurofibromin functions in drosophila growth and learning. PLoS Genet. 2011, 7, e1002281. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Norris, A.; Weiss, J.B.; Frasch, M. Jelly belly protein activates the receptor tyrosine kinase ALK to specify visceral muscle pioneers. Nature 2003, 425, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.L.; Eriksson, T.; Vernersson, E.; Vigny, M.; Hallberg, B.; Palmer, R.H. The ligand jelly belly (JEB) activates the drosophila ALKRTK to drive PC12 cell differentiation, but is unable to activate the mouse ALKRTK. J. Exp. Zool. Part B Mol. Dev. Evolut. 2007, 308, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Reiner, D.J.; Ailion, M.; Thomas, J.H.; Meyer, B.J. C. elegans anaplastic lymphoma kinase ortholog scd-2 controls dauer formation by modulating TGF-β signaling. Curr. Biol. CB 2008, 18, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Cheng, M.; Zhang, Q.; Wasik, M.; Kelsh, R.; Winkler, C. Anaplastic lymphoma kinase is required for neurogenesis in the developing central nervous system of zebrafish. PLoS ONE 2013, 8, e63757. [Google Scholar] [CrossRef] [PubMed]
- Hurley, S.P.; Clary, D.O.; Copie, V.; Lefcort, F. Anaplastic lymphoma kinase is dynamically expressed on subsets of motor neurons and in the peripheral nervous system. J. Comp. Neurol. 2006, 495, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Vernersson, E.; Khoo, N.K.; Henriksson, M.L.; Roos, G.; Palmer, R.H.; Hallberg, B. Characterization of the expression of the ALK receptor tyrosine kinase in mice. Gene Express. Patterns GEP 2006, 6, 448–461. [Google Scholar] [CrossRef] [PubMed]
- Bilsland, J.G.; Wheeldon, A.; Mead, A.; Znamenskiy, P.; Almond, S.; Waters, K.A.; Thakur, M.; Beaumont, V.; Bonnert, T.P.; Heavens, R.; et al. Behavioral and neurochemical alterations in mice deficient in anaplastic lymphoma kinase suggest therapeutic potential for psychiatric indications. Neuropsychopharmacology 2008, 33, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Lasek, A.W.; Lim, J.; Kliethermes, C.L.; Berger, K.H.; Joslyn, G.; Brush, G.; Xue, L.; Robertson, M.; Moore, M.S.; Vranizan, K.; et al. An evolutionary conserved role for anaplastic lymphoma kinase in behavioral responses to ethanol. PLoS ONE 2011, 6, e22636. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, npm, in non-hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, A.V.; Murray, P.B.; Shi, X.; Mo, E.S.; Mohanty, J.; Tome, F.; Bai, H.; Gunel, M.; Lax, I.; Schlessinger, J. Augmentor alpha and β (FAM150) are ligands of the receptor tyrosine kinases ALK and LTK: Hierarchy and specificity of ligand-receptor interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 15862–15867. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G. Adopting ALK and LTK. Proc. Natl. Acad. Sci. USA 2015, 112, 15783–15784. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Hsu, P.P.; Awad, M.M.; Engelman, J.A. Tyrosine kinase gene rearrangements in epithelial malignancies. Nat. Rev. Cancer 2013, 13, 772–787. [Google Scholar] [CrossRef] [PubMed]
- Toffalini, F.; Demoulin, J.B. New insights into the mechanisms of hematopoietic cell transformation by activated receptor tyrosine kinases. Blood 2010, 116, 2429–2437. [Google Scholar] [CrossRef] [PubMed]
- Ladanyi, M.; Cavalchire, G. Molecular variant of the npm-ALK rearrangement of KI-1 lymphoma involving a cryptic ALK splice site. Genes Chromosom. Cancer 1996, 15, 173–177. [Google Scholar] [CrossRef]
- Lamant, L.; Pulford, K.; Bischof, D.; Morris, S.W.; Mason, D.Y.; Delsol, G.; Mariame, B. Expression of the ALK tyrosine kinase gene in neuroblastoma. Am. J. Pathol. 2000, 156, 1711–1721. [Google Scholar] [CrossRef]
- Lovly, C.M.; Gupta, A.; Lipson, D.; Otto, G.; Brennan, T.; Chung, C.T.; Borinstein, S.C.; Ross, J.S.; Stephens, P.J.; Miller, V.A.; et al. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov. 2014, 4, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Yeap, B.Y.; Mino-Kenudson, M.; Digumarthy, S.R.; Costa, D.B.; Heist, R.S.; Solomon, B.; Stubbs, H.; Admane, S.; McDermott, U.; et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J. Clin. Oncol. 2009, 27, 4247–4253. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Engelman, J.A. ALK in lung cancer: Past, present, and future. J. Clin. Oncol. 2013, 31, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Heuckmann, J.M.; Balke-Want, H.; Malchers, F.; Peifer, M.; Sos, M.L.; Koker, M.; Meder, L.; Lovly, C.M.; Heukamp, L.C.; Pao, W.; et al. Differential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variants. Clin. Cancer Res. 2012, 18, 4682–4690. [Google Scholar] [CrossRef] [PubMed]
- Lamant, L.; Dastugue, N.; Pulford, K.; Delsol, G.; Mariame, B. A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood 1999, 93, 3088–3095. [Google Scholar] [PubMed]
- Cools, J.; Wlodarska, I.; Somers, R.; Mentens, N.; Pedeutour, F.; Maes, B.; De Wolf-Peeters, C.; Pauwels, P.; Hagemeijer, A.; Marynen, P. Identification of novel fusion partners of ALK, the anaplastic lymphoma kinase, in anaplastic large-cell lymphoma and inflammatory myofibroblastic tumor. Genes Chromosom. Cancer 2002, 34, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Lamant, L.; Gascoyne, R.D.; Duplantier, M.M.; Armstrong, F.; Raghab, A.; Chhanabhai, M.; Rajcan-Separovic, E.; Raghab, J.; Delsol, G.; Espinos, E. Non-muscle myosin heavy chain (MYH9): A new partner fused to ALK in anaplastic large cell lymphoma. Genes Chromosom. Cancer 2003, 37, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Meech, S.J.; McGavran, L.; Odom, L.F.; Liang, X.; Meltesen, L.; Gump, J.; Wei, Q.; Carlsen, S.; Hunger, S.P. Unusual childhood extramedullary hematologic malignancy with natural killer cell properties that contains tropomyosin 4—Anaplastic lymphoma kinase gene fusion. Blood 2001, 98, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Tort, F.; Pinyol, M.; Pulford, K.; Roncador, G.; Hernandez, L.; Nayach, I.; Kluin-Nelemans, H.C.; Kluin, P.; Touriol, C.; Delsol, G.; et al. Molecular characterization of a new ALK translocation involving moesin (MSN-ALK) in anaplastic large cell lymphoma. Lab. Investig. 2001, 81, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Tort, F.; Campo, E.; Pohlman, B.; Hsi, E. Heterogeneity of genomic breakpoints in MSN-ALK translocations in anaplastic large cell lymphoma. Hum. Pathol. 2004, 35, 1038–1041. [Google Scholar] [CrossRef] [PubMed]
- Colleoni, G.W.; Bridge, J.A.; Garicochea, B.; Liu, J.; Filippa, D.A.; Ladanyi, M. Atic-ALK: A novel variant ALK gene fusion in anaplastic large cell lymphoma resulting from the recurrent cryptic chromosomal inversion, inv(2)(p23q35). Am. J. Pathol. 2000, 156, 781–789. [Google Scholar] [CrossRef]
- Touriol, C.; Greenland, C.; Lamant, L.; Pulford, K.; Bernard, F.; Rousset, T.; Mason, D.Y.; Delsol, G. Further demonstration of the diversity of chromosomal changes involving 2p23 in ALK-positive lymphoma: 2 cases expressing ALK kinase fused to CLTCL (clathrin chain polypeptide-like). Blood 2000, 95, 3204–3207. [Google Scholar] [PubMed]
- Abate, F.; Todaro, M.; van der Krogt, J.A.; Boi, M.; Landra, I.; Machiorlatti, R.; Tabbo, F.; Messana, K.; Abele, C.; Barreca, A.; et al. A novel patient-derived tumorgraft model with TRAF1-ALK anaplastic large-cell lymphoma translocation. Leukemia 2015, 29, 1390–1401. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, L.; Bea, S.; Bellosillo, B.; Pinyol, M.; Falini, B.; Carbone, A.; Ott, G.; Rosenwald, A.; Fernandez, A.; Pulford, K.; et al. Diversity of genomic breakpoints in TFG-ALK translocations in anaplastic large cell lymphomas: Identification of a new TFG-ALK(XL) chimeric gene with transforming activity. Am. J. Pathol. 2002, 160, 1487–1494. [Google Scholar] [CrossRef]
- Lin, E.; Li, L.; Guan, Y.; Soriano, R.; Rivers, C.S.; Mohan, S.; Pandita, A.; Tang, J.; Modrusan, Z. Exon array profiling detects EML4-ALK fusion in breast, colorectal, and non-small cell lung cancers. Mol. Cancer Res. MCR 2009, 7, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Medico, E.; Russo, M.; Picco, G.; Cancelliere, C.; Valtorta, E.; Corti, G.; Buscarino, M.; Isella, C.; Lamba, S.; Martinoglio, B.; et al. The molecular landscape of colorectal cancer cell lines unveils clinically actionable kinase targets. Nat. Commun. 2015, 6, 7002. [Google Scholar] [CrossRef] [PubMed]
- Lipson, D.; Capelletti, M.; Yelensky, R.; Otto, G.; Parker, A.; Jarosz, M.; Curran, J.A.; Balasubramanian, S.; Bloom, T.; Brennan, K.W.; et al. Identification of new ALK and ret gene fusions from colorectal and lung cancer biopsies. Nat. Med. 2012, 18, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Aisner, D.L.; Nguyen, T.T.; Paskulin, D.D.; Le, A.T.; Haney, J.; Schulte, N.; Chionh, F.; Hardingham, J.; Mariadason, J.; Tebbutt, N.; et al. ROS1 and ALK fusions in colorectal cancer, with evidence of intratumoral heterogeneity for molecular drivers. Mol. Cancer Res. MCR 2014, 12, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Kang, S.Y.; Takeuchi, K.; Ko, Y.H. Identification of RANBP2-ALK fusion in ALK positive diffuse large B-cell lymphoma. Hematol. Oncol. 2014, 32, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Adam, P.; Katzenberger, T.; Seeberger, H.; Gattenlohner, S.; Wolf, J.; Steinlein, C.; Schmid, M.; Muller-Hermelink, H.K.; Ott, G. A case of a diffuse large B-cell lymphoma of plasmablastic type associated with the t(2;5)(p23;q35) chromosome translocation. Am. J. Surg. Pathol. 2003, 27, 1473–1476. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Nakasone, H.; Togashi, Y.; Sakata, S.; Tsuyama, N.; Baba, S.; Dobashi, A.; Asaka, R.; Tsai, C.C.; Chuang, S.S.; et al. ALK-positive large B-cell lymphoma: Identification of EML4-ALK and a review of the literature focusing on the ALK immunohistochemical staining pattern. Int. J. Hematol. 2016, 103, 399–408. [Google Scholar] [CrossRef] [PubMed]
- D’Amore, E.S.; Visco, C.; Menin, A.; Famengo, B.; Bonvini, P.; Lazzari, E. Stat3 pathway is activated in ALK-positive large B-cell lymphoma carrying sqstm1-ALK rearrangement and provides a possible therapeutic target. Am. J. Surg. Pathol. 2013, 37, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Onciu, M.; Behm, F.G.; Raimondi, S.C.; Moore, S.; Harwood, E.L.; Pui, C.H.; Sandlund, J.T. ALK-positive anaplastic large cell lymphoma with leukemic peripheral blood involvement is a clinicopathologic entity with an unfavorable prognosis. Report of three cases and review of the literature. Am. J. Clin. Pathol. 2003, 120, 617–625. [Google Scholar] [CrossRef] [PubMed]
- De Paepe, P.; Baens, M.; van Krieken, H.; Verhasselt, B.; Stul, M.; Simons, A.; Poppe, B.; Laureys, G.; Brons, P.; Vandenberghe, P.; et al. ALK activation by the cltc-ALK fusion is a recurrent event in large B-cell lymphoma. Blood 2003, 102, 2638–2641. [Google Scholar] [CrossRef] [PubMed]
- Van Roosbroeck, K.; Cools, J.; Dierickx, D.; Thomas, J.; Vandenberghe, P.; Stul, M.; Delabie, J.; De Wolf-Peeters, C.; Marynen, P.; Wlodarska, I. ALK-positive large B-cell lymphomas with cryptic sec31a-ALK and npm1-ALK fusions. Haematologica 2010, 95, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Du, X.L.; Hu, H.; Lin, D.C.; Xia, S.H.; Shen, X.M.; Zhang, Y.; Luo, M.L.; Feng, Y.B.; Cai, Y.; Xu, X.; et al. Proteomic profiling of proteins dysregulted in chinese esophageal squamous cell carcinoma. J. Mol. Med. (Berl.) 2007, 85, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Jazii, F.R.; Najafi, Z.; Malekzadeh, R.; Conrads, T.P.; Ziaee, A.A.; Abnet, C.; Yazdznbod, M.; Karkhane, A.A.; Salekdeh, G.H. Identification of squamous cell carcinoma associated proteins by proteomics and loss of β tropomyosin expression in esophageal cancer. World J. Gastroenterol. 2006, 12, 7104–7112. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Soda, M.; Togashi, Y.; Sugawara, E.; Hatano, S.; Asaka, R.; Okumura, S.; Nakagawa, K.; Mano, H.; Ishikawa, Y. Pulmonary inflammatory myofibroblastic tumor expressing a novel fusion, PPFIBP1-ALK: Reappraisal of anti-ALK immunohistochemistry as a tool for novel ALK fusion identification. Clin. Cancer Res. 2011, 17, 3341–3348. [Google Scholar] [CrossRef] [PubMed]
- Bridge, J.A.; Kanamori, M.; Ma, Z.; Pickering, D.; Hill, D.A.; Lydiatt, W.; Lui, M.Y.; Colleoni, G.W.; Antonescu, C.R.; Ladanyi, M.; et al. Fusion of the ALK gene to the clathrin heavy chain gene, CLTC, in inflammatory myofibroblastic tumor. Am. J. Pathol. 2001, 159, 411–415. [Google Scholar] [CrossRef]
- Debelenko, L.V.; Arthur, D.C.; Pack, S.D.; Helman, L.J.; Schrump, D.S.; Tsokos, M. Identification of cars-ALK fusion in primary and metastatic lesions of an inflammatory myofibroblastic tumor. Lab. Investig. 2003, 83, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, I.; Nilsson, T.; Domanski, H.A.; Isaksson, M.; Lindblom, P.; Mertens, F.; Mandahl, N. Fusion of the sec31l1 and ALK genes in an inflammatory myofibroblastic tumor. Int. J. Cancer 2006, 118, 1181–1186. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, B.; Perez-Atayde, A.; Hibbard, M.K.; Rubin, B.P.; Dal Cin, P.; Pinkus, J.L.; Pinkus, G.S.; Xiao, S.; Yi, E.S.; Fletcher, C.D.; et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am. J. Pathol. 2000, 157, 377–384. [Google Scholar] [CrossRef]
- Debiec-Rychter, M.; Marynen, P.; Hagemeijer, A.; Pauwels, P. ALK-atic fusion in urinary bladder inflammatory myofibroblastic tumor. Genes Chromosom. Cancer 2003, 38, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Hill, D.A.; Collins, M.H.; Morris, S.W.; Sumegi, J.; Zhou, M.; Zuppan, C.; Bridge, J.A. Fusion of ALK to the ran-binding protein 2 (RANBP2) gene in inflammatory myofibroblastic tumor. Genes Chromosom. Cancer 2003, 37, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yin, W.H.; Takeuchi, K.; Guan, H.; Huang, Y.H.; Chan, J.K. Inflammatory myofibroblastic tumor with RANBP2 and ALK gene rearrangement: A report of two cases and literature review. Diagn. Pathol. 2013, 8, 147. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.T.; Lee, J.C. An inflammatory myofibroblastic tumor in liver with ALK and RANBP2 gene rearrangement: Combination of distinct morphologic, immunohistochemical, and genetic features. Hum. Pathol. 2008, 39, 1854–1858. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.S.; Murphy, K.M.; Hawkins, A.L.; Cohen, J.S.; Long, P.P.; Perlman, E.J.; Griffin, C.A. RANBP2 and cltc are involved in ALK rearrangements in inflammatory myofibroblastic tumors. Cancer Genet. Cytogenet. 2007, 176, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Kim, P.; Keum, J.; Kim, S.N.; Choi, Y.S.; Do, I.G.; Lee, J.; Choi, S.J.; Kim, S.; Lee, J.E.; et al. Discovery of ALK-PTPN3 gene fusion from human non-small cell lung carcinoma cell line using next generation rna sequencing. Genes Chromosom. Cancer 2012, 51, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Soda, M.; Sakata, S.; Sugawara, E.; Hatano, S.; Asaka, R.; Nakajima, T.; Mano, H.; Takeuchi, K. KLC1-ALK: A novel fusion in lung cancer identified using a formalin-fixed paraffin-embedded tissue only. PLoS ONE 2012, 7, e31323. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Choi, Y.L.; Togashi, Y.; Soda, M.; Hatano, S.; Inamura, K.; Takada, S.; Ueno, T.; Yamashita, Y.; Satoh, Y.; et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin. Cancer Res. 2009, 15, 3143–3149. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.L.; Lira, M.E.; Hong, M.; Kim, R.N.; Choi, S.J.; Song, J.Y.; Pandy, K.; Mann, D.L.; Stahl, J.A.; Peckham, H.E.; et al. A novel fusion of tpr and ALK in lung adenocarcinoma. J. Thorac. Oncol. 2014, 9, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Tan, Z.P.; Zhu, X.; Crosby, K.; Haack, H.; Ren, J.M.; Beausoleil, S.; Moritz, A.; Innocenti, G.; Rush, J.; et al. Identification of anaplastic lymphoma kinase as a potential therapeutic target in ovarian cancer. Cancer Res. 2012, 72, 3312–3323. [Google Scholar] [CrossRef] [PubMed]
- Kusano, H.; Togashi, Y.; Akiba, J.; Moriya, F.; Baba, K.; Matsuzaki, N.; Yuba, Y.; Shiraishi, Y.; Kanamaru, H.; Kuroda, N.; et al. Two cases of renal cell carcinoma harboring a novel STRN-ALK fusion gene. Am. J. Surg. Pathol. 2016, 40, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, E.; Togashi, Y.; Kuroda, N.; Sakata, S.; Hatano, S.; Asaka, R.; Yuasa, T.; Yonese, J.; Kitagawa, M.; Mano, H.; et al. Identification of anaplastic lymphoma kinase fusions in renal cancer: Large-scale immunohistochemical screening by the intercalated antibody-enhanced polymer method. Cancer 2012, 118, 4427–4436. [Google Scholar] [CrossRef] [PubMed]
- Debelenko, L.V.; Raimondi, S.C.; Daw, N.; Shivakumar, B.R.; Huang, D.; Nelson, M.; Bridge, J.A. Renal cell carcinoma with novel VCL-ALK fusion: New representative of ALK-associated tumor spectrum. Mod. Pathol. 2011, 24, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Sukov, W.R.; Hodge, J.C.; Lohse, C.M.; Akre, M.K.; Leibovich, B.C.; Thompson, R.H.; Cheville, J.C. ALK alterations in adult renal cell carcinoma: Frequency, clinicopathologic features and outcome in a large series of consecutively treated patients. Mod. Pathol. 2012, 25, 1516–1525. [Google Scholar] [CrossRef] [PubMed]
- Marino-Enriquez, A.; Ou, W.B.; Weldon, C.B.; Fletcher, J.A.; Perez-Atayde, A.R. ALK rearrangement in sickle cell trait-associated renal medullary carcinoma. Genes Chromosom. Cancer 2011, 50, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, F.; Duplantier, M.M.; Trempat, P.; Hieblot, C.; Lamant, L.; Espinos, E.; Racaud-Sultan, C.; Allouche, M.; Campo, E.; Delsol, G.; et al. Differential effects of X-ALK fusion proteins on proliferation, transformation, and invasion properties of NIH3T3 cells. Oncogene 2004, 23, 6071–6082. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, F.; Lamant, L.; Hieblot, C.; Delsol, G.; Touriol, C. TPM3-ALK expression induces changes in cytoskeleton organisation and confers higher metastatic capacities than other ALK fusion proteins. Eur. J. Cancer 2007, 43, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Marino-Enriquez, A.; Dal Cin, P. ALK as a paradigm of oncogenic promiscuity: Different mechanisms of activation and different fusion partners drive tumors of different lineages. Cancer Genet. 2013, 206, 357–373. [Google Scholar] [CrossRef] [PubMed]
- De Brouwer, S.; De Preter, K.; Kumps, C.; Zabrocki, P.; Porcu, M.; Westerhout, E.M.; Lakeman, A.; Vandesompele, J.; Hoebeeck, J.; Van Maerken, T.; et al. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with mycn amplification. Clin. Cancer Res. 2010, 16, 4353–4362. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Schonherr, C.; Ruuth, K.; Eriksson, T.; Yamazaki, Y.; Ottmann, C.; Combaret, V.; Vigny, M.; Kamaraj, S.; Palmer, R.H.; Hallberg, B. The neuroblastoma ALK(I1250T) mutation is a kinase-dead RTK in vitro and in vivo. Transl. Oncol. 2011, 4, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Chand, D.; Yamazaki, Y.; Ruuth, K.; Schonherr, C.; Martinsson, T.; Kogner, P.; Attiyeh, E.F.; Maris, J.; Morozova, O.; Marra, M.A.; et al. Cell culture and drosophila model systems define three classes of anaplastic lymphoma kinase mutations in neuroblastoma. Dis. Models Mech. 2013, 6, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.Y.; Li, Q.; Lee, J.H.; Arango, M.E.; McDonnell, S.R.; Yamazaki, S.; Koudriakova, T.B.; Alton, G.; Cui, J.J.; Kung, P.P.; et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007, 67, 4408–4417. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.J.; Tran-Dube, M.; Shen, H.; Nambu, M.; Kung, P.P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-Met) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.G.; Zou, H.Y.; Arango, M.E.; Li, Q.; Lee, J.H.; McDonnell, S.R.; Yamazaki, S.; Alton, G.R.; Mroczkowski, B.; Los, G. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol. Cancer Ther. 2007, 6, 3314–3322. [Google Scholar] [CrossRef] [PubMed]
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.; Dezube, B.J.; Janne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Blackhall, F.; Ross Camidge, D.; Shaw, A.T.; Soria, J.C.; Solomon, B.J.; Mok, T.; Hirsh, V.; Janne, P.A.; Shi, Y.; Yang, P.C.; et al. Final results of the large-scale multinational trial profile 1005: Efficacy and safety of crizotinib in previously treated patients with advanced/metastatic ALK-positive non-small-cell lung cancer. ESMO Open 2017, 2, e000219. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti-Passerini, C.; Messa, C.; Pogliani, E.M. Crizotinib in anaplastic large-cell lymphoma. N. Engl. J. Med. 2011, 364, 775–776. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti-Passerini, C.; Orlov, S.; Zhang, L.; Braiteh, F.; Huang, H.; Esaki, T.; Horibe, K.; Ahn, J.S.; Beck, J.T.; Edenfield, W.J.; et al. Long-term effects of crizotinib in ALK-positive tumors (excluding NSCLC): A phase 1b open-label study. Am. J. Hematol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mosse, Y.P.; Lim, M.S.; Voss, S.D.; Wilner, K.; Ruffner, K.; Laliberte, J.; Rolland, D.; Balis, F.M.; Maris, J.M.; Weigel, B.J.; et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: A children’s oncology group phase 1 consortium study. Lancet Oncol. 2013, 14, 472–480. [Google Scholar] [CrossRef]
- Marsilje, T.H.; Pei, W.; Chen, B.; Lu, W.; Uno, T.; Jin, Y.; Jiang, T.; Kim, S.; Li, N.; Warmuth, M.; et al. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-n2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-n4-(2-(isopropylsulf onyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J. Med. Chem. 2013, 56, 5675–5690. [Google Scholar] [PubMed]
- Friboulet, L.; Li, N.; Katayama, R.; Lee, C.C.; Gainor, J.F.; Crystal, A.S.; Michellys, P.Y.; Awad, M.M.; Yanagitani, N.; Kim, S.; et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014, 4, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Galkin, A.V.; Melnick, J.S.; Kim, S.; Hood, T.L.; Li, N.; Li, L.; Xia, G.; Steensma, R.; Chopiuk, G.; Jiang, J.; et al. Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of npm-ALK. Proc. Natl. Acad. Sci. USA 2007, 104, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.; Spigel, D.; Felip, E.; de Marinis, F.; Ahn, M.J.; Groen, H.J.M.; Wakelee, H.A.; Hida, T.; Crino, L.; Nishio, M.; et al. Ascend-2: A single-arm, open-label, multicenter phase II study of ceritinib in adult patients (PTS) with ALK-rearranged (ALK plus) non-small cell lung cancer (NSCLC) previously treated with chemotherapy and crizotinib (CRZ). J. Clin. Oncol. 2015, 33. [Google Scholar] [CrossRef]
- Felip, E.; Orlov, S.; Park, K.; Yu, C.J.; Tsai, C.M.; Nishio, M.; Dols, M.C.; McKeage, M.J.; Su, W.C.; Mok, T.; et al. Ascend-3: A single-arm, open-label, multicenter phase ii study of ceritinib in ALKi-naive adult patients (PTS) with ALK-rearranged (ALK plus) non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2015, 33. [Google Scholar] [CrossRef]
- Soria, J.C.; Tan, D.S.W.; Chiari, R.; Wu, Y.L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.J.; et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ascend-4): A randomised, open-label, phase 3 study. Lancet 2017, 389, 917–929. [Google Scholar] [CrossRef]
- Novartis. Novartis Receives FDA Approval for Expanded Use of Zykadia® in First-Line ALK-Positive Metastatic Non-Small Cell Lung Cancer (NSCLC). Available online: https://www.novartis.com/news/media-releases/novartis-receives-fda-approval-expanded-use-zykadiar-first-line-ALK-positive (accessed on 12 December 2017).
- Cho, B.C.; Kim, D.W.; Bearz, A.; Laurie, S.A.; McKeage, M.; Borra, G.; Park, K.; Kim, S.W.; Ghosn, M.; Ardizzoni, A.; et al. Ascend-8: A randomized phase 1 study of ceritinib, 450 mg or 600 mg, taken with a low-fat meal versus 750 mg in fasted state in patients with anaplastic lymphoma kinase (ALK)-rearranged metastatic non-small cell lung cancer (NSCLC). J. Thorac. Oncol. 2017, 12, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, K.; Asoh, K.; Furuichi, N.; Ito, T.; Kawada, H.; Hara, S.; Ohwada, J.; Miyagi, T.; Kobayashi, T.; Takanashi, K.; et al. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (ch5424802). Bioorg. Med. Chem. 2012, 20, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Tsukaguchi, T.; Hiroshima, S.; Kodama, T.; Kobayashi, T.; Fukami, T.A.; Oikawa, N.; Tsukuda, T.; Ishii, N.; Aoki, Y. Ch5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011, 19, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Seto, T.; Kiura, K.; Nishio, M.; Nakagawa, K.; Maemondo, M.; Inoue, A.; Hida, T.; Yamamoto, N.; Yoshioka, H.; Harada, M.; et al. Ch5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001jp study): A single-arm, open-label, phase 1-2 study. Lancet Oncol. 2013, 14, 590–598. [Google Scholar] [CrossRef]
- Tamura, T.; Seto, T.; Nakagawa, K.; Maemondo, M.; Inoue, A.; Hida, T.; Yoshioka, H.; Harada, M.; Ohe, Y.; Nogami, N.; et al. Updated data of a phase 1/2 study (AF-001jp) of alectinib, a CNS-penetrant, highly selective ALK inhibitor in ALK-rearranged advanced NSCLC. Int. J. Radiat. Oncol. 2014, 90, S6. [Google Scholar] [CrossRef]
- Kodama, T.; Tsukaguchi, T.; Yoshida, M.; Kondoh, O.; Sakamoto, H. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett. 2014, 351, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Friboulet, L.; Koike, S.; Lockerman, E.L.; Khan, T.M.; Gainor, J.F.; Iafrate, A.J.; Takeuchi, K.; Taiji, M.; Okuno, Y.; et al. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin. Cancer Res. 2014, 20, 5686–5696. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Savooji, J.; Liu, D. Second- and third-generation ALK inhibitors for non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.S.; Liu, S.; Zou, D.; Thomas, M.; Wang, Y.; Zhou, T.; Romero, J.; Kohlmann, A.; Li, F.; Qi, J.; et al. Discovery of brigatinib (ap26113), a phosphine oxide-containing, potent, orally active inhibitor of anaplastic lymphoma kinase. J. Med. Chem. 2016, 59, 4948–4964. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Anjum, R.; Squillace, R.; Nadworny, S.; Zhou, T.; Keats, J.; Ning, Y.; Wardwell, S.D.; Miller, D.; Song, Y.; et al. The potent ALK inhibitor ap26113 can overcome mechanisms of resistance to first- and second-generation ALK TKIs in preclinical models. Clin. Cancer Res. 2015. [Google Scholar] [CrossRef]
- Fontana, D.; Ceccon, M.; Gambacorti-Passerini, C.; Mologni, L. Activity of second-generation ALK inhibitors against crizotinib-resistant mutants in an npm-ALK model compared to EML4-ALK. Cancer Med. 2015, 4, 953–965. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Anjum, R.; Squillace, R.; Nadworny, S.; Zhou, T.; Keats, J.; Ning, Y.; Wardwell, S.D.; Miller, D.; Song, Y.; et al. The potent ALK inhibitor brigatinib (ap26113) overcomes mechanisms of resistance to first- and second-generation ALK inhibitors in preclinical models. Clin. Cancer Res. 2016, 22, 5527–5538. [Google Scholar] [CrossRef] [PubMed]
- Gettinger, S.N.; Bazhenova, L.A.; Langer, C.J.; Salgia, R.; Gold, K.A.; Rosell, R.; Shaw, A.T.; Weiss, G.J.; Tugnait, M.; Narasimhan, N.I.; et al. Activity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: A single-arm, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 1683–1696. [Google Scholar] [CrossRef]
- Staff, N. FDA grants brigatinib accelerated approval for metastatic non-small cell lung cancer. In FDA Grants Brigatinib Accelerated Approval for Metastatic Non-Small Cell Lung Cancer; National Cancer Institute: Rockville, MD, USA, 2017; Volume 2017. [Google Scholar]
- Johnson, T.W.; Richardson, P.F.; Bailey, S.; Brooun, A.; Burke, B.J.; Collins, M.R.; Cui, J.J.; Deal, J.G.; Deng, Y.L.; Dinh, D.; et al. Discovery of (10R)-7-Amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(m etheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ros1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J. Med. Chem. 2014, 57, 4720–4744. [Google Scholar] [PubMed]
- Zou, H.Y.; Friboulet, L.; Kodack, D.P.; Engstrom, L.D.; Li, Q.; West, M.; Tang, R.W.; Wang, H.; Tsaparikos, K.; Wang, J.; et al. PF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical models. Cancer Cell 2015, 28, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Kennedy, E.; Sequist, L.V.; Brastianos, P.K.; Goodwin, K.E.; Stevens, S.; Wanat, A.C.; Stober, L.L.; Digumarthy, S.R.; Engelman, J.A.; et al. Clinical activity of alectinib in advanced ret-rearranged non-small cell lung cancer. J. Thorac. Oncol. 2016, 11, 2027–2032. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, F.; Keats, J.; Ning, Y.; Wardwell, S.D.; Moran, L.; Mohemmad, Q.K.; Anjum, R.; Wang, Y.; Zhu, X.; et al. Ap26113, a potent ALK inhibitor, overcomes mutations in EML4ALK that confer resistance to PF-02341066 (PF1066). Cancer Res. 2014. [Google Scholar] [CrossRef]
- Squillace, R.M.; Anjum, R.; Miller, D.; Vodala, S.; Moran, L.; Wang, F.; Clackson, T.; Garner, A.P.; Rivera, V.M. Ap26113 possesses pan-inhibitory activity versus crizotinib-resistant ALK mutants and oncogenic ROS1 fusions. Cancer Res. 2014. [Google Scholar] [CrossRef]
- Kim, D.W.; Tiseo, M.; Ahn, M.J.; Reckamp, K.L.; Holmskov Hansen, K.; Kim, S.W.; Huber, R.M.; West, H.J.; Groen, H.J.; Hochmair, M.J.; et al. Brigatinib (BRG) in patients (PTS) with crizotinib (CRZ)-refractory ALK+ non-small cell lung cancer (NSCLC): First report of effi cacy and safety from a pivotal randomized phase (PH) 2 trial (α). Clin. Oncol. 2016, 34. [Google Scholar] [CrossRef]
- Zou, H.Y.; Li, Q.; Engstrom, L.D.; West, M.; Appleman, V.; Wong, K.A.; McTigue, M.; Deng, Y.L.; Liu, W.; Brooun, A.; et al. PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations. Proc. Natl. Acad. Sci. USA 2015, 112, 3493–3498. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Ou, S.H.; Felip, E.; Bauer, T.M.; Besse, B.; Gadgeel, S.M.; Camidge, D.R.; Lin, C.C.; Seto, T.; Soo, R.A.; et al. Efficacy and safety of lorlatinib in patients (PTS) with ALK+ non-small cell lung cancer (NSCLC) with one or more prior ALK tyrosine kinase inhibitor (TKI): A phase I/II study. J. Clin. Oncol. 2017. [Google Scholar] [CrossRef]
- Solomon, B.; Shaw, A.; Ou, S.; Besse, B.; Felip, E.; Bauer, T.; Soo, R.; Bearz, A.; Lin, C.; Clancy, J.; et al. Oa 05.06 phase 2 study of lorlatinib in patients with advanced ALK+/ROS1+ non-small- cell lung cancer. J. Thorac. Oncol. 2017, 12, S1756. [Google Scholar] [CrossRef]
- Ardini, E.; Menichincheri, M.; Banfi, P.; Bosotti, R.; De Ponti, C.; Pulci, R.; Ballinari, D.; Ciomei, M.; Texido, G.; Degrassi, A.; et al. Entrectinib, a PAN-TRK, ROS1, and ALK inhibitor with activity in multiple molecularly defined cancer indications. Mol. Cancer Ther. 2016, 15, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Siena, S.; Ou, S.I.; Patel, M.; Ahn, M.J.; Lee, J.; Bauer, T.M.; Farago, A.F.; Wheler, J.J.; Liu, S.V.; et al. Safety and antitumor activity of the multitargeted PAN-TRK, ROS1, and ALK inhibitor entrectinib: Combined results from two phase I trials (ALKa-372-001 and startrk-1). Cancer Discov. 2017, 7, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Ueno, Y.; Konagai, S.; Fushiki, H.; Shimada, I.; Kondoh, Y.; Saito, R.; Mori, K.; Shindou, N.; Soga, T.; et al. The selective anaplastic lymphoma receptor tyrosine kinase inhibitor asp3026 induces tumor regression and prolongs survival in non-small cell lung cancer model mice. Mol. Cancer Ther. 2014, 13, 329–340. [Google Scholar] [CrossRef] [PubMed]
- George, S.K.; Vishwamitra, D.; Manshouri, R.; Shi, P.; Amin, H.M. The ALK inhibitor asp3026 eradicates NPM-ALK(+) T-cell anaplastic large-cell lymphoma in vitro and in a systemic xenograft lymphoma model. Oncotarget 2014, 5, 5750–5763. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; LoRusso, P.; Maitland, M.L.; Ou, S.H.; Bahceci, E.; Ball, H.A.; Park, J.W.; Yuen, G.; Tolcher, A. First-in-human, open-label dose-escalation and dose-expansion study of the safety, pharmacokinetics, and antitumor effects of an oral ALK inhibitor asp3026 in patients with advanced solid tumors. J. Hematol. Oncol. 2016, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Lovly, C.M.; Heuckmann, J.M.; de Stanchina, E.; Chen, H.; Thomas, R.K.; Liang, C.; Pao, W. Insights into ALK-driven cancers revealed through development of novel ALK tyrosine kinase inhibitors. Cancer Res. 2011, 71, 4920–4931. [Google Scholar] [CrossRef] [PubMed]
- Reckamp, K.L.; Infante, J.R.; Blumenschein, G.R.; Wakelee, H.; Carter, C.A.; Gockerman, J.P.; Lovly, C.; Dukart, G.; Harrow, K.; Liang, C.; et al. Phase I/II trial of x-396, a novel anaplastic lymphoma kinase (ALK) inhibitor, in patients with ALK+ non-small cell lung cancer (NSCLC). J. Thorac. Oncol. 2016, 11, S36–S37. [Google Scholar] [CrossRef]
- Cheng, M.; Quail, M.R.; Gingrich, D.E.; Ott, G.R.; Lu, L.; Wan, W.; Albom, M.S.; Angeles, T.S.; Aimone, L.D.; Cristofani, F.; et al. Cep-28122, a highly potent and selective orally active inhibitor of anaplastic lymphoma kinase with antitumor activity in experimental models of human cancers. Mol. Cancer Ther. 2012, 11, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Arkenau, H.T.; Sachdev, J.C.; Mita, M.M.; Dziadziuszko, R.; Lin, C.C.; Yang, J.C.; Infante, J.R.; Anthony, S.P.; Voskoboynik, M.; Su, W.C.; et al. Phase (PH) 1/2a study of TSR-011, a potent inhibitor of ALK and TRK, in advanced solid tumors including crizotinib-resistant ALK positive non-small cell lung cancer. J. Clin. Oncol. 2015. [Google Scholar] [CrossRef]
- Gettinger, S.N.; Zhang, S.; Hodgson, J.G.; Bazhenova, L.; Burgers, S.; Kim, D.W.; Tan, D.S.; Koh, H.A.; Ho, J.C.; Viteri Ramirez, S.; et al. Activity of brigatinib (BRG) in crizotinib (CRZ) resistant patients (PTS) according to ALK mutation status. J. Clin. Oncol. 2016, 34. [Google Scholar] [CrossRef]
- Gainor, J.F.; Chi, A.S.; Logan, J.; Hu, R.; Oh, K.S.; Brastianos, P.K.; Shih, H.A.; Shaw, A.T. Alectinib dose escalation reinduces central nervous system responses in patients with anaplastic lymphoma kinase-positive non-small cell lung cancer relapsing on standard dose alectinib. J. Thorac. Oncol. 2016, 11, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.L.; Soda, M.; Yamashita, Y.; Ueno, T.; Takashima, J.; Nakajima, T.; Yatabe, Y.; Takeuchi, K.; Hamada, T.; Haruta, H.; et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med. 2010, 363, 1734–1739. [Google Scholar] [CrossRef] [PubMed]
- Zuccotto, F.; Ardini, E.; Casale, E.; Angiolini, M. Through the “gatekeeper door”: Exploiting the active kinase conformation. J. Med. Chem. 2010, 53, 2681–2694. [Google Scholar] [CrossRef] [PubMed]
- Azam, M.; Seeliger, M.A.; Gray, N.S.; Kuriyan, J.; Daley, G.Q. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat. Struct. Mol. Biol. 2008, 15, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.Y.; Ji, F.Q. A molecular dynamics investigation on the crizotinib resistance mechanism of C1156Y mutation in ALK. Biochem. Biophys. Res. Commun. 2012, 423, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Ceccon, M.; Mologni, L.; Bisson, W.; Scapozza, L.; Gambacorti-Passerini, C. Crizotinib-resistant npm-ALK mutants confer differential sensitivity to unrelated ALK inhibitors. Mol. Cancer Res. MCR 2013, 11, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Okuda, K.; Zheng, W.; Butrynski, J.; Capelletti, M.; Wang, L.; Gray, N.S.; Wilner, K.; Christensen, J.G.; Demetri, G.; et al. The neuroblastoma-associated f1174l ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010, 70, 10038–10043. [Google Scholar] [CrossRef] [PubMed]
- Bresler, S.C.; Wood, A.C.; Haglund, E.A.; Courtright, J.; Belcastro, L.T.; Plegaria, J.S.; Cole, K.; Toporovskaya, Y.; Zhao, H.; Carpenter, E.L.; et al. Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma. Sci. Transl. Med. 2011, 3, 108ra114. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.H.; Klempner, S.J.; Greenbowe, J.R.; Azada, M.; Schrock, A.B.; Ali, S.M.; Ross, J.S.; Stephens, P.J.; Miller, V.A. Identification of a novel hip1-ALK fusion variant in non-small-cell lung cancer (NSCLC) and discovery of ALKI1171 (I1171N/S) mutations in two ALK-rearranged NSCLC patients with resistance to alectinib. J. Thorac. Oncol. 2014, 9, 1821–1825. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Koivunen, J.; Ogino, A.; Yanagita, M.; Nikiforow, S.; Zheng, W.; Lathan, C.; Marcoux, J.P.; Du, J.; Okuda, K.; et al. A novel ALK secondary mutation and egfr signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011, 71, 6051–6060. [Google Scholar] [CrossRef] [PubMed]
- Bossi, R.T.; Saccardo, M.B.; Ardini, E.; Menichincheri, M.; Rusconi, L.; Magnaghi, P.; Orsini, P.; Avanzi, N.; Borgia, A.L.; Nesi, M.; et al. Crystal structures of anaplastic lymphoma kinase in complex with atp competitive inhibitors. Biochemistry 2010, 49, 6813–6825. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci. Transl. Med. 2012, 4, 120ra117. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Pilling, A.B.; Aisner, D.L.; Kutateladze, T.G.; Le, A.T.; Weickhardt, A.J.; Kondo, K.L.; Linderman, D.J.; Heasley, L.E.; Franklin, W.A.; et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Toyokawa, G.; Inamasu, E.; Shimamatsu, S.; Yoshida, T.; Nosaki, K.; Hirai, F.; Yamaguchi, M.; Seto, T.; Takenoyama, M.; Ichinose, Y. Identification of a novel ALKG1123S mutation in a patient with ALK-rearranged non-small-cell lung cancer exhibiting resistance to ceritinib. J. Thorac. Oncol. 2015, 10, e55–e57. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.-H.I.; Schrock, A.B.; Gowen, K.; Stephens, P.J.; Ross, J.S.; Johnson, M.L.; Lovly, C.M.; Ali, S.M.; Miller, V.A.; Shaw, A.T. Association of ALK resistance mutations by EML4-ALK variant (V3 vs. Non-V3) in ALK+ non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2017, 35, 9010. [Google Scholar] [CrossRef]
- Heuckmann, J.M.; Holzel, M.; Sos, M.L.; Heynck, S.; Balke-Want, H.; Koker, M.; Peifer, M.; Weiss, J.; Lovly, C.M.; Grutter, C.; et al. ALK mutations conferring differential resistance to structurally diverse ALK inhibitors. Clin. Cancer Res. 2011, 17, 7394–7401. [Google Scholar] [CrossRef] [PubMed]
- Michels, S.Y.F.; Scheel, A.H.; Wündisch, T.; Heuckmann, J.M.; Menon, R.; Puesken, M.; Kobe, C.; Pasternack, H.; Heydt, C.; Scheffler, M.; et al. ALKG1269A mutation as a potential mechanism of acquired resistance to crizotinib in an ALK-rearranged inflammatory myofibroblastic tumor. Precis. Oncol. 2017, 1, 4. [Google Scholar] [CrossRef]
- Wang, H.Y.; Ho, C.C.; Shih, J.Y. Multiple acquired resistance mutations of the ALK tyrosine kinase domain after sequential use of ALK inhibitors. J. Thorac. Oncol. 2017, 12, e49–e51. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Sakashita, T.; Yanagitani, N.; Ninomiya, H.; Horiike, A.; Friboulet, L.; Gainor, J.F.; Motoi, N.; Dobashi, A.; Sakata, S.; et al. P-glycoprotein mediates ceritinib resistance in anaplastic lymphoma kinase-rearranged non-small cell lung cancer. eBioMedicine 2016, 3, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Ceccon, M.; Mologni, L.; Giudici, G.; Piazza, R.; Pirola, A.; Fontana, D.; Gambacorti-Passerini, C. Treatment efficacy and resistance mechanisms using the second-generation ALK inhibitor ap26113 in human npm-ALK-positive anaplastic large cell lymphoma. Mol. Cancer Res. MCR 2015, 13, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Bazhenova, L.; Hodgson, J.G.; Langer, C.J.; Simon, G.R.; Gettinger, S.N.; Ou, S.-H.I.; Reckamp, K.L.; West, H.J.; Chiappori, A.; Koh, H.A.; et al. Activity of brigatinib (BRG) in crizotinib (CRZ)-resistant ALK+ NSCLC patients (PTS) according to ALK plasma mutation status. J. Clin. Oncol. 2017, 35, 9065. [Google Scholar]
- Tchekmedyian, N.; Ali, S.M.; Miller, V.A.; Haura, E.B. Acquired ALK l1152r mutation confers resistance to ceritinib and predicts response to alectinib. J. Thorac. Oncol. 2016, 11, e87–e88. [Google Scholar] [CrossRef] [PubMed]
- Ignatius Ou, S.H.; Azada, M.; Hsiang, D.J.; Herman, J.M.; Kain, T.S.; Siwak-Tapp, C.; Casey, C.; He, J.; Ali, S.M.; Klempner, S.J.; et al. Next-generation sequencing reveals a novel NSCLCALK f1174v mutation and confirms ALKG1202R mutation confers high-level resistance to alectinib (ch5424802/ro5424802) in ALK-rearranged NSCLC patients who progressed on crizotinib. J. Thorac. Oncol. 2014, 9, 549–553. [Google Scholar] [PubMed]
- Shaw, A.T.; Engelman, J.A. Crizotinib resensitization by compound mutation. N. Engl. J. Med. 2016, 374, 1790–1791. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.D.; Rajan, S.S.; Liang, W.S.; Pongtornpipat, P.; Groysman, M.J.; Tapia, E.O.; Peters, T.L.; Cuyugan, L.; Adkins, J.; Rimsza, L.M.; et al. Evidence suggesting that discontinuous dosing of ALK kinase inhibitors may prolong control of ALK+ tumors. Cancer Res. 2015, 75, 2916–2927. [Google Scholar] [CrossRef] [PubMed]
- Ceccon, M.; Merlo, M.E.B.; Mologni, L.; Poggio, T.; Varesio, L.M.; Menotti, M.; Bombelli, S.; Rigolio, R.; Manazza, A.D.; Di Giacomo, F.; et al. Excess of NPM-ALK oncogenic signaling promotes cellular apoptosis and drug dependency. Oncogene 2016, 35, 3854–3865. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, M.; Yasuda, H.; Tani, T.; Hamamoto, J.; Arai, D.; Ishioka, K.; Ohgino, K.; Nukaga, S.; Hirano, T.; Kawada, I.; et al. Overcoming EGFR bypass signal-induced acquired resistance to ALK tyrosine kinase inhibitors in ALK-translocated lung cancer. Mol. Cancer Res. MCR 2017, 15, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Wilson, F.H.; Johannessen, C.M.; Piccioni, F.; Tamayo, P.; Kim, J.W.; Van Allen, E.M.; Corsello, S.M.; Capelletti, M.; Calles, A.; Butaney, M.; et al. A functional landscape of resistance to ALK inhibition in lung cancer. Cancer Cell 2015, 27, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Hrustanovic, G.; Olivas, V.; Pazarentzos, E.; Tulpule, A.; Asthana, S.; Blakely, C.M.; Okimoto, R.A.; Lin, L.; Neel, D.S.; Sabnis, A.; et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat. Med. 2015, 21, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Crystal, A.S.; Shaw, A.T.; Sequist, L.V.; Friboulet, L.; Niederst, M.J.; Lockerman, E.L.; Frias, R.L.; Gainor, J.F.; Amzallag, A.; Greninger, P.; et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014, 346, 1480–1486. [Google Scholar] [CrossRef] [PubMed]
- Laimer, D.; Dolznig, H.; Kollmann, K.; Vesely, P.W.; Schlederer, M.; Merkel, O.; Schiefer, A.I.; Hassler, M.R.; Heider, S.; Amenitsch, L.; et al. Pdgfr blockade is a rational and effective therapy for NPM-ALK-driven lymphomas. Nat. Med. 2012, 18, 1699–1704. [Google Scholar] [CrossRef] [PubMed]
- Lovly, C.M.; McDonald, N.T.; Chen, H.; Ortiz-Cuaran, S.; Heukamp, L.C.; Yan, Y.; Florin, A.; Ozretic, L.; Lim, D.; Wang, L.; et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat. Med. 2014, 20, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Kim, W.S.; Choi, Y.J.; Choi, C.M.; Rho, J.K.; Lee, J.C. Epithelial-mesenchymal transition leads to crizotinib resistance in H2228 lung cancer cells with EML4-ALK translocation. Mol. Oncol. 2013, 7, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.J.; Cho, B.C.; Kim, H.R.; Lee, H.J.; Shim, H.S. A case of ALK-rearranged adenocarcinoma with small cell carcinoma-like transformation and resistance to crizotinib. J. Thorac. Oncol. 2016, 11, e55–e58. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.C.; Liao, X.H.; Wang, W.X.; Xu, C.W.; Zhuang, W.; Zhong, L.H.; Du, K.Q.; Chen, Y.P.; Chen, G.; Fang, M.Y. Patients harboring ALK rearrangement adenocarcinoma after acquired resistance to crizotinib and transformation to small-cell lung cancer: A case report. OncoTargets Ther. 2017, 10, 3187–3192. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Ikushima, S.; Ono, R.; Awano, N.; Kondo, K.; Furuhata, Y.; Fukumoto, K.; Kumasaka, T. Transformation to small-cell lung cancer as a mechanism of acquired resistance to crizotinib and alectinib. Jpn. J. Clin. Oncol. 2016, 46, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Fujita, S.; Masago, K.; Katakami, N.; Yatabe, Y. Transformation to sclc after treatment with the ALK inhibitor alectinib. J. Thorac. Oncol. 2016, 11, e67–e72. [Google Scholar] [CrossRef] [PubMed]
- Takegawa, N.; Hayashi, H.; Iizuka, N.; Takahama, T.; Ueda, H.; Tanaka, K.; Takeda, M.; Nakagawa, K. Transformation of ALK rearrangement-positive adenocarcinoma to small-cell lung cancer in association with acquired resistance to alectinib. Ann. Oncol. 2016, 27, 953–955. [Google Scholar] [CrossRef] [PubMed]
- Levacq, D.; D’Haene, N.; de Wind, R.; Remmelink, M.; Berghmans, T. Histological transformation of ALK rearranged adenocarcinoma into small cell lung cancer: A new mechanism of resistance to ALK inhibitors. Lung Cancer 2016, 102, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. Met amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to egfr inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [PubMed]
- Gouji, T.; Takashi, S.; Mitsuhiro, T.; Yukito, I. Crizotinib can overcome acquired resistance to ch5424802: Is amplification of the met gene a key factor? J. Thorac. Oncol. 2014, 9, e27–e28. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crino, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Lou, Y.; Pellissier, J.; Burke, T.; Liu, F.X.; Xu, R.; Velcheti, V. Cost-effectiveness of pembrolizumab versus docetaxel for the treatment of previously treated PD-L1 positive advanced NSCLC patients in the united states. J. Med. Econ. 2017, 20, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Brahmer, J.R.; Juergens, R.A.; Borghaei, H.; Gettinger, S.; Chow, L.Q.; Gerber, D.E.; Laurie, S.A.; Goldman, J.W.; et al. Nivolumab in combination with platinum-based doublet chemotherapy for first-line treatment of advanced non-small-cell lung cancer. J. Clin. Oncol. 2016, 34, 2969–2979. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Health-related quality-of-life results for pembrolizumab versus chemotherapy in advanced, PD-L1-positive NSCLC (keynote-024): A multicentre, international, randomised, open-label phase 3 trial. Lancet Oncol. 2017, 18, 1600–1609. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Mazieres, J.; Planchard, D.; Stinchcombe, T.E.; Dy, G.K.; Antonia, S.J.; Horn, L.; Lena, H.; Minenza, E.; Mennecier, B.; et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (checkmate 063): A phase 2, single-arm trial. Lancet Oncol. 2015, 16, 257–265. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody mpdl3280a in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Song, Y.; Liu, D. Recent development in clinical applications of PD-1 and PD-L1 antibodies for cancer immunotherapy. J. Hematol. Oncol. 2017, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Govindan, R.; Ding, L.; Griffith, M.; Subramanian, J.; Dees, N.D.; Kanchi, K.L.; Maher, C.A.; Fulton, R.; Fulton, L.; Wallis, J.; et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 2012, 150, 1121–1134. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Shaw, A.T.; Sequist, L.V.; Fu, X.; Azzoli, C.G.; Piotrowska, Z.; Huynh, T.G.; Zhao, L.; Fulton, L.; Schultz, K.R.; et al. Egfr mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: A retrospective analysis. Clin. Cancer Res. 2016, 22, 4585–4593. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Mastini, C.; Martinengo, C.; Inghirami, G.; Chiarle, R. Anaplastic lymphoma kinase: An oncogene for tumor vaccination. J. Mol. Med. 2009, 87, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Pulford, K.; Falini, B.; Banham, A.H.; Codrington, D.; Roberton, H.; Hatton, C.; Mason, D.Y. Immune response to the ALK oncogenic tyrosine kinase in patients with anaplastic large-cell lymphoma. Blood 2000, 96, 1605–1607. [Google Scholar] [PubMed]
- Awad, M.M.; Mastini, C.; Blasco, R.B.; Mologni, L.; Voena, C.; Mussolin, L.; Mach, S.L.; Adeni, A.E.; Lydon, C.A.; Sholl, L.M.; et al. Epitope mapping of spontaneous autoantibodies to anaplastic lymphoma kinase (ALK) in non-small cell lung cancer. Oncotarget 2017, 8, 92265–92274. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Martinengo, C.; Mastini, C.; Ambrogio, C.; D’Escamard, V.; Forni, G.; Inghirami, G. The anaplastic lymphoma kinase is an effective oncoantigen for lymphoma vaccination. Nat. Med. 2008, 14, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Voena, C.; Menotti, M.; Mastini, C.; Di Giacomo, F.; Longo, D.L.; Castella, B.; Merlo, M.E.B.; Ambrogio, C.; Wang, Q.; Minero, V.G.; et al. Efficacy of a cancer vaccine against ALK-rearranged lung tumors. Cancer Immunol. Res. 2015, 3, 1333–1343. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cancer Type | ALK Fusion Partner (Chromosomal Localization) | Frequency % | References |
|---|---|---|---|
| ALCL | NPM1 (5q35.1) TPM3 (1q21.3) ATIC (2q35) TFG (3q12.2) TRAF1 (9q33.2) CLTC (17q23.1) RNF213 (17q25.3) TPM4 (19p13.1) MYH9 (22q12.3) MSN (Xq12) Aditional rare rearrangements | ~55% (in adults) | [36,48,49,50,51,52,53,54,55,56,57] |
| Breast cancer | EML4 (2p21) | N.D. | [58] |
| Colorectal cancer | EML4 (2p21) WDCP (2p23.3) | <1% | [58,59,60,61] |
| DLBCL | RANBP2 (2q13) EML4 (2p21) SEC31A (4q21.22) SQSTM1 (5q35) NPM1 (5q35.1) | <1% | [62,63,64,65,66,67,68] |
| Esophageal cancer | TPM4 (19p13.1) | N.D. | [69,70] |
| IMT | TPM3 (1q21.3) RANBP2 (2q13) ATIC (2q35) SEC31A (4q21.22) CARS (11p15.4) PPFIBP1 (12p11) CLTC (17q23.1) TPM4 (19p13.1) | Up to 50% | [43,49,71,72,73,74,75,76,77,78,79,80] |
| NSCLC | EML4 (2p21) TPR (1q31.1) CRIM1 (2p22.2) STRN (2p22.1) TFG (3q12.2) HIP1 (7q11.23) PTPN3 (9q31) KIF5B (10p11.22) KLC1 (14q32.3) CLTC (17q23.1) | 3–7% | [7,44,81,82,83,84] |
| Ovarian cancer | FN1 (2q35) | N.D. | [85] |
| RCC | VCL (10q22.2) TPM3 (1q21.2) EML4 (2p21) STRN (2p22.2) | <1% | [86,87,88,89] |
| RMC | VCL (10q22.2) | N.D. | [90] |
| Inhibitor | Targeted Kinase/s | Activity against Mutant Forms | Clinical Evidence | Brain Penetrance | References |
|---|---|---|---|---|---|
| Crizotinib * (Xalkori–Pfizer) | ALKc-MET sROS1 | EML4-ALKL1198F | Phase I Phase II Phase III s(Complete) | No | [9,10,101,102] |
| Ceritinib * s(Zykadia–Novartis) | ALK IGR-1R INSR STK22D | EML4-ALKI1171T/N, L1196M, S1206C/Y, G1269A/S | Phase I Phase II Phase III (NCT02393625) | Yes | [11,109,110,111] |
| Alectinib * (Alecensa–Roche) | ALK LTK GAK | EML4-ALKL1152P/R, C1156Y/T, L1196M, F1174C/Y, S1206C/YDCTN1-ALKG1269/S | Phase I Phase II Phase III (NCT02075840) | Yes | [14,116,117,127,129,130] |
| Brigatinib * (AP26113-Ariad) | ALK ROS1 | EML4-ALKI1151Tins, C1156Y/T, L1196M, L1152P/R, F1174C/L/V, G1269A/S1 EML4-ALKG1202R | Phase I Phase II Phase III (NCT02094573) | Yes | [16,121,123,124,125,131,132,133] |
| PF-06463922 (Lorlatinib-Pfizer) | ALK ROS1 | ROS1G2032R ROS1L2026M EML4-ALKL1196M, G1269A,S1206Y,C1156Y,F1174L,L1152R,1151Tins | Phase I Phase II (NCT01970865) Phase III (NCT03052608) | Yes (NCT02927340) | [8,127,134,135,136] |
| RXDX-101 (Entrectinib-Ignyta) | ALK ROS1 TrkA TrkB TrkC | EML4-ALKC1156Y, L1196M | Phase I (ALKA-372-001 and STARTRK-1; NCT02097810) | Yes | [137,138] |
| ASP3026 (Astellas Pharma) | ALK ACK ROS1 | EML4-ALKL1196M NPM-ALKI231N NPM-ALKL256Q | Phase I (NCT01284192) | N.D. | [139,140,141] |
| X-376 and X-396 (Xcovery) | ALK MET | EML4-ALKL1196M, C1156Y | Phase I/II (X-396) (NCT01625234) | Yes | [142,143] |
| CEP-28122 (Teva) | ALK FAK | N.D. | Phase I (NCT01922752) | N.D. | [144] |
| TSR-011 (Tesaro) | ALK TrkA TrkB TrkC | N.D. | Phase I/IIa (NCT02048488) | N.D. | [145] |
| TKI | Sensitive Mutants | Resistant Mutants | Disease | Evidence (In Vitro/In Vivo/Clinical) | Reference |
|---|---|---|---|---|---|
| Crizotinib | L1198F | I1151Tins L1152R C1156Y I1171T/N F1174L L1196M L1196Q L1198P G1202R D1203N S1206Y G1269A | NSCLC NSCLC NSCLC NSCLC IMT NSCLC NSCLC EML4-ALK BaF3 cells NSCLC NSCLC NSCLC NSCLC, IMT | Clinical Clinical Clinical Clinical Clinical Clinical Clinical In vitro Clinical Clinical Clinical Clinical | [158] [156] [148] [160] [156] [148] [161] [162] [158] [161] [158] [159,163] |
| Ceritinib | G1269A, I1171T, S1206Y, L1196M | R1275Q L1152P/R D1203 G1202R F1174C/V L1198F C1156Y/T | Neuroblastoma NSCLC NSCLC NSCLC NSCLC NSCLC NSCLC | In vitro In vitro Clinical Clinical Clinical In vitro In vitro | [94] [107] [164] [107] [107] [165] [107] |
| Alectinib | G1269A, S1206Y, L1152R, F1174L, 1151Tins | I1171T V1180L G1202R | NSCLC NSCLC NSCLC | Clinical In vitro Clinical | [119] [155] |
| Brigatinib | G1269A, S1206Y, L1152R, F1174C, 1151Tins, I1171T, D1203N, E1210K, F1245C | F1174V+L1198F G1202R S1206C/F | ALCL NSCLC NSCLC | In vitro Clinical Clinical | [166] [167] |
| Lorlatinib | L1196M, G1202R, G1269A | L1198F | NSCLC | Clinical | [147] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, G.G.; Mota, I.; Mologni, L.; Patrucco, E.; Gambacorti-Passerini, C.; Chiarle, R. Tumor Resistance against ALK Targeted Therapy-Where It Comes From and Where It Goes. Cancers 2018, 10, 62. https://doi.org/10.3390/cancers10030062
Sharma GG, Mota I, Mologni L, Patrucco E, Gambacorti-Passerini C, Chiarle R. Tumor Resistance against ALK Targeted Therapy-Where It Comes From and Where It Goes. Cancers. 2018; 10(3):62. https://doi.org/10.3390/cancers10030062
Chicago/Turabian StyleSharma, Geeta Geeta, Ines Mota, Luca Mologni, Enrico Patrucco, Carlo Gambacorti-Passerini, and Roberto Chiarle. 2018. "Tumor Resistance against ALK Targeted Therapy-Where It Comes From and Where It Goes" Cancers 10, no. 3: 62. https://doi.org/10.3390/cancers10030062
APA StyleSharma, G. G., Mota, I., Mologni, L., Patrucco, E., Gambacorti-Passerini, C., & Chiarle, R. (2018). Tumor Resistance against ALK Targeted Therapy-Where It Comes From and Where It Goes. Cancers, 10(3), 62. https://doi.org/10.3390/cancers10030062